
Abstract
Drought is an important abiotic stress that limits the production of tea in different regions of the world. Young roots of tea are responsible for nutrient and water uptake; hence, they are the first tissues to perceive drought stress. In this study, a forward suppression subtractive hybridization library was constructed from the tender roots of drought-tolerant tea (Camellia sinensis (L.) O. Kuntze) cultivar (TV-23) subjected to 21 days of drought stress. A total of 572 quality expressed sequence tags were generated by sequencing of 1,052 random clones which have resulted to 246 unigenes comprising 54 contigs and 192 singlets. The unigenes were assigned to various functional categories, i.e. cellular components, biological processes and molecular functions as defined for the Arabidopsis proteome. There were 13.04% of differentially regulated genes that have been associated to various stresses. A total of 123 putative drought-responsive genes were identified which include candidate genes of ubiquitin-proteasome, glutathione metabolism and sugar metabolism pathways and several transcription factors. In order to determine the possible expression, 10 genes associated to drought-responsive pathways were further analysed by reverse transcription polymerase chain reaction. This study provides a basis for studying the drought tolerance mechanism of this important commercial crop which will also be a valuable resource for the functional genomics study of woody plants in future.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Tea [Camellia sinensis (L.) O. Kuntze] is one of the most important plantation crops grown worldwide for the production of popular non-alcoholic beverage called ‘tea’. Since most of the world’s tea-growing areas are prone to drought, the tea plant is often subjected to water-deficit stress (Mondal 2008). Drought is the most important recurrent limiting factor of tea cultivation in India and other tea-growing countries which incurred around 40% of crop loss (Handique 1992). It severely affects the growth, crop yield and several biochemical processes of tea plantations (Zhu 2002). Although conventional breeding and propagation techniques contributed significantly to varietal improvement for the last several decades, yet due to the limitations of conventional breeding and lack of a distinct mutant, application of biotechnology becomes an alternative approach (Mondal et al. 2004).
Plant adaptations to harsh environmental conditions include synthesis of compatible solutes, exportation and compartmentalisation of ions and scavenging of reactive oxygen species (ROS) (Zhu 2002). These adaptations involve the expression of cascade of genes and interactions among their gene products (Shinozaki and Yamaguchi-Shinozaki 2007). There is very little information available on drought stress responses of tea plant at the molecular level which was basically studied in leaves. It has been reported that there are three drought-modulated expressed sequence tags (ESTs), namely dr1, dr2 and dr3 using differential display of mRNA (Sharma and Kumar 2005). The crop is being studied at the genomics level for secondary metabolites (Park et al. 2004), low temperature stress (Wang et al. 2009), transcriptome profiling (Shi et al. 2011), identification of conserved miRNAs (Das and Mondal 2010) and so on. However, no information exists on the drought stress responses of roots. Particularly in tea plants, young roots of current year growth play an important role for water and nutrient uptake (Konwar 2004). Since the roots are the primary site involved in the perception of drought stress, as such they are assumed to have a crucial role to trigger drought tolerance mechanism. The transcriptional changes can result in successful adaptations leading to stress tolerance by regulating gene expression and signal transduction in the stress response (regulatory proteins) or by directly protecting the plant against drought stress (functional proteins) (Shinozaki and Yamaguchi-Shinozaki 2007).
Recent efforts have demonstrated that analysis of ESTs is an appropriate strategy for identifying genes involved in specific biological functions in model plants (Yamamoto and Sasaki 1997) and even in non-model plants in which genomic data are not available (Xuxia et al. 2011; Xu et al. 2011b; Yu et al. 2011). The suppression subtractive hybridization (SSH) technique enables specific cloning of ESTs representing genes that are differentially expressed in different mRNA populations and isolates genes without prior knowledge of their sequence or identity (Diatchenko et al. 1996; Jin et al. 2011). Moreover, the availability of databases of known genes and proteins and gene ontology (GO) annotation provides an opportunity to predict the functions of newly isolated putative gene sequences (Ashburner et al. 2000). The present study was conducted for the identification of gene profiles in roots of tea plants under drought stress by analysing SSH cDNA library. Bioinformatics analyses of the ESTs allowed us to detect a wide spectrum of gene expression in tea roots that enabled the plant to withstand drought conditions. The differentially regulated genes in tea roots are potential candidate genes for engineering drought-tolerant crops and for revealing the molecular mechanisms of adaptation of plantation crops to water-deficit stress. Furthermore, possible expression patterns of superoxide dismutase (SOD), glutathione reductase (GR), glutathione S-transferase (GST), trehalose-6-phosphate synthase (TPS), calmodulin-like protein (CML), f-box protein, cullin, arginine decarboxylase (ADC), mitogen-activated protein kinase (MAPK) and S phase kinase-associated protein 1 (Skp-1) genes were confirmed under drought stress.
Materials and Methods
Plant Materials, Drought Stress Induction and Sampling
Two-year-old vegetatively propagated well-rooted tea plantlets (~36-in. height) of TV-23 cultivar (drought-tolerant) (Konwar 2004; Das 2012) were planted in earthen pots (12-in. diameter) under controlled greenhouse conditions (at a light intensity of 300 μmol m−2 s−1 and 25 ± 2°C with relative humidity of 65–70%) at Cooch Behar, West Bengal, India. Drought sensitivities of the cultivar were confirmed by breeders and commercially established on the basis of yield stability during drought periods over many years. Initially, the plantlets were watered regularly for 2 months to establish new growth, and subsequently, water was withheld in experimental ones for drought stress induction. Soil moisture content (SMC), relative water content (RWC) and physiological status of the plants were evaluated in every 2/3-day interval from the day of withholding water according to Black (1965) and Barr and Weatherley (1992) and using a Photosynthesis System (LCpro+, ADC, UK), respectively. On the 21st day of stress induction, tender roots were collected for the isolation of RNA.
