
Abstract
Jatropha (Jatropha curcas) and castor bean (Ricinus communis) possess several taxonomic similarities, and their seeds contain a high proportion of oil (up to 40%) which has been used in various industrial products, including diesel oil. Thirty-two candidate genes responsible for fatty acid biosynthesis were identified in the castor bean genome sequence. Testing of 48 primer pairs from candidate gene regions, including 12 SSRs from castor bean on 54 genotypes of J. curcas, 65% amplified successfully on Jatropha out of which 20% showed polymorphisms. Jatropha genotypes, categorized for oil content, were used in association analysis of candidate gene regions with high oil content. One marker–trait association for the oil trait was identified. Stearoyl desaturase amplicon (700 bp) consisting of intron and exon (P = 0.00013) showed association with high oil content in Jatropha genotypes. Sequencing of the 1.3-kb amplicon, including the 700-bp fragment of stearoyl desaturase, which had shown association with the high oil content, revealed SNPs in the exonic region. The SNPs resulted in substitution of leucine with glutamine in the open reading frame of stearoyl desaturase of low oil content genotypes. The molecular marker is expected to be useful in marker-assisted breeding of high oil content genotypes in Jatropha.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
In the present age of depletion of fossil fuels along with its environmental concerns, the search for alternate sources of fuels has gathered momentum in different parts of the world. Jatropha curcas is a promising energy crop with more than 40% oil content (Makkar et al. 1997; Divakara et al. 2010) in its seeds. The Jatropha oil contains 21% saturated fatty acids and 79% unsaturated fatty acids (palmitic acids 4.2%, stearic acid 6.9%, oleic acid 43.1%, linoleic acid 34.3%, and others 1.4%) (Akbar et al. 2009). J. curcas is native to tropical America with more than 200 species that are widely distributed in the tropics with a promise to be used as an oil crop for biodiesel. RAPD and ISSR analysis have shown narrow genetic base in J. curcas (Basha and Sujatha 2007; Tanya et al. 2011). Castor bean (Ricinus communis) is a plant species, which is taxonomically related to J. curcas such that both belong to Euphorbiaceae. The seeds of both contain a similar toxic protein, and more importantly, the seed oil from both the species is used in the production of biodiesel. The high level of synteny was reported at genomes level between these two plant species (Sato et al. 2011) which can be exploited to develop anchor markers by utilizing genome resources of castor bean.
Comparative genomics has been used to understand gene organization and genome evolution in different organisms (Keller and Feuillet 2000; Ellwood et al. 2008). Crop improvement programs can use comparative genetics to transfer information about genes from model species to their species of interest, to identify the genes controlling traits of interest, and to assess within-species allelic diversity so that the best alleles can be identified and assembled in superior varieties (Sorrells et al. 2003; Heesacker et al. 2009).
Candidate genes provide a valuable resource for mapping in various plants and further germplasm analysis using association genetics. Significant associations between candidate genes and traits of interest have been reported (Wilson et al. 2004; Casa et al. 2008; Cuesta-Marcos et al. 2010). Association mapping has been used to identify genes of interest in many plant species with varying degrees of success (Wilson et al. 2004; Aranzana et al. 2005; Breseghello and Sorrells 2006). Compared to the conventional linkage mapping, linkage disequilibrium (LD)-mapping, using the nonrandom associations of loci in haplotypes, is a powerful high-resolution mapping tool for complex quantitative traits. One difficulty, however, in applying association methods is that LD can be present because of genetic drift, selection, or population admixture. Thus, as sometimes seen in human populations, LD can contain the confounding effect of population substructure, resulting in a high frequency of false positive associations (Lander and Schork 1994). To control for this type of structure, Pritchard et al. (2000) developed a statistical approach that assigns the membership to various subpopulations by determining the amount of genetic correlation based on unlinked, random markers. Thornsberry et al. (2001) adapted Pritchard’s approach for use with quantitative variation and then successfully applied it to the evaluation of maize flowering time. By including estimates of population structure in this analysis, the risk of obtaining false positive associations was reduced (Thornsberry et al. 2001). The advantages of population-based association study, utilizing a sample of individuals from the germplasm collections or a natural population over traditional QTL-mapping in biparental crosses, primarily are due to the absence of need for the development of expensive and tedious biparental populations that saves time and is cost-effective.
