
Abstract
Aims
Many soil scientists think that soil phosphate exists as discrete compounds of iron, aluminium and calcium and, accordingly, use chemical fractionation schemes to identify these compounds.
Methods
We reacted a sample of goethite and a sample of aluminium oxide with a phosphate solution under conditions chosen to facilitate penetration of phosphate. Thus the sample of goethite had neither calcium nor aluminium present and similarly the sample of aluminium oxide had neither iron nor calcium. We included a sample of hydroxyapatite which had neither iron nor aluminium present. We subjected the samples to two fractionation procedures; the original Chang and Jackson (1957) method and a variant of it.
Results
For the phosphated goethite and aluminium oxide, energy dispersive X-ray spectra did not detect any discrete aluminium or iron phosphates; dissolution studies were consistent with penetration of phosphate. Both fractionation procedures detected discrete compounds even though none were present. They also detected iron, aluminium and calcium phosphates for samples for which they were not present. We also critically discuss other evidence for the existence of discrete iron, aluminium and calcium phosphates in soils.
Conclusions
Fractionation procedures designed to measure chemically specified phosphate fractions in soil are fallacious and should be abandoned.
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Some scientists think that soil phosphate largely occurs as discrete compounds of iron, aluminium and calcium (Price 2006; Penn and Camberto 2019). They then use this interpretation to predict the effects on availability. The origins of this precipitate-particulate theory may be traced back to the work of Hall and Amos (1906). They observed that repeated extractions with acid dissolved ever smaller amounts of phosphate and concluded “Reviewing the experiments as a whole, it may be concluded that the soil contains compounds of phosphoric acid of varying solubility”. These ideas were developed further especially by Lindsay (1979) who considered that that soil chemistry was largely a matter of precipitates, the solubility of which was determined by their solubility product.
Doubts about the precipitate-particulate theory were raised by Russell and Prescott (1916). In a remarkable paper, they produced the first sorption curves, and described them using an equation we now refer to as the Freundlich equation. Further, they showed that sorption depended on period of reaction and temperature. They wrote as follows “Hall and Amos considered that their results could be explained on the supposition that soil contains several phosphorus compounds of varying degrees of solubility.…We shall show that … something more is concerned than a mere mixture of phosphates… this simple conception of the constitution of the soil is inaccurate, and must be discarded.”
Theories about specific adsorption on variable charges surfaces, such as those provided by the oxides of iron and aluminium, were mostly developed in the surface science literature. For a comparison of theories see Barrow and Bowden (1987). The model of Bowden et al. (1977) was simplified by Posner and Barrow (1982). When heterogeneity was added to this simplified form (Barrow 1983), the shape of sorption curves first observed by Russell and Prescott (1916) and subsequently by many others, could be reproduced. Further, the effects of time and temperature, again first observed by Russell and Prescott (1916) and subsequently by many others, could be explained by assuming that adsorption was followed by diffusive penetration – the adsorption-diffusion theory. The reactions of phosphate, and many other specifically sorbed ions, can be quantitatively described by this theory (for a summary see Barrow (1999).
Support for the precipitate-particulate theory depends on three kinds of evidence. One of these derives from solubility product theories. A second derives from physical techniques, most recently from synchrotron-based X-ray Absorption Near-Edge Structure (XANES). The experiment we describe here deals with the third kind of evidence: chemical fractionations. Several variations of the method have been described but they may all be related back to that of Chang and Jackson (1957). This publication has had an extraordinary influence on soil science. According to Google scholar it had been cited 1816 times in January 2020, an increase of 37 over the previous six months. The scheme has been criticised many times, but always in terms of modifying the extraction procedures. This scheme, and its derivatives, purport to measure the amounts of phosphate present as discrete compounds of iron, aluminium and calcium plus other less-well defined fractions such as “occluded P”. Some scientists are sceptical of the literal interpretation of such fractions. Hedley et al. (1982) thought that it was impossible to identify Individual P compounds and, in reporting their results, describe them in terms of the procedure used; for example “resin P”. Nevertheless, literal interpretation of the fractions is common. In the experiment reported here, we tested not only whether the scepticism about the identity of the fractions is justified, but also whether the evidence supports the contention that discrete compounds are present.