Construction of SSH Library and Sequencing
Total RNA was isolated from collected roots (100 mg) of both drought-induced and well-watered plants (control) using a modified protocol based on sodium dodecyl sulphate-LiCl (Das 2012). Different aliquots of total RNA were pooled in order to extract the mRNA by PolyATtract® mRNA Isolation System (Promega, USA). The mRNA pool was precipitated with 0.1 volume of 3 M ammonium acetate (pH 5.2) and 1.0 volume of isopropanol at −70°C for 3 h. The concentrated mRNAs (4 μg) were used for the synthesis of cDNAs. A forward SSH library was constructed using the cDNAs of drought-induced plant as ‘tester’ and cDNAs of control plant as ‘driver’ according to the user manual of PCR-SelectTM cDNA subtraction kit (Clontech, USA). The subtracted cDNAs were cloned into pGEM®-T easy vector (Promega, USA) and transformed into electrocompetent DH10β Escherichia coli cells (Invitrogen). Plasmid DNAs were purified from the overnight grown transformed E. coli cells using the manufacturer’s protocol of GenElute plasmid miniprep kit (Sigma-Aldrich). A cycle sequencing PCR was performed using M13 forward primer (3.2 pmol) and Big Dye Terminator v 3.1 (Applied Biosystems) taking the recombinant plasmids as template. Single-pass sequencing was conducted at the 5′ end of cDNA clones with Genetic Analyzer 3130xl (Applied Biosystems).
Clustering and Functional Annotation
The raw ESTs were cleaned by removing the vector and adaptor sequences present at both 5′ and 3′ ends using Sequence scanner v 3.1 (Applied Biosystems). Trimmed sequences with no more than 178 bp in length were selected for BLASTn and BLASTx analyses against the non-redundant nucleotide and protein sequence databases of NCBI (www.blast.ncbi.nlm.nih.gov/Blast.cgi), respectively at an e-value threshold of 1e−05. Sequences with no significant hits were considered as novel. Sequences that passed through our set quality parameters were clustered using the dirty data algorithm considering ambiguous base calls for poor matches to exact base with the following criteria: (a) gap optimisation for small inserts and double-called bases through ReAligner algorithm, (b) prefer 3′ gap replacement, (c) minimum overlap of 20 bp and (d) minimum of 85% match using Sequencher 4.1 (Gene Codes Corporation). Both contigs and singletons were altogether defined as unigenes. Further, the unigenes were annotated for various functional properties.
Due to unavailability of a genome sequence for tea, we conducted GO analysis of unigenes against Arabidopsis genome by using TAIR 9 transcripts database (www.arabidopsis.org/index.jsp) through WU-BLAST. The corresponding locus hits of unigenes were selected at an e-value threshold of 1e−10 and further categorised on the basis of GOslim categories of TAIR (www.arabidopsis.org) based on: annotations to terms in GOslim category / total annotations to terms in this ontology × 100.
Identification of putative drought-responsive genes was performed on the basis of BLAST hits in non-redundant databases of NCBI and our knowledge on physiology and biochemistry. Wherever two or more genes were found in a single pathway, the corresponding pathway genes were analysed using the KEGG database (www.genome.jp/kegg). For the comparison of tissue-specific gene expression in roots and leaves, the available ESTs of tea leaves generated under drought stress were downloaded from NCBI (GenBank accession numbers GH623813–GH623574, GH738509–GH738781, by June 2011). The downloaded ESTs were vector-cleaned and passed through the quality parameters ((>100 bp length and e-value < 1e–05). Finally, sequences that passed through the quality parameters were clustered and used as query for searching the root’s homologue on the basis of bit score and identity percentage (minimum of 85%) by standalone BLASTn program of NCBI (Stephen et al. 1997).
RT-PCR Analysis
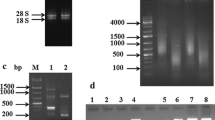
Total root RNAs isolated from control and drought-induced plants were used for reverse transcription polymerase chain reaction (RT-PCR) analysis. Genomic DNA was removed by DNase I. First-strand cDNA was synthesised from each 1 μg of total RNA sample using MMLV reverse transcriptase (Invitrogen, USA) according to the manufacturer’s protocol. The cDNAs were purified using a commercial column (Qiagen). To determine the expression of candidate genes, PCR was performed with 2 μl of the first-strand cDNA template and gene-specific primer pairs. Gene-specific RT-PCR primers were designed with Primer 3.0 (http://frodo.wi.mit.edu/primer3/) according to the EST sequences and were synthesised commercially (Sigma-Aldrich, USA). Tea 26S ribosomal RNA (rRNA) was used as inner control for RT-PCR analysis. All primers for the candidate genes and 26S rRNA are listed in Table 1. General PCR was conducted with the following programme: an initial denaturation at 94°C for 3 min, followed by 24 to 30 cycles with denaturation at 94°C for 30 s, annealing at 61°C (or as required for the specific primer pairs Table 1) for 45 s and extension at 72°C for 1 min, and a final extension at 72°C for 5 min. RT-PCR experiments were repeated three times, and the PCR products (8 μl) were detected by 1% agarose gel in 1× TAE with EtBr and viewed under ultraviolet light.