The present study reports identification of castor bean orthologous candidate gene-based markers and their association with high oil content in J. curcas.
Materials and Methods
In Silico Identification of Candidate Genes for Fatty Acid Biosynthesis in Castor Bean
Database search (BLASTn) was performed using previously identified fatty acid biosynthesis gene sequences from Arabidopsis, Brassica, Medicago, sunflower, soybean, and cotton available in GenBank. The nucleotide sequences were used as query sequences against castor bean genome sequence in The Institute of Genomic Research (TIGR) castor bean genome database (~400 Mb) consisting of 25,828 contigs (4X coverage) (http://castorbean.tigr.org/). Castor bean genome contig sequences showing identity to the fatty acid biosynthesis gene sequences from other plants (Arabidopsis, Brassica, Medicago, sunflower, soybean, and cotton) were downloaded and annotated for open reading frames, including the 5′ and 3′UTRs using gene prediction algorithms of FGenesH (http://sun1.softberry.com/berry.phtml?topic=fgenesh&group=programs&subgroup=gfind). The amino acid sequences of ORFs were subjected to BLASTp analysis to determine the identity of identified protein sequences to known fatty acid biosynthesis genes at the National Center for Biotechnology Information (NCBI) (www.ncbi.nlm.nih.gov/BLAST/). The candidate genes had multiple copies in the genome. The critical genes (genes involved in fatty acid biosynthesis in seeds) were, therefore, identified through in silico expression and comparing candidate genes with the critical genes of fatty acid biosynthesis pathway in Arabidopsis, Brassica, soybean, and cotton. The in silico expression status of candidate genes was searched in ESTdb of NCBI at http://www.ncbi.nlm.nih.gov/BLAST and TIGR at http://compbio.dfci.harvard.edu/tgi/cgiin/tgi/Blast/index.cgi. Annotation of candidate genes was checked with the coding DNA sequence (CDS) of fatty acid biosynthesis gene sequences for J. curcas available in GenBank. There are several taxonomic and biochemical similarities between Jatropha and castor bean, and it is assumed that there could be similarities in the number and type of genes involved in the biosynthesis of fatty acids in the seed oil. Forty-eight primer pairs were designed from intron, exon, 5′UTR, and 3′UTR regions of fatty acid biosynthesis genes (Supplementary Table 1).
Gene-Based Marker Designing
Simple sequence repeat (SSR) motifs were identified in the exons, introns, 3′UTR, and 5′UTR regions of candidate genes using SSR identification tool (SSRIT) at (http://www.gramene.org/db/searches/ssrtool). Primer pairs were designed from the flanking sequences of microsatellite repeat motifs using Primer 3.0 (http://frodo.wi.mit.edu/cgi-bin/primer3/primer 3.cgi) (Supplementary Table 2). Primer pairs were also designed from exons, introns, 3′UTR, and 5′UTR regions of fatty acid biosynthesis from the castor bean to test amplification and detection of polymorphisms between high versus low oil content genotypes of Jatropha.
Plant Material and Marker Genotyping
J. curcas genotypes used in the study were obtained from the Himalayan Forest Research Institute, Shimla and the National Bureau of Plant Genetic Resources (NBPGR), New Delhi, India (Supplementary Table 3). A network program run by the Department of Biotechnology, Government of India has identified a collection of genotypes with high versus low oil content in their seeds. Total genomic DNA was isolated from leaves and seeds according to a modified CTAB-based procedure (Murray and Thompson 1980). The quality of DNA was checked on 1% agarose gels. PCR reactions for all the primer pairs were performed in 25 μl reaction volumes. The PCR was performed on 30 ng of genomic DNA with primer pairs, Mg2+, dNTPs (Intron Technologies), and Taq DNA polymerase (Intron Technologies). Amplification programs included 94°C for 5 min, 30 cycles of 94°C for 45 s, annealing temperature of 54.6°C(for gene based primer pairs) and −55.7°C (for SSR primes) for 45 s, 72°C for 2 min, and a final extension of 7 min at 72°C. Ten microliters of each PCR product was mixed with 2 μl of 10X gel loading dye (0.2% bromophenol blue, 0.2% xylene cyanol dye, and 30% glycerol in a TA buffer) and was electrophoresed in a 4% agarose gel prepared in 0.5X Tris–borate–EDTA buffer (0.05 M Tris, 0.05 M boric acid, 1 mM EDTA pH 8.0). The gel was run at a constant voltage of 80 V for 1.5 to 2 h, stained with ethidium bromide, and analyzed using the gel documentation system AlphaImager EP (Alpha Innotech Corp., USA). The amplified products were gel eluted, cloned in pGEM-T vector, and transferred to the DH5α chemically competent cells of Escherichia coli for multiplication. The transformants were selected on the basis of blue/white colonies formed on LA plates containing IPTG, X-gal, and ampicillin. The transformants were confirmed by setting a PCR with T7SP6 universal primer and also with gene specific primer pairs. Isolation of recombinant plasmid DNA was done by using plasmid DNA isolation kits (GenAxy). Ocimum Biosolutions, Hyderabad, India, sequenced PCR products. Sequence data were subjected to homology search by using the default settings of BLASTn with the nonredundant database of GenBank (www.http://www.ncbi.nlm.nih.gov).
