
Abstract
Enhancers are known to be capable of overriding the specificity of nearby promoters in a distance-dependent manner, which is problematic when multiple promoters coexist in a single transgene unit. In an attempt to determine whether enhancer activation function is inversely related to its distance from the target promoter, we inserted 1-, 2-, and 4-kb bacteriophage λ fragments, respectively, between the cauliflower mosaic virus 35S enhancer and a flower-specific AGAMOUS second intron-derived promoter (AGIP) fused to the β-glucuronidase (GUS) coding region. In the absence of an insert sequence, the 35S enhancer activates AGIP-driven GUS expression in vegetative tissues of transgenic Arabidopsis thaliana lines. Moreover, neither the 2-kb nor the 4-kb λ fragment was able to block GUS expression in transgenic leaves, implying that the 35S enhancer can override a distance barrier of at least 4 kb in our system. Unexpectedly, insertion of the 1-kb λ insert into the same site resulted in diminished GUS expression in transgenic leaves. Our data indicate that this fragment functions as a true enhancer-blocking insulator that could potentially be utilized to minimize enhancer–promoter interference between multiple transcriptional units within a plasmid vector during plant transformation experiments.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Transgenic plant technology is an important tool for both basic plant biological research and the improvement of agronomic traits in a wide variety of crop species through the stable expression of foreign genes (Lanfranco 2003). Often, this necessitates the use of tissue-, organ-, or developmental stage-specific and strong constitutive promoters (Li et al. 2001; Liu and Liu 2008; Savidge et al. 1995) to drive transgene expression exclusively in targeted tissues. Until recently, the majority of transgenic research has been directed towards the improvement of a single trait (for example virus resistance, herbicide resistance) despite the fact that crops in field conditions must cope with diverse biotic and abiotic challenges and, thus, often require a comprehensive approach to enhance the performance of multiple traits. This demands the use of a transformation vector with the ability to harbor multiple transcriptional gene units, a feat that has been demonstrated to be feasible with the introduction of six transcriptional cassettes into plants using a single pPZP-based vector (Goderis et al. 2002).
Unfortunately, the presence of multiple promoter and enhancer elements within a single vector might, due to the inherent orientation-independent nature of enhancers, provoke enhancer–promoter or promoter–promoter interference, thereby altering the specificity and strength of discrete promoters in transgenic plants. This crosstalk phenomenon is commonly observed in transgenic plant-bearing vectors in which the enhancer derived from the constitutive 35S promoter (Odell et al. 1988) is inserted near plant-derived promoters, resulting in a drastic augmentation of their transcriptional activity in nontargeted tissues. For example, the 35S enhancer has been shown to activate the root-specific LRP1 (Smith and Fedoroff 1995), vascular tissue-specific AAP2 (Hirner et al. 1998), carpel-specific AGL5 (Savidge et al. 1995), tapetum-specific TA29 (Koltunow et al. 1990), and embryogenesis-specific PAB5 (Belostotsky and Meagher 1996) promoters in leaf, stem, and root tissues (Jagannath et al. 2001; Yoo et al. 2005; Zheng et al. 2007), resulting in an expression pattern that is indistinguishable from that of the 35S promoter (Benfey et al. 1989, 1990). Since the 35S promoter is used to drive the expression of the selectable marker gene in a broad range of plant transformation vectors, including pPZP, pCAMBIA, and pINDEX1 family vectors (Hajdukiewicz et al. 1994; Ouwerkerk et al. 2001), its strong enhancer function has the potential to be an enormous hindrance to transgenic research when precisely controlled expression of the transgene is essential.
While a number of elements have been identified in animals that are able to impede interactions between an enhancer and promoter when situated between them (Chung et al. 1993), very few such enhancer-blocking insulators have been described in plants. Recently, we developed an enhancer-blocking assay system and used it to successfully identify the first true enhancer-blocking insulator in plants (Hily et al. 2009). In this study, we used the same system to investigate the relationship between insulation function and the length of inserted DNA fragments. We found that a specific 1 kb EcoRI/SalI fragment of bacteriophage λ origin, but not a 2 kb HindIII or 4 kb NcoI fragment, respectively, functions as an enhancer-blocking insulator when inserted between the 35S promoter and flower-specific AGIP (Hily et al. 2009; Liu and Liu 2008) fused to the GUS gene in transgenic Arabidopsis lines, consequently permitting the proper tissue-specific expression of the transgene. Since insulators are often able to function in a broad range of species (Chung et al. 1993; Namciu et al. 1998), the 1-kb λ fragment has a potential use in the future of transgenic research.