Results
Response of Tea Plants to Drought Stress Induction
To confirm an appropriate duration of drought stress induction to be applied for stress treatment of plants prior to SSH library construction, SMC, RWC and physiological parameters, i.e. photosynthesis rate and stomatal conductance were monitored till the plants get wilted permanently. The visible wilting symptoms were observed in the plants from the 18th day onwards. As shown in Fig. 1, after 18 days of drought stress induction, SMC and RWC were decreased significantly compared to the control (P < 0.05), while there was no significant difference between the 24th and 27th day of stress induction. The plants showed severe wilting symptoms from the 24th day onwards. After 27 days of drought stress induction, the plants were getting permanently wilted and they could not be revived even after sufficient rewatering. Thus, we decided to induce 21 days of drought stress to the plants for constructing the SSH library. On the 21st day of stress induction, SMC and RWC were found as 7% (−1.2 ± 0.20 MPa) and 63%, respectively (Fig. 1). Moreover, the photosynthesis rate and stomatal conductance were recorded as 8.73 μmol m−2 s−1 and 0.42 mmol m−2 s−1, respectively.

Relative water content (RWC) in leaves and soil moisture content (SMC) in 3-day interval of drought induction from 0 to 27 days. Values are means ± S.E. based on three replicates. Means with the same letters are not significantly different at P < 0.05
Analyses of the ESTs and Functional Annotation
In order to identify up-regulated genes during drought stress, a forward SSH library was constructed with 21 days of drought-induced plant cDNAs as the ‘tester’ set and control plant cDNAs as the ‘driver’ set. A total of 1,052 bacterial colonies were randomly picked from the library and sequenced. The analysis of cDNA sequences showed that 572 clones were passed through the set quality parameters and found to contain inserts ranging from 178 to 832 bp. The total of 572 up-regulated ESTs assembled into 246 unigenes comprises 54 contigs and 192 singlets. The contigs were composed of 2 to 201 individual ESTs (Fig. 2). Analyses of the SSH library are summarised in Table 2. BLASTx analysis of deduced amino acid sequences was performed to identify any encoded proteins. Using BLASTx, out of 572 ESTs, 248 (43.36%) were identified encoding different enzymes and functional proteins, 207 (36.19%) similar to unknown proteins, 40 (6.99%) similar to predicted proteins, 22 (3.85%) homologue to small heat shock proteins, 21 (3.67%) encoding molecular chaperone and 34 (5.94%) ESTs failed to match any sequences in the public database (Supplementary material 1/ESM1). There were most abundant ESTs for ribosomal protein, cysteine protease and chaperone followed by heat shock proteins (Table 3). There were 14 non-redundant ESTs (5.69%) without significant hits in Arabidopsis genome. The nucleotide sequence data of our EST collection have been deposited to the GenBank dbEST database under the accession numbers from GT968791 to GT969386, GW316843 to GW317159 and GW315010 to GW315149.

Distribution and numbers of assembled ESTs
For the comparison of in silico tissue-specific gene expression under drought stress, 665 available drought-responsive ESTs of tea leaf were downloaded from the GenBank dbEST database. After vector trimming and removing of low quality sequences, finally, 321 ESTs were considered for further analysis. The ESTs were clustered into 116 unigenes containing 45 contigs and 71 singlets. Out of 116, there were 12 unigenes found to be homologue with root-specific unigenes (Fig. 3). The homologue unigenes were encoding dehydrin, sugar transporter, histone, unknown proteins as well as predicted proteins (Supplementary material 2/ESM2).

Venn diagram of drought-induced genes in roots and leaves. The genes were put in three categories: drought induced in roots (total unigenes 246), leaves (total unigenes 116) and in both (total unigenes 12)
A total of 187 (76%) differentially expressed unigenes could be assigned to three broad functional categories, i.e. cellular components, biological processes and molecular functions as established for the Arabidopsis proteome (Fig. 4). In the cellular components category, genes assigned to the ‘other intracellular components’ category accounted for the largest group (19.65%), followed by ‘other cytoplasmic components’ associated genes (16.78%), and genes of ‘endoplasmic reticulum related’ represented least in the group (0.88%). In the biological process category, the largest group was ‘other cellular processes’ (20.83%) followed by other metabolic process (18.28%), and the signal transduction-associated genes were the least in the group (0.95%). Interestingly, 13.04% of genes represented the ‘stress responsive’ category, while 11.45% of genes were in the ‘abiotic or biotic stimulus’ responsive group. In the molecular functions category, the highest percentage was covered by ‘other binding’ related genes (12.90%) followed by ‘transferase activity’ related genes (12.20%), and the ‘transcription factor’ activity-associated genes were the least in the group (2.44%). Based on GO annotation, we found ESTs that are associated to ubiquitin-proteasome degradation pathways, glutathione synthesis and metabolism pathways as well as genes associated to sugar synthesis, transport and metabolism (Table 4; Fig. 5).