Statistical Analysis for Detection of Linkage Disequilibrium
PowerMarker ver. 3.25 (Liu and Muse 2005) and GenAlEx 6.2 (Peakall and Smouse 2006) were used to measure the variability at each locus, the deviation from Hardy–Weinberg equilibrium (HW). Deviations from HW equilibrium and tests for linkage disequilibrium were evaluated using Fisher’s exact tests and sequential Bonferroni corrections.
Principal components analysis (PCA) was conducted using 11 markers and GenAlex 6.2 (Peakall and Smouse 2006) software to structure the genotypes and generate a PC-matrix. Linkage disequilibrium measures r 2 (Hill and Robertson 1968) and absolute D′ (Lewontin 1964) were calculated using the TASSEL software ver. 2.1 (http://www.maizegenetics.net/tassel). Fisher’s exact test was applied to calculate the significance of pairwise LD. For LD mapping, genetic map of Jatropha (Wang et al. 2011) was used. FSTAT (Goudet 1995) was used to estimate pairwise F st values among the resulting structural populations. Analysis of molecular variances (AMOVA) was conducted using GenAlex 6.2 (Peakall and Smouse 2006). Structured associations were calculated by mixed model approach (Yu et al. 2006; Zhao et al. 2007). The estimated Q-matrix was used in the subsequent association analysis, which was carried out by logistic regression in the TASSEL ver. 1.4 software (http://www.maizegenetics.net/tassel). The model used was of the form Y = Xα + Qβ + Zu + ε, with Y as a vector of phenotypes, X as a vector of single locus genotypes that were considered as fixed effects, α as a vector of fixed effects of the n – 1 genotype classes, Q as a matrix of the K – 1 subpopulation ancestry estimates for each individual from STRUCTURE, β as a vector of the fixed effects for each of the K – 1 subpopulations, Z as an identity matrix, u as a matrix of random deviates due to genome wide relatedness (as inferred from K), and ε as a vector of residual errors. For the linked marker, one-way AMOVA was conducted to test if markers were associated with differences in oil content across the genotypes. Markers exhibiting a P value less than 0.001 were considered significantly associated with a phenotypic trait.