Materials and Methods
Generation of Stable Transgenic Lines
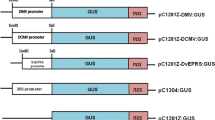
Figure 1a includes a schematic diagram of the transformation vectors utilized in this study. All vectors were generated using standard protocols and are present in a pBIN19 (Bevan 1984) background. The construction of vectors JM79, JM69, and JM85, which incorporates a 3,967-bp NcoI fragment of bacteriophage λ (GenBank accession number J02459; nucleotides 23901–27868) between head-to-head 35S::green fluorescent protein (GFP) and AGIP::GUS cassettes, has been described previously (Hily et al. 2009). To generate the JM86 and JM87 constructs, bacteriophage λ was initially digested with HindIII to generate a 2,027-bp fragment (nucleotides 23130–25157) or EcoRI and SalI to produce a 998-bp fragment (nucleotides 31747–32745; Fig. 1b), which were isolated and cloned into pAUX3131 (Goderis et al. 2002). The resulting plasmids were digested with I-SceI, and the λ fragments were inserted into the pPZP-RSC1 multiple cloning site (Hajdukiewicz et al. 1994) between the 35S enhancer and AGIP of JM69 to yield JM86 (2,027-bp spacer) and JM87 (998-bp spacer). The presence of the desired insert sequence between enhancer and promoter in each of the final vectors was confirmed by sequencing.

Schematic representation of transformation constructs. a Plasmid constructs. Construct JM79 is a control plasmid for GUS expression driven by the AGIP. Two transcriptional fusion units, 35S::GFP and AGIP::GUS, are present in a head-to-head orientation in JM69. The JM85, JM86, and JM87 constructs are derivatives of JM69 with a 4-kb NcoI, 2-kb HindIII, or 1-kb EcoRI/SalI fragment of the bacteriophage λ genome inserted between the 35S promoter and AGIP. The direction of transcription in each case is indicated by an arrow. T 1 nopaline synthase transcriptional terminator; T 2 procyclin-associated gene 7 transcriptional terminator; 35S cauliflower mosaic virus 35S promoter; GUS β-glucuronidase coding sequence; GFP enhanced green fluorescent protein coding sequence; AGIP carpel- and stamen-specific promoter derived from the second intron of the Arabidopsis AGAMOUS gene; λ lambda phage spacer sequence. All constructs shown are in a pBIN19 background. b Schematic diagram of the 1-kb EcoRI/SalI λ fragment. Restriction sites (EcoRI and SalI) utilized to clone the fragment and the lengths of each partial coding sequence are indicated. The direction of transcription that would occur within the λ genome, as well as the lengths of the resulting endogenous transcripts, is also shown. Regions of the transcripts contained within the 1-kb region are denoted by a solid line while missing sequences are depicted by dashed lines. Exo exonuclease-encoding gene; Bet beta protein-encoding gene
Each vector was introduced into A. thaliana ecotype C10, and the presence of the λ inserts was confirmed in a selection of JM85, JM86, and JM87 lines by polymerase chain reaction (PCR) analysis and sequencing according to Hily et al. (2009). While the exact number of transgene copies in each line was not determined, it has been found previously that the floral dip method utilized for transformation in this study (Clough and Bent 1998) results in the introduction of a single T-DNA insert in over 50% of transgenic lines, with an average of 1.5 inserts per line (Rosso et al. 2003). In each case, only those lines with a normal phenotype were chosen for further analysis.