Gene ontology (GO) analysis of C. sinensis unigenes. The relative frequencies of GO hits for unigenes assigned to the GO functional categories: cellular components, biological processes and molecular functions, as defined for the Arabidopsis proteome

Scheme of known major metabolic pathways representing drought-induced ESTs: 1 = GR (GT969327), 2 = GST (GT969164), 3 = ECS (GT969335), 4 = CML (GT969228), 5 = calcium-binding EF hand family protein (GT969175), 6 = MAPK (GT969241), 7 = LEA (GT969372), 8 = dehydrin (GT969343), 9 = HSP 83 (GT969070), 10 = Mt HSP (GT969378), 11 = HSP 70 (GT969253), 12 = SOD (GT969381), 13 = POX (GT969365), 14 = thioredoxin peroxidase (GT969216), 15 = zinc finger protein (GT969021), 16 = transcription initiation factor IIB (GT969286), 17 = global transcription factor group (GT969161), 18 = ethylene-responsive element-binding protein (GT969363), 19 = NAM-like protein (GT969278), 20 = TPS (GT969159), 21 = sugar transporter (GT969292), 22 = hexose transporter (GT969219), 23 = UDP-glucose 4-epimerase (GT969354), 24 = glucose-6-phosphate (GT969341), 25 = glyceraldehyde 3-phosphate dehydrogenase (GT969248), 26 = phosphoenolpyruvate carboxykinase (GT969259), 27 = ADC (GT969323), 28 = ubiquitin (GT969157), 29 = F-box (GT969218), 30 = skp1 (GT969255), 31 = cullin (GT969138 and GT969139), 32 = ring finger (GT969226), 33 = E2 (GT969274), 34 = polyubiquitin (GT969158)
Comparison of Tea Root ESTs with Published Abiotic Stress-Responsive Genes
Drought, low temperature and high salinity are common stress conditions that adversely affect plant growth and crop production (Xiong et al. 2002). In our collection of nucleotide sequences, there are ESTs having homology to transcripts known to be associated with these abiotic stress tolerances. In order to identify drought-responsive gene profiles in tea roots, we presented 123 putative drought-responsive genes in Table 4. It includes a diverse set of genes involved in synthesis, metabolism, signalling and transcription (Fig. 5). There are transcripts related to polyamine synthesis (arginine decarboxylase); sugar synthesis, transportation and metabolism (trehalose-6-phosphate synthase, UDP-glucose 4-epimerase, sugar transporter, hexose transporter, glucose-6-phosphate, glyceraldehyde 3-phosphate dehydrogenase, phosphoenolpyruvate carboxykinase); glutathione synthesis and metabolism (γ-glutamylcysteine synthetase and glutathione reductase); and molecular chaperone and HSPs (late embryogenesis abundant protein, dehydrin, heat shock protein 70, heat shock protein 80 and mitochondrial heat shock protein). These genes are involved in reducing the initial impact of water-deficit stress. There are also some genes encoding proteins to detoxify the ROS generated by drought stress, such as SOD, peroxidase (POX), thioredoxin peroxidase and probable phospholipid hydroperoxide, GST. Several genes involved in proteasome degrading pathways were also reported such as ubiquitin, f-box protein, skp1, cullin, ring finger protein, enzyme 2 and polyubiquitin. These genes are mostly involved in the SCF complex-mediated protein degradation through ubiquitination. Leaf senescence-associated genes such as ACC oxidase were also found in our SSH library.
In addition, the EST set contains some transcripts encoding proteins with signalling functions such as CML and MAPK. The CML functions as transducers of Ca2+ signals in plants. It provides a sense to activate MAPKs, a family of ser/thr protein kinases functioning in many signal transduction pathways (Ranty et al. 2006; Xu et al. 2010). In our SSH library, other transcripts homologue to signalling proteins are protein kinase APK1A, chloroplast precursor, NAD + kinase, cyclin-dependent kinase 8 (cdk8) and nucleoside diphosphate kinase. In general, the signalling molecules act by interaction with nuclear transcription factors which induce the expression of a specific set of genes (Sahi et al. 2006).
In our SSH library, several transcripts were found encoding transcription factors (TFs). Transcription factors are critical regulators of the changes in gene expression and abiotic stress responses. Ethylene-responsive factors (ERFs) belong to the large APETALA2 (AP2)/ERF transcription factor superfamily that is unique to plants (Song et al. 2005). NAC family of transcription factor represents one of the largest families of transcription factor in plants (Hu et al. 2006). The ERF and NAC domain protein factors were found to be induced in our library. The other TFs found in our SSH library were global transcription factor, NAM-like proteins, YIPF1 and arginine-serine-rich factors.
RT-PCR Analysis for the Confirmation of Differential Expressed Genes
In order to determine the validity of the library with respect to drought-induced expression, 10 interested genes from different drought-responsive pathways were analysed by RT-PCR. RNA was isolated from the roots of drought-induced and control plants. The amount of cDNA was adjusted to yield equivalent levels of 26S rRNA amplification. RT-PCR results showed that the expression levels of seven candidate genes were more highly expressed in drought-stressed roots (Fig. 6). The GST and GR, glutathione synthesis and metabolism pathway-associated genes were found to be up-regulated. The genes involved in trehalose and polyamine synthesis, namely TPS and ADC, were also found to be up-regulated. The other up-regulated genes include antioxidative enzyme, SOD; calcium sensor, CML; and regulatory protein, MAPK. However, the ubiquitination pathway-associated genes, i.e. F-box, Cullin and Skp-1 were found to be down-regulated (Fig. 6). It was concluded that overall, there was a good agreement between the SSH library data and the RT-PCR results.

Verification of SSH library results by RT-PCR analysis. The plants were drought-induced for 21 days. 26S rRNA used as loading control
Discussion
Drought stress is one of the major factors limiting the growth and crop yield of tea plantations in India and other tea-growing countries. Drought is a complex quantitative trait regulated by a large number of genes (Shinozaki and Yamaguchi-Shinozaki 2007). Exploitation and enrichment of drought tolerance-associated genes are very important to accelerate research in this area. Although several studies have been made to explore the transcripts in the leaves of tea plants, no information is available on the expression of genes involved in responses to drought stress in roots. The SSH approach has been proven to be a powerful tool to enrich the differentially expressed genes (Diatchenko et al. 1996). This article reports the construction of subtracted cDNA library from root tissues of tea plant under drought stress and the analysis of 572 putative drought-responsive genes. The differential expression of 10 genes of interest upon drought stress was further confirmed by RT-PCR analysis. The study was focussed on generating the gene profiles at the early stages of wilting of the plants due to important roles of early wilting-responsive genes in mediating the effects of drought stress.