Results
Amplification of Castor Bean Fatty Acid Biosynthesis Genes in J. curcas
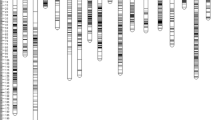
Thirty-two genes responsible for fatty acid biosynthesis in seeds of castor bean were identified (see “Materials and Methods”). The CDS varied from 326 bp for one homolog of stearoyl ACP desaturase to 6,723 bp for ACCase). The exon number for different genes varied from 1 to 30. The complete FGenesH annotation, including gene length, gene CDS, castor bean genome sequence contigs, intron–exon coordinates, etc. are given in Supplementary Table 4. Out of 48 primer pairs designed to check the transferability of exons, introns, 5′UTR and 3′UTR regions of candidate genes, 32 amplified on both castor bean and Jatropha whereas 10 primer pairs amplified only in castor bean. All the candidate gene regions amplified successfully in J. curcas with maximum amplification (~80%) from exons followed by exon–intron junctions (~78%), 5′UTR and 3′UTR regions (~ 44%), respectively. Eleven primer pairs (7 from exon–intron junctions, 2 from UTRs, and 2 from exonic regions) showed polymorphisms in 54 genotypes of J. curcas. Seven primer pairs from exon–introns of ACCase (6th and 11th exon–intron junctions), linoleoyl desaturase (2nd exon–intron junction), FatA (1st exon–intron junction), KAS III (2nd exon–intron junction), stearoyl desaturase (2nd exon–intron junction), and KAS II (1st exon–intron junction) genes were polymorphic among J. curcas genotypes. Out of 11 polymorphic candidate gene regions, 1 amplicon of 700 bp designated as JJM1from exon–intron junction of stearoyl desaturase gene (Fig. 1) showed amplification only in 36 high oil content genotypes, whereas no amplicons in 11 low oil content genotypes of J. curcas. Sequencing of stearoyl desaturase amplicon (1.3 kb) encompassing the JJM1 (700-bp fragment) showed 2 SNPs (TT → AA) in the exonic region and an insertion of 5 bp (AAAAC) in the intron of low oil content genotypes. Comparison of sequences for this candidate gene region from low to high oil content genotypes with the stearoyl desaturase DNA sequence of J. curcas in the GenBank (DQ084491) showed functional polymorphism in the low oil content genotypes (Fig. 2). SNPs identified in the exonic region lead to a substitution of leucine with glutamine in stearoyl desaturase polypepetide of low oil content genotypes. Twelve primer pairs were designed to amplify SSRs in candidate genes out of which seven amplified in castor bean and Jatropha whereas three amplified only in castor bean. One SSR primer pair with (ag)15 repeat from 5′UTR of stearoyl desaturase showed polymorphism in Jatropha genotypes, but no association with seed oil content.

Amplification of SD_intron2 marker from exon and intron of stearoyl desaturase in high oil content genotypes (H1–H11) and no amplification in low oil content genotypes (L1–L6)

Comparison of DNA sequence of JJM1 region (small exonic region of Steroyl desaturase) of high versus low oil content genotypes of J. curcas, with available sequence of stearoyl desaturase in GenBank; DQ084491: GenBank accession of J. curcas stearoyl desaturase, J. curcas_H: high oil content (>35%), J. curcas_L: low oil content (<30%)
Linkage Disequilibrium
Extent of genome-wide LD was evaluated through pairwise comparisons among 12 marker loci. For LD mapping, genetic map of Jatropha by Wang et al. (2011) was used. The r 2 averaged 0.4952 ± 0.0013 ranged from 0.0004 to 0.76. One marker loci maintained a strong LD index with each other (D′ > 0.78 and r 2 > 0.76). The r 2 value corresponding to the 95th percentile of the distribution was 0.0152, and this value was used as a population-specific threshold for r 2 as evidence of genetic linkage.
Association Testing
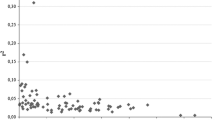
We used the model-based approach of Pritchard et al. (2000) for the SSR, and gene-specific marker sets ability to detect population structure and assigns individuals to populations. Analysis of population structure using STRUCTURE provides correct number of subpopulations for association mapping. For all data sets, the likelihood increased most when K (the number of populations in the model) was increased from two to three; results were very consistent across runs with K = 3 but became less consistent at higher values of K. Interestingly, the percent individuals assigned to the populations did not increase with K, as might be expected. Maximal assignment occurred at K = 3 and then decreased steadily, suggesting that the genetic diversity should be structured into subpopulations in this sampled collection. The three Ks structure corresponded to the genetic populations revealed by PCA (Fig. 3). PCA analysis of the 12 marker loci suggested that 11 principal components were extracted (Table 1). The high oil content genotypes were structured in K1, and low oil content genotypes were structured in K2 and K3. This validated the results from PCA including population structure and PC-matrix for following association mapping. For these K populations, F st of K1 was 0.383 with F st of K2 and was 0.221 with K3 and others. Up to 92% of the LD parameter r 2 were significant (P < 0.05) in pairwise comparisons of the 12 markers, indicative of a validation for further analysis of association mapping. Based on the criterion P value less than 0.0001 with 12 markers for high and low oil traits, we identified one significant marker–trait association. Two SNPs (TT → AA) of stearoyl desaturase (P = 0.00013) showed significant association with high oil content.