In developing the adsorption-diffusion theory, specific minerals, especially goethite, were used as models. It was shown by Strauss et al. (1997) that, if goethite crystals are imperfect, phosphate was initially adsorbed and then penetrated by diffusion. Goethite into which phosphate has diffused can be prepared so that the phosphate is not present as a discrete iron compound and further neither calcium nor aluminium is present. Similarly aluminium oxide into which phosphate has diffused can be prepared so that there is neither calcium nor iron present. We think that these materials are an appropriate model for soil phosphate. We also included a sample of hydroxyapatite for which there are neither iron nor aluminium compounds present. We tested whether two procedures could correctly detect that there were no discrete phosphate compounds present in the phosphated goethite and aluminium oxide, and whether they would identify the cations present. We used the original Chang and Jackson (1957) method and also the variant described by Zhang and Kovar (2009). We used this method because its authors reported that it was chosen after comparing previously-reported versions of the method.
Methods
Preparation of materials
We prepared goethite using the method described by Strauss et al. (1997) for the sample they described as Goe-132. We oxidised a 0.05 M FeCl2 solution with atmospheric oxygen in the presence of CO2. The reaction with oxygen was stopped after 2 days; the sample was washed with distilled water, dried at 60 °C and gently broken up in an agate mortar.
To prepare aluminium oxide, we reacted metallic aluminium with ethyl alcohol to form aluminium alkoxide (Shirai et al. 2009). We hydrolyzed aluminium alkoxide by excess water (at 80 °C) under vigorous stirring. The material was dried at 110 °C for 24 h and then heat treated at 1200 °C to transform to Al2O3 powder (Yoldas 1973).
We prepared poorly-crystallized hydroxyapatite using the method of Alobeedallah et al. (2011). A 0.5 M solution of di-ammonium phosphate was slowly added at a rate 5 mL per minute to a vigorously-stirred solution of 0.5 M calcium nitrate, adjusted to pH 10–12 by concentrated ammonium solution, at room temperature to reach a Ca/P ratio to 1.6. The slow rate of addition resulted in formation of a poorly crystallized form of hydroxyapatite and this was also favoured at room temperature. The reaction was allowed to proceed for 24 h with constant stirring. The solution was centrifuged to collect the precipitate, washed several times and dried at 40 °C overnight.
Reaction with phosphate
We loaded samples of goethite and of aluminium oxide with P by a process similar to that of Strauss et al. (1997). We mixed the samples with a 0.01 M NaNO3 solution containing 1.25 g P kg−1 as KH2PO4 at a pH close to 4.0 and a solution to solid ratio of 50:1. We incubated the suspension at 70 °C for four days with vigorous shaking once a day. After incubation, the suspension was separated by centrifugation but 0.5 mL remained entrapped. We measured the phosphorus in the suspension and after allowance for the phosphorus in the entrapped solution, calculated the P retained by goethite. The samples were dried at 40 °C for 24 h before fractionation and dissolution studies. A similar process was followed for loading the aluminium oxide with P.
Characterising the materials
We characterized samples of the original materials and their phosphated forms by powder XRD and by scanning electron microscopy followed by energy dispersive X-ray spectroscopy (EDS) analysis.
Some samples were crushed and ground into fine powder with an agate mortar and pestle in order to improve the sensitivity. We collected data of powder X-ray diffraction at room temperature using BRUKER AXS D8 diffractometer with Cu-Kα X-ray radiation (λ = 1.5418 Å). Data measurement was performed with a step size of 0.02°.