Drought stress imposes osmotic stress, oxidative stress and ionic stress to plants. There are cascades of gene networks involved in stress perception, signal transduction, transcriptional control and scavenging of ROS (Zhu 2002). A number of genes get up-regulated or down-regulated during drought by increasing or decreasing the levels of several metabolites and proteins to fight against cellular damages. Under drought stress, genes encoding key enzymes regulating biosynthesis of compatible solutes such as amino acids (e.g. proline) and amines (e.g. polyamines) and a variety of sugars and sugar alcohols (e.g. trehalose, galactinol) play important roles to reduce the damage to plants. Several of these genes introduced into transgenic plants could demonstratively enhance drought stress tolerance (Zhang et al. 2004; Umezawa et al. 2006). In the present study, genes of trehalose (trehalose-6-phosphate synthase), polyamine synthesis (arginine decarboxylase), sugar transporters (hexose transporter, sugar transporter) and several genes of glycolysis pathway (glucose-6-phosphate, glyceraldehyde 3-phosphate dehydrogenase, phosphoenolpyruvate carboxykinase) were induced under drought stress. The trehalose-6-phosphate synthase gene was reported for conferring drought tolerance in cotton (Kosmas et al. 2006). An arginine decarboxylase gene (PtADC) from Poncirus trifoliata demonstrated abiotic stress tolerance and promotes primary root growth in Arabidopsis (Wang et al. 2010). Prior analysis also demonstrated that drought stress induced the up-regulation of genes encoding enzymes involved in the glycolytic pathway in tobacco and foxtail millet (Rizhsky et al. 2002; Zhang et al. 2007). One of the possible reasons for the up-regulation of genes involved in glycolysis may be to generate more ATP, which is the primary source of energy for cellular metabolism.
Plants have evolved a complex signalling network for the perception of and responses to different abiotic stresses. A generic signal transduction pathway starts with signal perception, followed by the generation of second messengers. Second messengers can modulate intracellular Ca2+ levels, often initiating a protein phosphorylation cascade that finally targets proteins directly involved in cellular protection or transcription factors controlling specific sets of stress-regulated genes (Xiong et al. 2002). Ca2+ signals are deciphered by various Ca2+-binding proteins that convert the signals into a wide variety of biochemical changes. The calmodulin-like proteins (CMLs) are a group of calcium-binding proteins with two to six predicted EF hand motif. There are 50 CML genes identified in Arabidopsis genome (Ranty et al. 2006). Recently, a novel calmodulin-like gene, OsMSR2, was demonstrated for drought and salt tolerance in Arabidopsis (Xu et al. 2011a). A number of plant protein kinases have now also been found to be activated by drought stress (Zhu 2002). MAPK was serine/threonine protein kinase, which was known to be one of the major pathways by which extracellular signals such as growth factors, hormones and abiotic stress stimuli were transduced into intracellular responses in plants (Munnik and Meijer 2001). An enhanced tolerance to drought and salt stress was reported by overexpressing a MAPK gene, CsNMAPK, in tobacco plant (Xu et al. 2010). In our SSH library, CLM, MAPK, protein kinase APK1A, NAD + kinase, cdk8 and nucleoside diphosphate kinase were induced indicating that these genes may play important roles in transduction of signalling during drought stress in tea plants. Furthermore, the CLM and MAPK genes were also found to be up-regulated.
The ATPases, water channel proteins and ion transporters play a crucial role in maintaining ion homeostasis under drought stress. The proton electrochemical gradient formed by the vacuolar ATPase provides the primary driving force for the transport of numerous ions and metabolites against their electrochemical gradients which is the fundamental requirement of many cellular processes, such as osmoregulation, signal transduction and metabolic regulation (Santos 2006). Late embryogenesis abundant (LEA) proteins are involved in protecting from damage caused by environmental stresses, especially drought (Goyal et al. 2005). Heat-shock proteins (Hsps) or chaperones such as Hsp70 and Hsp90 are responsible for protein folding, assembly, translocation and degradation in many normal cellular processes; stabilise proteins and membranes; and are actively involved in protein refolding under stress conditions including drought (Wang et al. 2004; Liu et al. 2011). Metallothionein, a superfamily of low molecular weight proteins that are involved in metal detoxification and scavenging of oxygen-free radicals, can decrease injury caused due to drought stress (Talame et al. 2007). Phospholipase D and its products, phosphatidic acid, exert their effect by functioning in signal transduction cascades and by influencing the biophysical state of lipid membranes. It has been implicated in multiple plant stress responses in plants including drought (Bargmann and Munnik 2006). In our SSH library, vacuolar ATPase, dehydrin, LEA, Hsp70, Hsp90, metallothionein and phospholipase D were obtained indicating the roles of these genes in maintaining ion homeostasis in tea plants under drought stress.