PCA for 54 accessions genotyped with 11 molecular markers
Discussion
The development of molecular markers from candidate genes encoding a particular phenotype has been pursued vigorously in plants with the long-term goals of using candidate gene markers in molecular breeding and for genetic manipulation of crop plants for economically important traits. Candidate genes often present low levels of polymorphism because they are relatively conserved, and it is generally difficult to map these regions using markers based on size polymorphism. Genomic resources of castor bean were successfully used for the identification of candidate genes involved in fatty acid biosynthesis in J. curcas and for establishing that the candidate gene markers are valuable genetic resources for investigating relationships and comparative mapping in Euphorbiaceae. In higher plants, gene families encode many proteins and enzymes, and the existence of gene families can sometimes reflect additional levels of genetic control or isoforms of proteins with specific functions (Chi et al. 2011). Therefore, it is of interest to detect potential gene families involved in the fatty acid biosynthesis pathway. The high (~80% from exonic regions) transferability from the castor bean to Jatropha showed a high level of sequence identity between two plant species. Sato et al. (2011) have also observed high level of synteny between castor bean and Jatropha.
Out of 11 candidate gene markers from different regions of fatty acid biosynthesis genes, one marker–trait association for high oil trait was identified in Jatropha. A marker from exon–intron junction of stearoyl desaturase showed association with high oil content in Jatropha genotypes. The occurrence of sequence differences in stearoyl desaturase amplicons from high to low oil content genotypes of J. curcas may be due to the existence of different copies of fatty acid biosynthesis genes in low oil content genotypes that gives leaky phenotypes as in the case of starch biosynthesis pathway where different copies of genes were responsible for low, medium, and high amylase contents in rice (Tian et al. 2009). Association mapping has been used to detect the degree to which a gene or genotype and phenotype vary together in a sampled population based on linkage disequilibrium (Zondervan and Cardon 2004). When a molecular marker is associated with a phenotypic trait, it should associate with others that highly correlate with this trait in theory. Sequencing of a 1.3-kb amplicon, including the 700-bp fragment of stearoyl desaturase associated with the high oil content, revealed SNPs in the exonic region. Comparison of stearoyl desaturase sequences from low to high oil content genotypes, including the GenBank sequence (DQ084491), showed functional polymorphism (nonpolar amino acid to polar amino acid) in low oil content genotypes. The SNPs (TT → AA) in the exonic region of stearoyl desaturase gene resulted in substitution of leucine (nonpolar) with glutamine (polar) in the open reading frame of stearoyl desaturase in low oil content genotypes of J. curcas. The low oil content in Jatropha genotypes might be because of lower amount of oleic acid in the seed oil (low oil yielding genotypes) which is otherwise present in high amount (~43%) in Jatropha genotypes (high oil yielding genotypes). The in silico sequence comparison of stearoyl desaturase genes from low to high oil content Jatropha genotypes with the sequences of stearoyl desaturase gene from soybean, castor bean, and Arabidopsis revealed SNPs (TT → AA) between castor bean and Arabidopsis (Sharma and Chauhan 2011). The same SNPs were identified in the low oil content Jatropha genotypes. Both plant species, castor bean and Arabidopsis, have low oleic acid content of 15% and 7%, respectively (Akbar et al. 2009; Bonaventure et al. 2003). Variations in expression of stearoyl desaturase genes at various stages from fruit formation to ripening were also observed between high and low oil content genotypes of J. curcas (Sharma and Chauhan 2011). Chen et al. (2010) identified two key mutations within the Δ12 fatty acid desaturase gene in high-oleate peanut cultivars which include a 1-bp substitution of G:C → A:T in the A genome and a 1-bp insertion of A:T in the B genome. Both of these mutations contribute to abolishing or reducing the desaturase activity, leading to accumulation of oleate versus linoleate in peanut. Guan et al. (2011) compared the coding regions of FAD2-1 and FAD2-1′ of safflower revealing that a deletion of cytosine exists at the position +603 bp of FAD2-1′, which results in low transcript level, showed that the expression of CtFAD2-1′ in high oleic acid genotype is significantly lower than that in normal genotypes during seed development. These results suggested that the change of CtFAD2-1′ gene sequence results in the deactivation and lower transcription of Δ12 fatty acid desaturase in high oleic safflower genotypes.
The present study identified a molecular marker associated with high oil content in Jatropha. The comparative genomics approach used in this study can be used in the species where there is a lack of genetic resources.