The phosphated alumina and goethite was investigated by Energy Dispersive X-ray Spectra (EDS) analysis using a ZEISS EVO 60 Scanning Electron Microscope equipped with an Oxford EDS Detector with an accelerating voltage of 10–15 kV. The data were analysed by the Lebail refinement using the Jana2006 software-package (Petrícek et al. 2014).
Dissolution with acid
We used the method of Strauss et al. (1997) to study the rate of dissolution of the phosphated goethite and aluminium oxide. We gently shook twelve 45 mg samples with 30 mL of 5 M hydrochloric acid. We measured the phosphate and the iron/aluminium concentrations in the solution in each of the twelve samples after the following periods: 1, 2, 5, 10, 15, 20, 30, 45, 60, 120, 180, and 240 min.
Fractionation procedures
For the Chang and Jackson (1957) method, 1 g of material was placed in a centrifuge tube and shaken with 50 mL M NH4Cl for 30 min, then centrifuged at 11380 G for 10 min. This step is supposed to remove water-soluble, loosely-bound phosphorus. The amount of phosphorus removed is supposed to be small and was discarded. The solid in the tube was then shaken with 50 mL 0.5 M NH4F for one hour, centrifuged and the clear supernatant collected for analysis of phosphorus. This is supposed to be aluminium phosphate. The solid saved was washed twice with 25 mL of saturated NaCl and then mixed with 50 mL 0.1 M NaOH on a shaker for 17 h. The suspension was centrifuged and P in the clear supernatant solution was determined. This is supposed to be iron phosphate. The solid saved after this extraction was washed twice with 25 mL portions of saturated NaCl. It was then extracted with 50 mL 0.25 M H2SO4 for one hour on a shaker. The suspension was centrifuged and phosphate was estimated in the supernatant solution. This is supposed to be calcium phosphate. The solid saved after this extraction was washed twice with 25 mL portions of saturated NaCl. It was then suspended in 40 mL of 0.3 M sodium citrate solution plus 1 g solid Na2S2O4. The suspension was heated in a water bath at 85 °C with constant stirring for 15 min. The supernatant solution after centrifugation was collected. The solid was washed twice with 25 mL portions of saturated NaCl and washings were mixed with the supernatant solution and recorded as reductant soluble P. The residue was extracted with 50 mL 0.1 M NaOH. The phosphate dissolved was recorded as occluded phosphate.
The Zhang and Kovar (2009) method is similar to that of Chang and Jackson (1957) but different in the following aspects. In the first step, the extract with 50 mL of M NH4Cl which was discarded in the Chang and Jackson method was included and designated as soluble and loosely bound P. The sequence of steps differed in that treatment with reducing agents followed extraction with sodium hydroxide, and preceded treatment with sulphuric acid. The reducing mixture differed in that it also contained 5 mL 1 M sodium bicarbonate. Further, washings with 25 mL saturated solutions of NaCl were not discarded but mixed with the preceding fraction. There was no estimation of “occluded P” in this method.
Analytical
Phosphate was determined by the method of Murphy and Riley (1962). Aluminium and iron were determined by Atomic Absorption Spectrophotometer (AAS, Perkin Elmer).
Further data
We used data from two previously published sources. In Barrow (1972) the effect of calcium concentration was studied. Two soils of high sorption capacity and different pH were mixed with different concentrations of CaCl2 and a range of concentrations of phosphate, molybdate or sulphate at 25 °C for 24 h. Sorption at constant solution concentration of the anions was interpolated. For phosphate and molybdate the concentration used was 0.2 mg L−1. For sulfate, for which sorption was weaker, the concentration was 10 mg L−1.
In Barrow and Shaw (1979), desorption of previously added phosphate into 0.03 M solutions of the chloride salts of monovalent cations ranging from Li+ to Cs+ was measured.