Plants have developed an efficient antioxidative system comprising enzymatic and non-enzymatic components to fight against ROS during stress. The SOD is a well-studied antioxidative enzyme providing the first line of cellular defence against oxidative stress by early scavenging of superoxide radicals and then converting them to hydrogen peroxide (Chatzidimitriadou et al. 2009). The peroxidases catalyse the conversion of hydrogen peroxide to water using ascorbate, glutathione or thioredoxin as substrate (Rossel et al. 2006). In our SSH library, SOD, peroxidase and thioredoxin peroxidase were obtained. Expression of SOD demonstrated higher drought tolerance and shoot regeneration in transgenic pepper (Chatzidimitriadou et al. 2009). Glutathione (GSH) is an important non-enzymatic antioxidant which reduces the active oxygen radicals generated due to stress (Shao et al. 2008). Glutathione is synthesised from its constituent amino acids in an adenosine triphosphate (ATP)-dependent, two step reaction catalysed by the enzymes γ-glutamylcysteine synthetase (γ-ECS) and glutathione synthetase. Glutathione reductase (GR) mediates the reduction of GSSG to GSH by using NADPH as an electron donor, and thus a highly reduced state of GSH/GSSG ratio is maintained during stress (Carvalho and Contour-Ansel 2008). Overexpression of GR for conferring drought tolerance was reported earlier in cowpea and common bean (Carvalho and Contour-Ansel 2008; Torres-Franklin et al. 2008). Glutathione S-transferase (GST) detoxifies organic hydroperoxides generated under dehydration stress and removed by covalently linking glutathione to a hydrophobic substrate, forming less reactive and more polar glutathione S-conjugate (Neuefeind et al. 1997). Overexpression of the GST gene under drought condition was demonstrated in transgenic tobacco plant transformed with a GsGST from wild soybean (Ji et al. 2010). The up-regulation of SOD, GR and GST genes in the present investigation corroborated with the earlier reports cited above and suggested the role of enzymatic antioxidants in drought tolerance mechanism in tea plants.
TFs have also been proven quite useful in improving stress tolerance in transgenic plants, through influencing expression of a number of stress-associated target genes (Shinozaki and Yamaguchi-Shinozaki 2007). There are many TFs that have been successfully demonstrated to play key regulatory roles in drought tolerance. Three Arabidopsis NAC genes, ANAC019, ANAC055 and ANAC072, were shown to bind to the promoter region of ERD1 which was characterised as a stress-responsive gene and overexpression of these three genes in Arabidopsis resulted in enhanced tolerance to drought stress (Tran et al. 2004). Transgenic tobacco plants expressing SodERF3 of sugarcane under the transcriptional control of constitutive CaMV 35S promoter showed remarkably enhanced drought tolerance (Trujillo et al. 2009). Overexpression of a NAM, ATAF, and CUC (NAC) transcription factor was reported for the enhancement of drought resistance and salt tolerance in rice (Hu et al. 2006). In our SSH library, the presence of several transcripts of TFs indicates their possible role in drought tolerance mechanism of tea plants.
The response to drought stress involves not only the up-regulation of genes, but also the down-regulation of functional genes. For the last several years, much research has been focussed on how the relevant genes are up-regulated during drought stress. There were 79 genes found to be down-regulated during drought in Arabidopsis (Seki et al. 2002). As such, there were 37 genes found to be down-regulated during drought in foxtail millet (Zhang et al. 2007). In our SSH library, 10 unigenes of proteasome degrading pathways were induced which include ubiquitin conjugating enzyme (E2), ubiquitin, polyubiquitin, ubiquitin ligase (E3), f-box, skp1, cullin, ubiquitin specific protease, 26S proteasome and zinc finger protein. Possible expression of three of them, i.e. f-box, cullin and skp1, was validated and found to be down-regulated. These three genes are the components of the SCF complex of E3 ligase. Out of all the ubiquitin ligases, SCF class of E3 ligases has been thoroughly studied in plants (Ciechanover et al. 2000). However, whether these genes are false positive in our SSH library or play an important role for drought tolerance by down-regulating their expression in tea plants need to be further confirmed. It was reported that transgenic tobacco with wheat (Triticum aestivum) ubiquitin gene (Ta-Ub2) demonstrated to be an effective strategy for enhancing drought tolerance (Guo et al. 2009).
The conclusion of this study is that, in addition to the expression of several well-characterised drought-responsive genes, a high-level constitutive expression of molecular chaperone and heat shock proteins, transcription factor and proteinase inhibitor seems to be a major factor in rendering tea plant resistant to drought. This study is the first global analysis of transcripts in tea under drought stress. The challenge remains to demonstrate which (if any) of these genes has the potential to improve the drought tolerance of plants, or alternatively, if they can be used to develop useful markers for drought resistance in the tea-breeding program. The ESTs identified in this study should provide a useful genomic resource for biologists and plant breeders in developing new strategies for improving drought-tolerant tea cultivar.