References
Akbar E, Yaakob J, Kamarudin SK, Ismail M, Salimon J (2009) Characteristic and composition of Jatropha Curcas oil seed from Malaysia and its potential as biodiesel feedstock. Eur J Sci Res 29:396–403
Aranzana MJ, Kim S, Zhao K, Bakker E, Horton M et al (2005) Genome-wide association mapping in Arabidopsis identifies previously known flowering time and pathogen resistance genes. PLoS Genet 1:e60
Basha SD, Sujatha M (2007) Inter and intra-population variability of Jatropha curcas (L.) characterized by RAPD and ISSR markers and development of population-specific SCAR markers. Euphytica 156:375–386
Bonaventure G, Salas JJ, Pollard MR, Ohlrogge JB (2003) Disruption of the FATB Gene in Arabidopsis Demonstrates an Essential Role of Saturated Fatty Acids in Plant Growth. The Plant Cell 15:1020–1033
Breseghello F, Sorrells ME (2006) Association mapping of kernel size and milling quality in wheat (Triticum aestivum L.) cultivars. Genetics 172:1165–1177
Casa AM, Pressoir G, Brown PJ, Mitchell SE, Rooney WL, Tuinstra MR, Franks CD, Kresovich S (2008) Community resources and strategies for association mapping in sorghum. Crop Sci 48:30–40
Chen Z, Wang ML, Barkley NA, Pittman RN (2010) A simple allele-specific PCR assay for detecting FAD2 alleles in both A and B genomes of the cultivated peanut for high-oleate trait selection. Plant Mol Biol Rep 28(3):542–548
Chi X, Yang Q, Lu Y, Wang J, Zhang Q, Pan L, Chen M, He Y, Yu S (2011) Genome-wide analysis of fatty acid desaturases in soybean (Glycine max). Plant Mol Biol Rep 29(4):769–783
Cuesta-Marcos A, Szőcs P, Close TJ, Filichkin T, Muehlbauer GJ, Smith KP, Hayes PM (2010) Genome-wide SNPs and re-sequencing of growth habit and inflorescence genes in barley: implications for association mapping in germplasm arrays varying in size and structure. BMC Genomics 11:707
Divakara BN, Upadhyaya HD, Wani SP, Laxmipathi Gowda CL (2010) Biology and genetic improvement of Jatropha curcas L.: a review. Appl Energy 87:732–742
Ellwood SR, Phan HTT, Jordan M, Hane J, Torres AM, Avila CM, Serafín Cruz-Izquierdo S, Oliver RP (2008) Construction of a comparative genetic map in faba bean (Vicia faba L.); conservation of genome structure with Lens culinaris. BMC Genomics 9:380–391
Goudet J (1995) FSTAT (Version 1.2): a computer program to calculate F-statistics. J Hered 86:485–489
Guan LL, Xu YW, Wang YB, Chen L, Shao JF, Wu W (2011) Isolation and characterization of a temperature-regulated microsomal oleate desaturase gene (CtFAD2-1) from safflower (Carthamus tinctorius L.). Plant Mol Biol Rep. doi:10.1007/s11105-011-0349-7
Heesacker A, Venkata KK, Gao W, Tang S, Kolkman JM, Gingle A, Matvienko M, Kozik A, Michelmore RM, Lai Z, Rieseberg LH, Knapp SJ (2009) SSRs and INDELs mined from the sunflower EST database: abundance, polymorphisms, and cross-taxa utility. Theor Appl Genet 117:1021–1029
Hill WG, Robertson A (1968) Linkage disequilibrium in finite populations. Theor Appl Genet 38:226–231
Keller B, Feuillet C (2000) Collinearity and gene density in grass genomes. Trends Plant Sci 5:246–251
Lander ES, Schork NJ (1994) The genetic dissection of complex traits. Science 265:2037–2048
Lewontin RC (1964) The interaction of selection and linkage. I. General considerations; heterotic models. Genetics 49:49–67
Liu K, Muse S (2005) PowerMarker: an integrated analysis environment for genetic marker analysis. Bioinformatics 21:2128–2129
Makkar HPS, Becker K, Sporen F, Wink M (1997) Studies on nutritive potential and toxic constituents of different provenances of Jatropha curcas L. J Agric Food Chem 45:3152–3157