Results
Characteristics of the materials
The powder XRD plots showed that the hydroxyapatite and the goethite were single-phase, with no indication of any phase impurity. For the aluminium oxide, all peaks with significant intensity matched the cubic form (Fd-3 m space group). There were some low-intensity peaks coming from hexagonal alumina (space group P63/mmc) indicating that a small percentage was present as the hexagonal form (On-line resource 1).
Energy dispersive X-ray spectra (EDS) (On-line resource 1) is a semi-quantitative technique. Further the surfaces were rough and hence exact ratios of elements cannot be expected. Table 1 shows that, for the samples of goethite, the ratio Fe:O varied. We think this was partly because the crystal faces of goethite have different ratios and partly because poorly-crystalized goethite was used in order to better represent soil goethite. For both samples of aluminium oxide the ratios were about 2:3 as would be expected from the formula. In all cases, only a small portion of phosphate was present (Table 1). Potassium (derived from the KH2PO4 used) was detected on two ground samples of goethite showing that reaction with phosphate had conveyed negative charge to the surface. If precipitates of iron or of aluminium were present, the ratio of Metal:P would be close to 1:1, and no negative charge would have been present. Thus the phosphate molecules were present on the oxide surfaces rather than as precipitates.
Dissolution with acid
For goethite, phosphate was released faster than iron; however, it was not until 25 to 30% of the iron had been dissolved that most of the phosphate was released (Fig. 1a). Hence appreciable phosphate was not close to the surface but had penetrated the goethite. For aluminium oxide (Fig. 1b), dissolution was slower than for goethite with about 23% of the aluminium released by the end of the experiment (240 min) compared to nearly 40% of the iron. Release of phosphate from the aluminium oxide was not quite complete by the end of the experiment again providing evidence that phosphate had similarly penetrated the aluminium oxide.

Rate of dissolution of goethite (a) and of aluminium oxide (b) in 5 M hydrochloric acid. Both materials had been reacted with phosphate at 1250 mg kg−1 at 70 °C for 4 days
For a given fraction of oxide dissolved, more phosphate was released from aluminium oxide than from goethite (Fig. 2), indicating that penetration was deeper for goethite.

Relation between the fraction of phosphate dissolved and the fraction of the iron dissolved from goethite, or aluminium dissolved from aluminium oxide
Fractionation
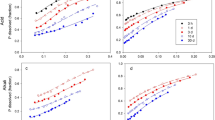
Both the Chang and Jackson (1957) method and the Zhang and Kovar (2009) method detected discrete phosphate compounds when there were none (Fig. 3). Further, when tested against hydroxyapatite (which is indeed a discrete phosphate compound but containing no aluminium), both methods record the presence of aluminium phosphate.

Results from applying two fractionation procedures to samples of goethite and of aluminium oxide which had been reacted with phosphate at 1250 mg kg−1 at 70 °C for 4 days and to hydroxyapatite. The fractionation procedure of Chang and Jackson (1957) is indicated by “a”; the procedure of Zhang and Kovar (2009) is indicated by “b”
Both methods detected a small amount of aluminium phosphate in goethite where there was none present; both detected appreciable iron phosphate in aluminium oxide where none was present; and both detected calcium phosphates in both goethite and aluminium oxide where no calcium was present.
In all cases, recovery of the phosphate present was incomplete. This appears to contrast with Chang and Jackson (1957) who reported complete recovery. However, they used discrete iron and aluminium phosphates whereas we used oxides into which phosphate had diffused. Further the rate at which apatite dissolves is known to differ between sources. They describe their source as “apatite from Florida ground to pass 100 mesh sieve”.
Discussion
Interpretation of our results
Consider the steps involved in the fractionation methods. The first extraction step involves ammonium fluoride solutions with the concentration, pH and duration varying between different versions. This step is supposed to dissolve discrete aluminium compounds. However fluoride would be expected to competitively displace some adsorbed phosphate. In the procedures tested here, the first extraction step involves neutral 0.5 M ammonium fluoride solution for one hour. Fluoride is most strongly sorbed by soil at pH 5.5, but appreciable sorption occurs at pH 7 (Barrow and Ellis 1986). Appreciable phosphate was indeed desorbed from the aluminium oxide but rather less from the goethite. If these procedures were applied to soil, the results would be recorded as discrete aluminium phosphate, even if none were present.