PCR Analysis of Transgenic Plants
To ascertain that no significant rearrangements or deletions had occurred within 35S promoter, GUS, and AGIP sequences in plant-bearing constructs with a functional insulator, PCR tests were conducted on DNA from three independent lines of JM69, JM86, and JM87, respectively, as well as an untransformed control. Genomic DNA was extracted from young leaves of various transgenic lines as described by Kobayashi et al. (1998). PCR analyses were performed using approximately 100 ng template and GoTaq DNA Polymerase (Promega, Madison, WI) according to the manufacturer's instructions. Primers PZPR1 and eGFPR2 were utilized to amplify the entire 35S promoter sequence; AGI-IIF11 and pBINF1 were employed to amplify the entire GUS sequence; and PZPF2 and GUSR1 were utilized to amplify the entire AGIP. Primer sequences have been described elsewhere (Hily et al. 2009). The amplification regime consisted of 94°C for 5 min, 30 cycles of 94°C for 45 s, 55°C for 1 min, and 72°C for either 1.5 min (for the 35S promoter-specific product), 2.5 min (for the GUS-specific product), or 3.5 min (for the AGIP-specific product), followed by a single iteration of 72°C for 7 min.
Histochemical Staining and Fluorometric Assays of GUS Activity
Histochemical assays of GUS activity were carried out using leaf and floral tissues from transgenic T1 lines carrying each construct, respectively, as well as an untransformed control. Each sample was incubated in 1 mM 5-bromo-4-chloro-3-indolyl-β-d-glucuronide (X-gluc; in 100 mM phosphate buffer, pH 7.0, 10 mM EDTA, 0.5 mM potassium ferrocyanide, and 0.1% Triton X-100) at 37°C for 24–48 h depending on the tissue analyzed and subsequently depigmented in 70% ethanol.
Fluorometric assays of GUS activity were executed as described by Hily et al. (2009). Leaf tissue from 15 independent T1 JM69, JM79, and JM87 lines, respectively, as well as an untransformed control, was analyzed in duplicate. GUS activities were calculated as the mean value of pmol MU generated per minute per mg protein, and statistical analyses were conducted using the Mann–Whitney test for nonparametric data.
Quantitative Real-Time RT-PCR
Total RNA was isolated from leaf tissues of three independent lines transformed with JM69, JM79, and JM87, respectively, as well as an untransformed control, using the RNeasy Plant Mini kit according to the manufacturer's instructions (Qiagen, Valencia, CA). Contaminating DNA was removed using the DNA-free system (Ambion, Austin, TX). For each line, 500 ng of RNA was reverse transcribed with the Superscript VILO cDNA synthesis kit (Invitrogen, San Diego, CA), and cDNA products were utilized for quantitative real-time PCR assays with GUS-specific and actin2-specific primers according to Hily et al. (2009). Assays including no template or template from reverse transcription reactions lacking reverse transcriptase were included in each trial. All GUS expression data were normalized to that of actin, and each assay was performed twice in triplicate.
GFP Detection
A Typhoon Trio fluorescence scanner with the control v5.0 software (Amersham Bioscience, Piscataway, NJ) was used to scan shoot apices from JM69, JM79, and JM87 lines, as well as an untransformed control, for visualization of fluorescence emitted from either GFP or chlorophyll as described by Hily and Liu (2009). Blue light (488 nm, blue, PMT 300V) was used for excitation in both cases. A 520-nm emission filter (BP 40) was used to scan for green fluorescence emitted from GFP while a 670-nm filter (BP 30) was used to scan for red fluorescence emitted from chlorophyll. The resulting images were overlaid for the simultaneous visualization of both GFP and chlorophyll fluorescence.
Bioinformatic Sequence Analyses
Analyses of the λ spacer sequences were conducted using various computational programs and databases available online. Specifically, prediction of CTCF binding sites was carried out using the CTCFBSDB database (http://insulatordb.utmem.edu; Bao et al. 2008); the presence of matrix attachment region (MAR) motifs was investigated with MARSCAN (http://mobyle.pasteur.fr/cgi-bin/MobylePortal/portal.py?form=marscan) and MAR-Wiz (www.futuresoft.org/MAR-Wiz; Singh et al. 1997); a search for palindromic sequences was conducted using CRISPRFinder (http://crispr.u-psud.fr/Server/CRISPRfinder.php; Grissa et al. 2007) and PALINDROME (http://mobyle.pasteur.fr/cgi-bin/MobylePortal/portal.py?form=palindrome); and a search for inverted and tandem repeat sequences was carried out using EINVERTED (http://mobyle.pasteur.fr/cgi-bin/MobylePortal/portal.py?form=einverted), REPFIND (http://zlab.bu.edu/mfrith/cgi-bin/repfind.cgi), ETANDEM (http://mobyle.pasteur.fr/cgi-bin/MobylePortal/portal.py?form=etandem), and TANDEM REPEATS FINDER (http://tandem.bu.edu/trf/trf.html; Benson 1999). Promoter prediction was carried out using the Promoter Scan program (www.bimas.cit.nih.gov/molbio/proscan; Prestridge 1995).