Murray MG, Thompson WF (1980) Rapid isolation of high molecular weight plant DNA. Nucleic Acids Res 8:4321–4325
Peakall R, Smouse PE (2006) GENALEX 6: genetic analysis in excel population genetic software for teaching and research. Mol Ecol Notes 6:288–295
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959
Sato S, Hirakawa H, Isobe S, Fukai E, Watanabe A, Kato M, Kawashima K, Minami C, Muraki A, Nakazaki N, Takahashi C, Nakayama S, Kishida Y, Kohara M, Yamada M, Tsuruoka H, Sasamoto S, Tabata S, Aizu T, Toyoda A, Shin-I T, Minakuchi Y, Kohara Y, Fujiyama A, Tsuchimoto S, Kajiyama S, Makigano E, Ohmido N, Shibagaki N, Cartagena JA, Wada N, Kohinata T, Atefeh A, Yuasa S, Matsunaga S, Fukui K (2011) Sequence analysis of the genome of an oil-bearing tree, Jatropha curcas L. DNA Res 18:65–76
Sharma A, Chauhan RS (2011) In-silico identification and comparative genomics of candidate genes involved in biosynthesis and accumulation of seed oil in plants. Comparative and Functional Genomics (in press)
Sorrells ME, Rota ML, Bermudez-Kandianis CE et al (2003) Comparative DNA sequence analysis of wheat and rice genomes. Genome Res 13:1818–1827
Tanya P, Taeprayoon P, Hadkam Y, Srinives P (2011) Genetic diversity among Jatropha and Jatropha-related species based on ISSR markers. Plant Mol Biol Rep 29:252–264
Thornsberry JM, Goodman MM, Doebley J, Kresovich S, Nielsen D, Buckler ES (2001) Dwarf8 polymorphisms associate with variation in flowering time. Nat Genet 28:286–289
Tian Z, Qian Q, Liu Q, Yan M, Liu X, Yan C, Liu G, Gao Z, Tang S, Zeng D, Wang Y, Yu J, Gu M, Li J (2009) Allelic diversities in rice starch biosynthesis lead to a diverse array of rice eating and cooking qualities. Proc Natl Acad Sci USA 106:21760–21765
Wang CM, Liu P, Yi C, Gu K, Sun F et al (2011) A first generation microsatellite- and SNP-based linkage map of Jatropha. PLoS One 6(8):e23632
Wilson LM, Whitt SR, Ibanez AM, Rocheford TR, Goodman MM et al (2004) Dissection of maize kernel composition and starch production by candidate gene association. Plant Cell 16:2719–2733
Yu J, Pressoir G, Briggs W, Vroh BI, Yamasaki M, Doebley JF, McMullen MD, Gaut BS, Nielsen DM, Holland J, Kresovich S, Buckler ES (2006) A unified mixed-model method for association mapping that accounts for multiple levels of relatedness. Nat Genet 38:203–208
Zhao K, Aranzana MJ, Kim S, Lister C, Shindo C, Tang C, Toomajian C, Zheng H, Dean C, Marjoram P, Nordborg M (2007) An Arabidopsis example of association mapping in structured samples. PLoS Genet 31:e4
Zondervan KT, Cardon LR (2004) The complex interplay among factors that influence allelic association. Nat Rev Genet 5:89–100
Acknowledgment
The authors are thankful to the Department of Biotechnology, Ministry of Science and Technology, Government of India for providing a research grant to RSC.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Supplementary Table 1
Primers used to amplify candidate genes for fatty acid biosynthesis in Jatropha (DOCX 42 kb)
Supplementary Table 2
SSRs identified in the candidate genes for fatty acid biosynthesis in castor bean (DOCX 36 kb)
Supplementary Table 3
List of genotypes used in the present study (DOCX 37 kb)
Supplementary Table 4
Annotation of candidate genes for fatty acid biosynthesis in the castor bean genome (DOCX 49 kb)
Rights and permissions
About this article
Cite this article
Sharma, A., Chauhan, R.S. Identification and Association Analysis of Castor Bean Orthologous Candidate Gene-Based Markers for High Oil Content in Jatropha curcas . Plant Mol Biol Rep 30, 1025–1031 (2012). https://doi.org/10.1007/s11105-011-0408-0
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11105-011-0408-0