The second extraction step uses sodium hydroxide with the concentration and duration also varying between versions. The Hedley et al. (1982) method also uses sodium hydroxide. At the resulting high pH, considerable desorption of phosphate would be expected because phosphate is weakly adsorbed at high pH (Strauss et al. 1997). Indeed considerable desorption of phosphate occurred for both the goethite and the aluminium oxide. This would be recorded as discrete iron phosphate, again even though none was present.
The third extraction is with 0.25 M sulfuric acid for one hour. Other methods (Hedley et al. 1982; Beauchemin et al. 2003) use M hydrochloric acid. We have shown that treatment with 5 M hydrochloric acid dissolves both goethite and aluminium oxide and releases phosphate. We think that 0.25 M sulfuric acid also dissolved appreciable iron and aluminium oxides and so released phosphate. Similarly, this is recorded as calcium phosphate. We suggest that the amount of calcium phosphates present in soil has been over-estimated when detection and measurement is by acid extraction in fractionation procedures.
Other evidence for the existence of discrete calcium phosphate compounds in soils
When calcium phosphate fertilisers are added to soil, discrete calcium phosphate compounds must be present and will persist for some time. Nevertheless, we think that the importance of calcium phosphates in acid soils has been exaggerated, for example by Penn and Camberto (2019). Belief in the importance of calcium phosphates rests on three kinds of evidence. One is the results from soil phosphate fractionation schemes. We have shown this to be ill founded. Gu et al. (2020) have recently also shown that fractionation methods overestimate calcium bound P.
The second is from observations that the presence of calcium ions increases phosphate sorption compared to say sodium ions. This effect of calcium ions has been accepted as strong evidence for the presence of calcium phosphates by Penn and Camberto (2019). However, this effect is not confined to phosphate: similar effects of calcium concentration occur for both molybdate and sulfate over a wide range of pH values (Fig. 4). It is difficult to explain these effects by the formation of precipitates. They are better explained in terms of the electric potential of the surface. As pointed out by Barrow and Shaw (1979), this potential develops because of the separation of charge at the soil surface. It is affected by the concentration of electrolyte, because when this changes, the distribution of the charge changes. The activity of an adsorbing ion at the surface (and hence the amount of adsorption), depends not only on its activity in the bulk solution but also on the potential of the surface. If the potential of the surface changes because of a change in electrolyte concentration and consequent change in the distribution of charge, the activity of the ion in solution must also change in order to maintain the same activity of the adsorbed ion at the surface and thus the same amount of adsorption. Part of the change in activity in solution arises because of changes in the activity coefficient but a change in concentration is also involved.

Effect of initial calcium concentration on sorption of phosphate, molybdate and sulfate. Values for calcium concentration are plotted on a square root scale
Further, the monovalent cations form a sequence with desorption decreasing as the atomic number increased (Fig. 5). It is also difficult to explain these effects by the formation of precipitates. For these ions, differences in the hydration energy and in polarizability mean that the higher the atomic number the greater the proportion of ions in the Stern layer and the smaller the proportion which have a water molecule between them and the surface (Shainberg and Kemper 1966, 1967). These differences between species of cation in the distribution of charge near the surface induce an effect on the potential of the surface which is analogous to that caused by changes in the concentration of an individual electrolyte.