Results
The 1-kb λ EcoRI/SalI Fragment, But Not 2-kb HindIII or 4-kb NcoI Fragments, Represses 35S Enhancer-Mediated Activation of AGIP-Driven GUS Expression in Vegetative Tissues
To gain insight into the relationship between 35S enhancer activation function and the distance to its target promoter, we inserted three bacteriophage λ DNA fragments between the enhancer and AGIP in JM69 to create JM85, JM86, and JM87 (Fig. 1a, b), respectively. The selection of λ DNA fragments as spacer sequences was based on the assumption that the λ genome is free of DNA-binding motifs that might be recognized by plant regulatory factors.
Histochemical staining of leaf tissue from 15 independent T1 lines containing each construct, respectively (Fig. 2), demonstrated that unlike JM79-transformed plants (harboring the flower-specific AGIP::GUS cassette), in which only 13.3% of leaves tested exhibited weak GUS activity, 93.3% of JM69 lines (harboring AGIP::GUS and 35S::GFP cassettes in a head-to-head orientation) and 100% of JM85 and JM86 lines (harboring a 4-kb NcoI or 2-kb HindIII λ insert, respectively, between 35S promoter and AGIP) displayed GUS activity in the leaves. These results confirm our previous findings that the 35S enhancer overrides AGIP-specific expression patterns when present within the same construct (Hily et al. 2009) and are consistent with earlier reports in which the 35S enhancer was found to activate tissue-specific promoters in nontargeted plant tissues (Yoo et al. 2005; Zheng et al. 2007). Interestingly, only one of the 15 JM87-transformed lines tested (harboring the 1-kb λ insert between 35S promoter and AGIP) exhibited weak GUS staining in the leaves, resembling JM79 lines.

Histochemical GUS staining in the leaves of transgenic lines. Images display the leaves of representative lines transformed with each construct or the untransformed control
In accordance with the histochemical data, fluorometric assays (Fig. 3a) of leaf tissue from 15 independent lines of JM87, JM69, and JM79 revealed that the mean GUS activity in the leaves of JM87-transformed plants (84 ± 19 pmol MU/min/mg protein) was reduced to less than 1% of JM69-transformed plants (8905 ± 3818 pmol MU/min/mg protein), a difference that is significant even at the 1% level as determined by the Mann–Whitney test for nonparametric data. No significant divergence was observed between the GUS activities of JM87 plants and those bearing the JM79 construct (75 ± 28 pmol MU/min/mg protein). Quantitative real-time RT-PCR assays displayed a similar pattern in that both JM79 and JM87 leaves exhibited a substantial reduction in the mean normalized levels of GUS mRNA as compared to lines bearing the JM69 vector (Fig. 3b). Taken together, these results indicate that the 1-kb λ EcoRI/SalI fragment is able to prevent 35S enhancer-induced GUS transcription in nontarget tissues.

Analyses of GUS activity in the leaves of transgenic lines. a Each block represents the mean GUS activity in pmol MU generated per minute per mg protein from three leaves of 15 independent T1 lines containing each construct, respectively, or the untransformed control. Bars indicate standard errors. b Five hundred nanograms total RNA from the leaves of three lines bearing each construct, respectively, or the untransformed control was assayed for levels of GUS transcript and the internal control, actin2. Each block represents the mean normalized value of GUS mRNA for each construct. Bars indicate standard errors
The 1-kb λ EcoRI/SalI Fragment Does Not Silence the 35S Promoter or AGIP
It is possible that the lack of GUS expression in JM87 lines may have resulted from a 1-kb λ EcoRI/SalI fragment-induced destabilization of surrounding transgene sequence, which could cause the deletion or truncation of 35S enhancer, AGIP, or GUS coding regions in transgenic plants. To address this possibility, we tested three independent JM86, JM87, and JM69 lines, respectively, by PCR to analyze the integrity of the 35S enhancer, GUS coding region, and AGIP (Fig. 4a). In each case, fragments of identical size were amplified for the 35S enhancer, GUS coding region, and AGIP, respectively, indicating that transgene deletion or truncation had not taken place in any of the plants analyzed.