Abbreviations
- ESTs:
-
Expressed sequence tags
- ROS:
-
Reactive oxygen species
- RT-PCR:
-
Reverse transcription polymerase chain reaction
- SSH:
-
Suppression subtractive hybridization
- HSPs:
-
Heat shock proteins
References
Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM et al (2000) The gene ontology consortium. Nat Genet 25:25–29
Bargmann BOR, Munnik T (2006) The role of phospholipase D in plant stress responses. Curr Opin Plant Biol 9:515–522
Barr HD, Weatherley PE (1992) A re-examination of the relative turgidity technique for estimating water deficit in leaves. Aust J Biol Sci 15:413–428
Black CA (1965) Methods of soil analysis: part I physical and mineralogical properties. American Society Agronomy, Madison
Carvalho MHC, Contour-Ansel D (2008) GR, beans and drought stress. Plant Signal Behav 3:834–835
Chatzidimitriadou K, Nianiou-Obeidat I, Madesis P, Perl-Treves R, Tsaftaris A (2009) Expression of SOD transgene in pepper confer stress tolerance and improve shoot regeneration. Electron J Biotechnol 12:1–9
Ciechanover A, Orian A, Schwartz AL (2000) Ubiquitin-mediated proteolysis: biological regulation via destruction. Bioessays 22:442–451
Das A (2012) Generation and characterization of expressed sequence tags of tea (Camellia sinensis (L). O. Kuntze). PhD thesis, University of North Bengal, Siliguri, Darjeeling, India
Das A, Mondal TK (2010) Computational identification of conserved microRNAs and their targets in tea (Camellia sinensis). Am J Plant Sci 1:77–86
Diatchenko L, Lau YF, Campbell AP, Chenchik A, Moqadam F, Huang B, Lukyanov S, Lukyanov K, Gurskaya N, Sverdlov ED, Siebert PD (1996) Suppression subtractive hybridization: a method for generating differentially regulated or tissue-specific cDNA probes and libraries. Proc Natl Acad Sci 93:6025–6030
Goyal K, Walton LJ, Tunnacliffe A (2005) LEA proteins prevent protein aggregation due to water stress. Biochem J 388:151–157
Guo P, Baum M, Grando S, Ceccarelli S, Bai G, Li R, Korff MV, Varshney RK, Graner A, Valkoun J (2009) Differentially expressed genes between drought-tolerant and drought-sensitive barley genotypes in response to drought stress during the reproductive stage. J Exp Bot 60:3531–3544
Handique AC (1992) Some silent features in the study of drought resistance in tea. Two Bud 39:16–18
Hu H, Dai M, Yao J, Xiao B, Li X, Zhang Q, Xiong L (2006) Overexpressing a NAM, ATAF, and CUC (NAC) transcription factor enhances drought resistance and salt tolerance in rice. Proc Natl Acad Sci 35:12987–12992
Ji W, Zhu Y, Li Y, Yang L, Zhao X, Cai H, Bai X (2010) Over-expression of a glutathione S-transferase gene, GsGST, from wild soybean (Glycine soja) enhances drought and salt tolerance in transgenic tobacco. Biotechnol Lett 32:1173–1179
Jin X, Qin J, Wu T, Zhou X (2011) Identification of ethylene-responsive genes in ethrel-treated shoot apices of cucumber by suppression subtractive hybridization. Plant Mol Biol Rep 4:875–884. doi:10.1007/s11105-011-0304-7
Konwar BK (2004) Evaluation and development of planting material. In: Notes on tea management. Tea Research Association, Assam, India, pp 1–6
Kosmas SA, Loukas AAMG, Eliopoulos E, Kaltsikes STPJ (2006) Isolation and characterization of drought-related trehalose 6-phosphate-synthase gene from cultivated cotton (Gossypium hirsutum L.). Planta 223:329–339
Liu CC, Li CM, Liu BG, Ge SJ, Dong XM, Li W, Zhu HY, Wang BC, Yang CP (2011) Genome-wide identification and characterization of a dehydrin gene family in poplar (Populus trichocarpa). Plant Mol Biol Rep. doi:10.1007/s11105-011-0395-1
Mondal TK (2008) Tea breeding. In: Priyadarshan PM, Jain SM (eds) Tropical crops. Springer, Berlin, pp 547–587
Mondal TK, Bhattacharya A, Laxmikumaran M, Ahuja PS (2004) Recent advances in tea biotechnology. Plant Cell Tissue Organ Cult 75:795–856
Munnik T, Meijer HJG (2001) Osmotic stress activates distinct lipid and MAPK signalling pathways in plants. FEBS Lett 498:172–178
Neuefeind T, Reinemer P, Bieseler B (1997) Plant glutathione S-transferases and herbicide detoxification. Biol Chem 378:199–205
Park JS, Kim JB, Haha BS, Kim KH, Ha SH, Kim JB, Kim YH (2004) EST analysis of genes involved in secondary metabolism in Camellia sinensis (tea), using suppression subtractive hybridization. Plant Sci 166:953–961
Ranty B, Aldon D, Galaud JP (2006) Plant calmodulins and calmodulin-related proteins. Plant Signal Behav 1:96–104
Rizhsky L, Liang H, Mittler R (2002) The combined effect of drought stress and heat shock on gene expression in tobacco. Plant Physiol 130:1143–1151
Rossel JB, Walter PB, Hendrickson L, Chow WS, Poole A, Mullineaux PM, Pogson BJ (2006) A mutation affecting ascorbate peroxidase 2 gene expression reveals a link between responses to high light and drought tolerance. Plant Cell Environ 29:269–281
Sahi CH, Singh A, Blumwaldb E, Grover A (2006) Beyond osmolytes and transporters: novel plant salt-stress tolerance-related genes from transcriptional profiling data. Physiol Plant 127:1–9
Santos L (2006) Molecular mechanisms of the AAA proteins in plants. Adv Agril Food Biotechnol 37:1–15
Seki M, Narusaka M, Ishida J et al (2002) Monitoring the expression profiles of 7000 Arabidopsis genes under drought, cold and high-salinity stresses using a full-length cDNA microarray. Plant J 31:279–292
Shao HB, Chu LY, Lu ZH, Kang CM (2008) Primary antioxidant free radical scavenging and redox signaling pathways in higher plant cells. Int J Biol Sci 4:8–14