The third comes from physical techniques, most recently from synchrotron-based X-ray Absorption Near-Edge Structure (XANES). In such techniques the signal from soil samples is compared with that from standard substances. For example Eriksson et al. (2015) detected significant signal which they interpreted as coming from calcium phosphates. We query whether these signals necessarily came from discrete calcium phosphates. In one sense, calcium phosphates should be present in almost all soils; adsorbed phosphate always carries some negative charge; calcium ions are usually the main cations present; hence many of the adsorbed phosphate ions will form an ionic bond with calcium ions. Possibly these give rise to a signal similar to that from discrete calcium phosphate compounds.
The phosphate particle hypothesis
As pointed out by Russell and Prescott (1916), the main problem with this hypothesis is that it does not explain observations. They were the first to produce phosphate sorption curves and to show that sorption depended on period of reaction. They argued that such observations could not be matched by the hypothesis that phosphate existed in a series of discrete particles of different solubilities. Since then we have learned that phosphate reaction also depends on temperature, ionic strength and pH and that desorption does not necessarily follow the same track as adsorption. None of these effects can be quantitatively explained by the particle hypothesis, but can be explained by the specific adsorption-penetration hypothesis: for a summary see Barrow (1999).
Despite these considerations, many defend the particle hypothesis. Consider, for example, the following equation reproduced from Penn and Camberto (2019):
Al(OH3)(s) + H+ + H2PO4−(aq) ↔ AlPO4 · 2H2O(s) (variscite) + H2O (1).
This equation says that phosphate ions detach aluminium atoms from a crystal of aluminium hydroxide producing a compound (variscite) which then forms a separate precipitate. It is not clear how the presence of phosphate ions causes the aluminium atoms to break their bond with oxygen atoms, one of the strongest bonds in chemistry, nor how the sparingly-soluble aluminium phosphate molecule could combine with other aluminium phosphate molecules to form a separate particle. It is more probable that the phosphate molecule would simply react with the aluminium hydroxide surface and penetrate it. Analogous arguments apply to reaction of phosphate with iron (hyr)oxides. In the present work, we reacted aluminium oxide and goethite with phosphate solution at a pH close to 4, a pH supposed to favour eq. (1). We did not find any evidence of a separate phase in energy dispersive X-ray spectra studies. Further, in our dissolution studies, if a precipitate (variscite or strengite) had been formed, we would expect the ratio of phosphate to metal released to be approximately constant. This was not what happened (Fig. 6). Rather, the ratio decreased as the fraction dissolved increased. This is consistent with penetration of the phosphate.

Ratio of instantaneous slopes calculated from the fitted lines in Fig.1 plotted against the fraction of the metal dissolved
There have been many studies in which soils were reacted with phosphate at a range of concentrations and at different values for pH. If iron or aluminium phosphate precipitation occurred, in such studies, that would be most likely at low pH and at high phosphate concentration. As the appropriate pH or concentration of phosphate was exceeded there would be a sharp decrease in concentration and a concomitant increase in apparent sorption. We are not aware of this behaviour under any circumstances.
Physical techniques have also been used in attempts to identify phosphorus compounds present in soil. In such techniques a spectrum obtained from soil is matched by adding spectra obtained from mixtures of known phosphorus compounds. In most cases, several different combinations of possible constituents describe the observations almost equally well. However the main problem with such techniques is, like chemical fractionation, assumptions in give rise to assumptions out. That is, the conclusions depend on the standard spectra used. For example, Prietzel et al. (2013) used discrete aluminium and iron phosphates and concluded that they were important constituents. On the other hand, the standards used by Eriksson et al. (2015) included phosphate adsorbed to aluminium and iron oxides. They concluded that a large proportion of the phosphate was in this form rather than as being present as discrete iron or aluminium.
Conclusions
In the introduction, we noted that there are two theories about the nature of soil phosphate. We call one the adsorption-diffusion theory and note that it quantitatively describes observations not only of phosphate reaction but also the reaction of other specifically adsorbed anions and cations. We call the other the precipitate-particulate theory and note that it has not been shown to quantitatively describe such observations. In the work reported here we question the three lines of evidence on which this theory is based and especially we provide data to show that fractionation schemes identify discrete compounds where none are present. The question we now address is whether the two theories are mutually exclusive or whether they can coexist.