The 1-kb λ EcoRI/SalI fragment functions as a true enhancer-blocking insulator in transgenic plants. a Amplification of 35S, GUS, and AGIP fragments from leaf genomic DNA samples from three independent lines containing JM69, JM86, and JM87 transgenes, respectively. b Histochemical GUS staining of floral tissue from JM69, JM79, and JM87 lines, as well as an untransformed control. The flower-specific GUS staining pattern seen in JM87 lines is reminiscent of JM79 lines, confirming that the λ insert sequence does not interfere with expression from the AGIP. c GFP fluorescence in shoot apices was detected in those lines containing the 35S::eGFP cassette (JM69 and JM87), indicating that the λ fragment does not interfere with expression from the 35S promoter. Representative lines are shown in each case. Wt wild type; M DNA marker
To rule out the alternative possibility that the obstruction of GUS transcription in JM87 leaves is due to silencing of AGIP and/or 35S promoter activity by the λ EcoRI/SalI fragment, rather than its enhancer-blocking function, we subjected floral tissue to histochemical GUS assays (Fig. 4b) and GFP fluorescence analyses (Fig. 4c) to determine whether these two promoters maintain their inherent activities in JM87 lines. While JM87 plants displayed diminished GUS expression in leaf tissue, they retained the same flower-specific GUS expression as control JM79 lines (Fig. 4b). Furthermore, we confirmed that both JM69 and JM87 lines exhibited GFP fluorescence (Fig. 4c). Evidently, the activities of the 35S promoter and AGIP were not hampered by the 1-kb λ fragment in JM87 lines, suggesting that the insert fragment does not function by silencing nearby regulatory elements but instead acts as a true enhancer-blocking insulator.
The λ EcoRI/SalI Fragment Does Not Contain Any Predicted Structural, Insulator, or MAR Elements
Although we have provided evidence that a 1-kb λ EcoRI/SalI fragment is able to block interference by the 35S enhancer, the mechanism by which it functions is unknown. The 1-kb λ sequence utilized in this study consists of two partial coding regions: the first encodes an exonuclease (Exo) that is truncated by 400 bp at its 3′ terminus and the second encodes the single-stranded binding protein, beta (Bet), which is truncated by 65 bp at its 5′ end (Fig. 1b). While unlikely, even if partial Exo and Bet transcripts were generated by a nearby plant promoter, they would not be expected to yield functional products or cause the specific repression of GUS expression in JM87 leaves. Instead, the λ EcoRI/SalI fragment may contain unique sequence motifs that are recognized or interpreted as insulation signals in plants. To search for putative sequences that might be recognized as insulation signals in plants, we performed thorough bioinformatic analyses of the 1-kb EcoRI/SalI λ fragment using a variety of computational programs and databases. We first investigated the possibility of similarities between the λ EcoRI/SalI fragment and sequences in the CTCFBSDB database. Our results indicated that the λ sequence exhibits sequence identity with at least 65 CTCF binding sites ranging from 14 to 19 nt. However, further analyses with various other DNA fragments derived from the λ phage revealed comparable frequencies of CTCF binding sites. Therefore, whether the predicted CTCF binding sites in this λ fragment are truly functional or simply false positives remains to be elucidated. We also searched for MAR motifs, palindromes, inverted and tandem repeats, as well as predicted promoter elements, but no such sequences could be identified in the 1-kb λ fragment.