Sharma P, Kumar S (2005) Differential display-mediated identification of three drought-responsive expressed sequence tags in tea [Camellia sinensis (L.) O. Kuntze]. J Biosci 30:231–235
Shi CY, Yang H, Wei CL, Yu O, Zhang ZZ, Jiang CJ, Sun J, Li YY, Chen Q, Xia T, Wan XC (2011) Deep sequencing of the Camellia sinensis transcriptome revealed candidate genes for major metabolic pathways of tea-specific compounds. BMC Genomics 12:131. doi:10.1186/1471-2164-12-131
Shinozaki K, Yamaguchi-Shinozaki K (2007) Gene networks involved in drought stress response and tolerance. J Exp Bot 58:221–227
Song CP, Agarwal M, Ohta M, Guo Y, Halfter U, Wang P, Zhua JK (2005) Role of an Arabidopsis AP2/EREBP-type transcriptional repressor in abscisic acid and drought stress responses. Plant Cell 17:2384–2396
Stephen FA, Thomas LM, Alejandro AS, Jinghui Z, Zheng Z, Webb M, David JL (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25:3389–3402
Talame V, Ozturk NZ, Bohnert HJ, Tuberosa R (2007) Barley transcript profiles under dehydration shock and drought stress treatments: a comparative analysis. J Exp Bot 58:229–240
Torres-Franklin ML, Contour-Ansel D, Zuily-Fodil Y, Pham-Thi AT (2008) Molecular cloning of glutathione reductase cDNAs and analysis of GR gene expression in cowpea and common bean leaves during recovery from moderate drought stress. J Plant Physiol 165:514–521
Tran LS, Nakashima K, Sakuma Y et al (2004) Isolation and functional analysis of Arabidopsis stress inducible NAC transcription factors that bind to a drought responsive cis-element in the early responsive to dehydration stress 1 promoter. Plant Cell 16:2481–2498
Trujillo L, Menendez C, Ochogavia ME, Hernandez I, Borras O, Rodriguez R, Coll Y, Arrieta JG, Banguela A, Ramirez R, Hernandez L (2009) Engineering drought and salt tolerance in plants using SodERF3, a novel sugarcane ethylene responsive factor. Biotechnologia Aplicada 26:168–171
Umezawa T, Fujita M, Fujita Y, Yamaguchi-Shinozaki K, Shinozaki K (2006) Engineering drought tolerance in plants: discovering and tailoring genes unlock the future. Curr Opin Biotechnol 17:113–122
Wang J, Sun PP, Chen CL, Wang Y, Fu XZ, Liu JH (2010) An arginine decarboxylase gene PtADC from Poncirus trifoliata confers abiotic stress tolerance and promotes primary root growth in Arabidopsis. J Exp Bot 62:2899–2914
Wang L, Li X, Zhao Q, Jing S, Chen S, Yuan H (2009) Identification of genes induced in response to low-temperature treatment in tea leaves. Plant Mol Biol Rep 27:257–265
Wang W, Vinocur B, Shoseyov O, Altman A (2004) Role of plant heat-shock proteins and molecular chaperones in the abiotic stress response. Trends Plant Sci 9:244–252
Xiong L, Schumaker KS, Zhu JK (2002) Cell signaling during cold, drought, and salt stress. Plant Cell 14(Suppl):S165–S183. doi:org/cgi/doi/10.1105/tpc.000596
Xu GY, Rocha PSCF, Wang ML, Xu ML, Li Luo-Ye, Zhu YX, Xia X (2011a) A novel rice calmodulin-like gene, OsMSR2, enhances drought and salt tolerance and increases ABA sensitivity in Arabidopsis. Planta 234:47–59
Xu H, He X, Wang K, Chen L, Li K (2011b) Identification of early nitrate stress response genes in spinach roots by suppression subtractive hybridization. Plant Mol Biol Rep. doi:10.1007/s11105-011-0376-4
Xu HN, Li KZ, Yang FJ, Shi QH, Wang XF (2010) Overexpression of CsNMAPK in tobacco enhanced seed germination under salt and osmotic stresses. Mol Biol Rep 37:3157–3163
Xuxia W, Jie C, Bo W, Lijun L, Hui J, Diluo T, Dingxiang P (2011) Characterization by suppression subtractive hybridization of transcripts that are differentially expressed in leaves of anthracnose-resistant ramie cultivar. Plant Mol Biol Rep. doi:10.1007/s11105-011-0376-4
Yamamoto K, Sasaki T (1997) Large-scale EST sequencing in rice. Plant Mol Biol 35:135–144
Yu S, Zhang F, Yu Y, Zhang D, Zhao X, Wang W (2011) Transcriptome profiling of dehydration stress in the Chinese cabbage (Brassica rapa L. ssp. pekinensis) by tag sequencing. Plant Mol Biol Rep. doi:10.1007/s11105-011-0313-6
Zhang J, Liu T, Fu J, Zhu Y, Jia J, Zheng J, Zhao Y, Zhang Y, Wang G (2007) Construction and application of EST library from Setaria italica in response to dehydration stress. Genomics 90:121–131
Zhang JZ, Creelman RA, Zhu JK (2004) From laboratory to field. Using information from Arabidopsis to engineer salt, cold, and drought tolerance in crops. Plant Physiol 135:615–621
Zhu JK (2002) Salt and drought stress signal transduction in plants. Annu Rev Plant Biol 53:247–273
Acknowledgements
We thank Prof. Swapan Kumar Ghosh of Uttar Banga Krishi Viswavidyalaya for encouraging us in pursuing the work and Dr. Mridul Hazarika, Director, Tocklai Experimental Station, Tea Research Association, Jorhat, Assam (India) for providing us the sequencing facility. We also thank Mr. Kamal Das for his technical help. The work was funded by the Department of Biotechnology, Govt. of India, New Delhi.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Das, A., Das, S. & Mondal, T.K. Identification of Differentially Expressed Gene Profiles in Young Roots of Tea [Camellia sinensis (L.) O. Kuntze] Subjected to Drought Stress Using Suppression Subtractive Hybridization. Plant Mol Biol Rep 30, 1088–1101 (2012). https://doi.org/10.1007/s11105-012-0422-x
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11105-012-0422-x