Penn and Camberto (2019) recently pictured sorption and precipitation as processes that do coexist. As pointed out above, we do not think this is realistic. However, there appear to be two situations in which both forms of phosphate may occur. One is for young soils in which the primary minerals derived from the rock are still present. It has been put to us that another possibility exists; that after sufficient time some of the penetrated phosphate might again form minerals, in effect, a reversal of normal soil development. We cannot exclude this possibility but note that we have studied soils that have been fertilised for over 100 years and found that all of the observations were consistent with the adsorption-diffusion theory (Barrow and Debnath 2014, 2015; Barrow et al. 2016).
The differences between the precipitate-particulate theory and the adsorption-diffusion theory are not merely academic. The adsorption-diffusion theory explains why each application of phosphate makes all subsequent applications more effective (Barrow et al. 2018). This effect is very important in determining appropriate rates of application of phosphate. We think that under-appreciation of this causes over-application of phosphate and consequent problems with contamination of water bodies. The precipitate-particulate theory does not seem to have any mechanism to explain this and we suggest that adherence to this theory contributes to this problem.
Finally, we note that methods for studying soil phosphate are based on the notion that discrete fractions are present. This is also true of the Hedley scheme even though the fractions are not identified. If instead, soil phosphate is thought to be present as a continuum, then a different approach is needed; one reflecting this continuum. Perhaps it would be profitable to explore whether measuring the rate of dissolution in some standard chemical would reflect that continuum.
References
Alobeedallah H, Ellis JL, Rohanizadeh R, Coster H, Dehghani F (2011) Preparation of nanostructured hydroxyapatite in organic solvents for clinical applications. Trends Biomater Artif Organs 25:12–19
Barrow NJ (1999) The four laws of soil chemistry: the Leeper lecture 1998. Aust J Soil Res 37:787–829
Barrow NJ (1972) Influence of solution concentration of calcium on the adsorption of phosphate, sulfate and molybdate by soils. Soil Sci 113:175–180
Barrow NJ (1983) A mechanistic model for describing the sorption and desorption of phosphate by soil. J Soil Sci 34:733─750
Barrow NJ, Barman P, Debnath A (2018) Three residual benefits of applying phosphate fertilizer. Soil Sci Soc Am J 82:1168–1176
Barrow NJ, Bowden JW (1987) A comparison of models for describing the adsorption of anions on a variable charge mineral surface. J Colloid Interface Sci 119:236–250
Barrow NJ, Debnath A (2014) Effect of phosphate status on the sorption and desorption properties of some soils of northern India. Plant Soil 378:383–395
Barrow NJ, Debnath A (2015) Effect of phosphate status and pH on sulphate sorption and desorption. Europ J Soil Sci 66:286–297
Barrow NJ, Debnath A, Chattergee S (2016) Effect of pH and prior phosphate application on the reaction of fluoride with soils from northern India. Europ J Soil Sci 67:294–230
Barrow NJ, Ellis AS (1986) Testing a mechanistic model 3. The effects of pH on fluoride retention by a soil. J Soil Sci 37:287–293
Barrow NJ, Shaw TC (1979) Effect of ionic strength and nature of the cation on desorption of phosphate from soil. J Soil Sci 30:53–65
Beauchemin S, Hesterberg D, Chou J, Beauchemin M, Simard RR, Sayers DE (2003) Speciation of phosphorus in phosphorus-enriched agricultural soils using X-ray absorption near-edge structure spectroscopy and chemical fractionation. J Environ Qual 32:1809–1819