Discussion
We have analyzed the ability of various bacteriophage λ fragments to block enhancer-mediated activation of a target promoter in Arabidopsis by analyzing GUS activity in the leaves of stable transformants containing transgenes with a 1-kb λ EcoRI/SalI (comprising partial coding regions of the exonuclease-encoding gene, Exo, and the single-stranded DNA binding protein-encoding gene, Bet), 2-kb HindIII, and 4-kb NcoI fragment, respectively, inserted between a 35S enhancer and AGIP::GUS cassette (Fig. 1). In the absence of any intervening sequence, the 35S enhancer interfered with the stamen- and carpel-specific activity of the AGIP, resulting in GUS expression within vegetative tissues. These findings are consistent with earlier reports, which found that the 35S enhancer can effectively activate tissue-specific promoters in nontargeted tissues in plants (Hily et al. 2009; Yoo et al. 2005, Zheng et al. 2007). While the insertion of 2- and 4-kb λ spacer fragments did not result in the inhibition of enhancer-mediated activation of the AGIP in leaf tissues, unexpectedly, the 1-kb λ sequence effectively blocked interactions between the 35S enhancer and AGIP (Figs. 2 and 3).
While it is possible that the 1-kb λ fragment had an effect on GUS expression by destabilizing the introduced transgene or by silencing the nearby 35S promoter or AGIP, our analyses suggest otherwise. This was made apparent by the fact that no transgene deletions or rearrangements were evident in any of the lines analyzed (Fig. 4a), and both the flower-specific activity of the AGIP and ubiquitous function of the 35S promoter in insulated lines were not compromised (Fig. 4b, c). Thus, our results support the conclusion that the 1-kb λ fragment functions as a true enhancer-blocking insulator.
Whereas knowledge concerning enhancer-blocking insulators is accumulating in animal systems (for example Chung et al. 1993; Geyer et al. 1986; Hebbes et al. 1994; Kellum and Schedl 1991; 1992), very little is known about the function of these elements in plants. It has been suggested that the 35S enhancer is only able to act at a relatively close range and that virtually any sequence could be used as an insulator as long as the length was sufficient to block enhancer–promoter interactions (Jagannath et al. 2001; van der Geest and Hall 1997). However, we have found that the 35S enhancer can override a length barrier of at least 4 kb in our assay system (Figs. 2 and 3), indicating that the capacity of the 1-kb λ EcoRI/SalI fragment to inhibit enhancer–promoter communication is not simply be the result of its length. Therefore, it is tempting to surmise that this sequence, like the recently identified TBS fragment (Hily et al. 2009), possesses as of yet unidentified properties that prevent interactions between an enhancer and promoter when situated between them.
It is feasible that these “properties” may include regions that provide a steric effect through modifications to higher order chromatin conformation, which inhibit direct contact between enhancer and promoter, or sequences that enable the physical obstruction or attenuation of a signal sent from the 35S enhancer to the AGIP. Such sequences could very well be sequence elements that have been found previously in animal enhancer-blocking insulators, such as MAR regions (Dunn et al. 2003; Nabirochkin et al. 1998; Stief et al. 1989) or promoter sequences (Kellum and Schedl 1992). Alternatively, the EcoRI/SalI fragment might possess palindromic sequences or repeat structures that could pose a structural hindrance on enhancer–promoter interactions. However, our bioinformatic analyses failed to identify any such characteristic sequence motifs within this fragment.
It is also possible that the presence of unanticipated protein-binding sites within the 1-kb λ sequence are involved in its ability to impede enhancer–promoter crosstalk, since interactions between the highly conserved zinc-finger protein, CTCF, and a 20-nt consensus sequence is essential for the function of many enhancer-blocking insulators in metazoans (Kim et al. 2007). As of yet, no functional equivalents of CTCF binding sites have been identified in plants. However, a number of plant zinc-finger gene families collectively share some degree of identity at the amino acid level with the zinc-finger domains of vertebrate CTCF proteins, suggesting that a CTCF-dependent insulator system might also exist in plants. Despite the fact that we were able to identify numerous putative CTCF binding sites in the 1-kb λ EcoRI/SalI fragment, the presence of a similar number of predicted sites within other λ DNA fragments suggests that they are probably not functional.
Further detailed molecular analyses should provide insight concerning the mechanism driving the enhancer-blocking insulator function of the 1-kb λ EcoRI/SalI fragment in plants. Nonetheless, regardless of the manner in which this 1-kb λ fragment functions, our study has identified an effective enhancer-blocking insulator that, like the recently characterized petunia TBS fragment (Hily et al. 2009), may provide a novel means for minimizing enhancer–promoter crosstalk during plant transformation experiments with composite vectors.
References
Bao L, Zhou M, Cui Y (2008) CTCFBSDB: a CTCF-binding site database for characterization of vertebrate genomic insulators. Nucleic Acids Res 36:D83–D87
Belostotsky DA, Meagher RB (1996) A pollen-, ovule-, and early embryo-specific poly(A) binding protein from Arabidopsis complements essential functions in yeast. Plant Cell 8:1261–1275
Benfey PN, Ren L, Chua NH (1989) The CaMV 35S enhancer contains at least two domains which can confer different developmental and tissue-specific expression patterns. EMBO J 8:2195–2202
Benfey PN, Ren L, Chua NH (1990) Tissue-specific expression from CaMV 35S enhancer subdomains in early stages of plant development. EMBO J 9:1677–1684
Benson G (1999) Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res 27:573–580
Bevan M (1984) Agrobacterium vectors for plant transformation. Nucleic Acids Res 12:8711–8721
Chung JH, Whiteley M, Felsenfeld G (1993) A 5′ element of the chicken β-globin domain serves as an insulator in human erythroid cells and protects against position effect in Drosophila. Cell 74:505–514
Clough SJ, Bent AF (1998) Floral dip: a simplified method for Agrobacterium-mediated transformation of Arabidopsis thaliana. Plant J 16:735–743
Dunn KL, Zhao H, Davie JR (2003) The insulator binding protein CTCF associates with the nuclear matrix. Exp Cell Res 288:218–223
Geyer PK, Spana C, Corces VG (1986) On the molecular mechanism of gypsy-induced mutations at the yellow locus of Drosophila melanogaster. EMBO J 5:2657–2662
Goderis IJWM, De Bolle MFC, François IEJA, Wouters PFJ, Broekaert WF, Cammue BPA (2002) A set of modular plant transformation vectors allowing flexible insertion of up to six expression units. Plant Mol Biol 50:17–27
Grissa I, Vergnaud G, Pourcel C (2007) CRISPRFinder: a web tool to identify clustered regularly interspaced short palindromic repeats. Nucleic Acids Res 35:W52–W57. doi:10.1093/nar/gkm360:1-6
Hajdukiewicz P, Svab Z, Maliga P (1994) The small, versatile pPZP family of Agrobacterium binary vectors for plant transformation. Plant Mol Biol 25:989–994
Hebbes TR, Clayton AL, Thorne AW, Crane-Robinson C (1994) Core histone hyperacetylation co-maps with generalized DNase I sensitivity in the chicken β-globin chromosomal domain. EMBO J 13:1823–1830
Hily JM, Liu Z (2009) A simple and sensitive high-throughput GFP screening in woody and herbaceous plants. Plant Cell Rep 28:493–501
Hily JM, Singer SD, Yang Y, Liu Z (2009) A transformation booster sequence (TBS) from Petunia hybrida functions as an enhancer-blocking insulator in Arabidopsis thaliana. Plant Cell Rep 28(7):1095–1104
Hirner B, Fischer WN, Rentsch D, Kwart M, Frommer WB (1998) Developmental control of H+/amino acid permease gene expression during seed development of Arabidopsis. Plant J 14:535–544
Jagannath A, Bandyopadhyay P, Arumugam N, Gupta V, Kumar P, Pental D (2001) The use of a Spacer DNA fragment insulates the tissue-specific expression of a cytotoxic gene (barnase) and allow high-frequency generation of transgenic male sterile lines in Brassica juncea L. Mol Breeding 8:11–23
Kellum R, Schedl P (1991) A position-effect assay for boundaries of higher order chromosomal domains. Cell 64:941–950
Kellum R, Schedl P (1992) A group of scs elements function as domain boundaries in an enhancer-blocking assay. Mol Cell Biol 12:2424–2431
Kim T, Abdullaev Z, Smith A, Ching K, Loukinov D, Green R, Zhang M, Lobanenkov V, Ren B (2007) Analysis of the vertebrate insulator protein CTCF-binding sites in the human genome. Cell 128:1231–1245
Kobayashi N, Horikoshi T, Katsuyama H, Handa T, Takayanagi K (1998) A simple and efficient DNA extraction method for plants, especially woody plants. Plant Tissue Cult Biotechnol 4:76–80