Bowden JW, Posner AM, Quirk JP (1977) Ionic adsorption on variable charge mineral surfaces. Theoretical charge development and titration curves. Aust J Soil Res 15:121–136
Chang SC, Jackson ML (1957) Fractionation of soil phosphorus. Soil Sci 84:133–144
Eriksson AK, Gustafsson JP, Hesterberg D (2015) Phosphorus speciation of clay fractions from long-term fertility experiments in Sweden. Geoderma 241:68–74
Gu C, Dam T, Hart S, Turner B. L, Chadwick O, Berhe A. A, Hu Y, Zhu M (2020) Quantifying uncertainties in sequential chemical extraction of soil phosphorus using XANES spectroscopy. Env Sc Just accepted
Hall AD, Amos A (1906) XXVII.—the determination of available plant food in soils by the use of weak acid solvents. Part II. J Chem Soc Trans 89:205–222
Hedley MJ, Stewart JWB, Chauhan BS (1982) Changes in inorganic and organic soil phosphorus fractions induced by cultivation soil practices and by laboratory incubations. Sci Soc Am J 46:970–976
Lindsay WL (1979) Chemical equilibria in soils. Wiley
Marcus Y (1988) Ionic radii in aqueous solutions. Chem Rev 88:1475–1498
Murphy J, Riley JP (1962) A modified single solution method for the determination of phosphate in natural waters. Anal Chim Acta 27:31–36
Penn CJ, Camberto JJ (2019) A critical review on soil chemical processes that control how soil pH affects phosphorus availability to plants. Agriculture 9:120–138
Petrícek V, Dušek M, Palatinus L (2014) Crystallographic computing system JANA2006: general features. Zeitschrift für Kristall - Crystalline Materials 229:345–352. https://doi.org/10.1515/zkri-2014-1737
Posner AM, Barrow NJ (1982) Simplification of a model for ion adsorption on oxide surfaces. J Soil Sci 33:211–231
Price G (ed) (2006) Australian soil fertility manual, 3rd edition, fertilizer industry Federation of Australia and CSIRO, p 45
Prietzel J, Dümig A, Wu Y, Zhou J, Klysubun W (2013) Synchrotron-based P K-edge XANES spectroscopy reveals rapid changes of phosphorus speciation in the topsoil of two glacier foreland chronosequences. Geochim Cosmochim Acta 108:154–171
Russell EJ, Prescott JA (1916) The reaction between dilute acids and the phosphorus compounds of the soil. J Agric Sci 8:65–110
Shirai T, Watanabe H, Fuji M, Takahasi M (2009) Structural properties and surface characteristics on aluminium oxide powders. Ann Rep Ceram Res 9:23–31
Shainberg I, Kemper WD (1966) Hydration status of adsorbed cations. Proc Soil Sci Soc Amer 30:707–713
Shainberg I, Kemper WD (1967) Ion exchange equilibrium on montmorillonite. Soil Sc 103:4–9
Strauss R, Brümmer GW, Barrow NJ (1997) Effects of crystallinity of goethite II rates of sorption and desorption of phosphate. Europ J Soil Sci 48:101–114
Yoldas BE (1973) Hydrolysis of aluminium alkoxides and bayerite conversion. J Appl Chem Biotechnol 23:803–809
Zhang H, Kovar JL (2009) Fractionation of soil phosphorus. In: Kovar JL, Pierzyhski GM (eds) Methods of phosphorus analysis in soils, 2nd edn. North Carolina State University, Raleigh, pp 50–60
Acknowledgements
The authors acknowledge Dr. Partha Pratim Jana for his help and support. Nilanjan Roy acknowledges CRF, IIT Kharagpur for instrumental facility and CSIR for a junior research fellowship.
Author information
Authors and Affiliations
Corresponding author
Additional information
Responsible Editor: Hans Lambers.
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
ESM 1
(PDF 515 kb)
Rights and permissions
About this article

Cite this article
Barrow, N.J., Sen, A., Roy, N. et al. The soil phosphate fractionation fallacy. Plant Soil 459, 1–11 (2021). https://doi.org/10.1007/s11104-020-04476-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11104-020-04476-6