Koltunow AM, Truettner J, Cox KH, Wallroth M, Goldberg RB (1990) Different temporal and spatial gene expression patterns occur during anther development. Plant Cell 2:1201–1224
Lanfranco L (2003) Engineering crops, a deserving venture. Riv Biol 96:31–54
Li Z, Jayasankar S, Gray DJ (2001) Expression of a bifunctional green fluorescent protein (GFP) fusion marker under the control of three constitutive promoters and enhanced derivatives in transgenic grape (Vitis vinifera). Plant Sci 160:877–887
Liu Z, Liu Z (2008) The second intron of AGAMOUS drives carpel- and stamen-specific expression sufficient to induce complete sterility in Arabidopsis. Plant Cell Rep 27:855–863
Nabirochkin S, Ossokina M, Heidmann T (1998) A nuclear matrix/scaffold attachment region co-localizes with the gypsy retrotransposon insulator sequence. J Biol Chem 273:2473–2479
Namciu SJ, Blochlinger KB, Fournier REK (1998) Human matrix attachment regions insulate transgene expression from chromosomal position effects in Drosophila melanogaster. Mol Cell Biol 18:2382–2391
Odell JT, Knowlton S, Lin W, Mauvais J (1988) Properties of an isolated transcription stimulating sequence derived from the cauliflower mosaic virus 35S promoter. Plant Mol Biol 10:263–272
Ouwerkerk PBF, de Kam RJ, Hoge JHC, Meijer AH (2001) Glucocorticoid-inducible gene expression in rice. Planta 213:370–378
Prestridge DS (1995) Predicting pol II promoter sequences using transcription factor binding sites. J Mol Biol 249:923–932
Rosso MG, Li Y, Strizhov N, Reiss B, Dekker K, Weisshar B (2003) An Arabidopsis thaliana T-DNA mutagenized population (GABI-Kat) for flanking sequence tag-based reverse genetics. Plant Mol Biol 53:247–259
Savidge B, Rounsley SD, Yanofsky MF (1995) Temporal relationship between the transcription of two Arabidopsis MADS box genes and the floral organ identity genes. Plant Cell 7:721–733
Singh GB, Kramer JA, Krawetz SA (1997) Mathematical model to predict regions of chromatin attachment to the nuclear matrix. Nucleic Acids Res 25:1419–1425
Smith DL, Fedoroff NV (1995) LRP1, a gene expressed in lateral and adventitious root primordia of Arabidopsis. Plant Cell 7:735–745
Stief A, Winter DM, Strätling WH, Sippel AE (1989) A nuclear DNA attachment element mediates elevated and position-independent gene activity. Nature 341:343–345
van der Geest AHM, Hall TC (1997) The β-phaseolin 5′ matrix attachment region acts as an enhancer facilitator. Plant Mol Biol 33:553–557
Yoo SY, Bomblies K, Yoo SK, Yang JW, Choi MS, Lee JS, Weigel D, Ahn JH (2005) The 35S promoter used in a selectable marker gene of a plant transformation vector affects the expression of the transgene. Planta 221:523–530
Zheng X, Deng W, Luo K, Duan H, Chen Y, McAvoy R, Song S, Pei Y, Li Y (2007) The cauliflower mosaic virus (CaMV) 35S promoter sequence alters the level and patterns of activity of adjacent tissue- and organ-specific gene promoters. Plant Cell Rep 26:1195–1203
Acknowledgments
We thank Mr. Dennis Bennett for technical assistance. This study was funded by the United States Department of Agriculture (USDA)-Agricultural Research Service Headquarter 2005 and 2007 classes of postdoctoral grants and a USDA Cooperative State Research, Education, and Extension Service Biotechnology Risk Assessment Research grant (2006-03701).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Singer, S.D., Hily, JM. & Liu, Z. A 1-kb Bacteriophage Lambda Fragment Functions as an Insulator to Effectively Block Enhancer–Promoter Interactions in Arabidopsis thaliana . Plant Mol Biol Rep 28, 69–76 (2010). https://doi.org/10.1007/s11105-009-0122-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11105-009-0122-3