
Abstract
Background and aims
Take-all, caused by the soilborne pathogen Gaeumannomyces graminis var. tritici, (Ggt), is an important root disease of wheat. Continuous wheat cropping has been shown to induce take-all decline (TAD). This research investigated the mechanisms of TAD in 13 New Zealand soils in two experiments and identified the associated microorganisms using denaturing gradient gel electrophoresis (DGGE).
Methods
In Experiment 1, a sterile sand/maize-meal mixture inoculated or not inoculated with Ggt, was added at 4 % (w/w) to sterilised and non-sterilised soils to determine their ability to suppress take-all, and to help identify the nature of suppression. Experiment 2 investigated the transferability of suppressive properties in five of the soils from Experiment 1. The microbial communities of these five soils were analysed using PCR-DGGE.
Results
Ten of the soils were able to suppress take-all but the suppression was biological in nature in only four of these soils. The suppressive properties of two of the soils were transferred to a γ-irradiated base soil amended with Ggt, indicating that suppression could be specific in nature (i.e., attributed to a specific microorganism or group of microorganisms). The suppressive properties in one soil were not transferrable, suggesting a general form of suppression, most probably because the conditions in the soil were suitable for other microorganisms to compete with Ggt. DGGE analyses of the microbial communities for five of the soils showed similar banding patterns for those with similar forms of suppression (specific, general and non-suppressive) and identified the potential microorganisms that distinguished them.
Conclusion
These distinguishing microorganisms are likely to independently or interactively have a function in suppressing take-all.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Take-all, caused by the soilborne pathogenic fungus Gaeumannomyces graminis (Sacc.) von Arx & Oliver var. tritici Walker, is one of the most important root diseases of wheat (Triticum aestivum L.) worldwide (Weller et al. 1985; Cook 2003). Controlling this disease is difficult, mainly because breeding take-all resistant wheat cultivars has been unsuccessful and fungicides offer limited control (Weller et al. 2002). In the absence of effective control methods, growing non-susceptible crop hosts in rotation for 1–2 years is commonly used to manage the disease (Yarham 1981; Cook 2003).
Use of real-time quantitative polymerase chain reaction (qPCR) to estimate Ggt DNA concentrations in soils from wheat fields in New Zealand (Cromey et al. 2004) showed that pre-sowing Ggt DNA concentrations increased (from 106 to 1419 pg g−1 soil) with increasing years of successive wheat cropping, probably due to inoculum carry-over. However, where soils had 3–4 years of successive wheat crops and relatively high inoculum concentrations (>325 pg g−1 soil) before sowing, subsequent crops had low severity of take-all, indicating that disease development may have been suppressed. The phenomenon whereby take-all reduces when wheat is grown continuously in the same field is known as take-all decline (TAD), and is one of the most common forms of natural suppression of this disease (Hornby et al. 1998). It is characterised by an initially low take-all level during the early cropping year(s), an outbreak of the disease between 2 and 7 years and subsequently a decline of the disease with increased yields (Rovira and Cook 1981; Mazzola 2002; Weller et al. 2002). Therefore, it seemed likely that the New Zealand wheat soils described above could be demonstrating TAD.
The mechanisms involved in TAD have often been defined as ‘general’ and ‘specific’ (Graham and Mitchell 1999; Cook 2003). General suppression develops when soil conditions (e.g., soil types and soil physico-chemical conditions) are suitable for many antagonistic activities of the total microbial biomass in the soil (Weller et al. 2002; Weller 2007). This includes competition for resources, antagonism in lesions by secondary colonists, and stimulation of host defence mechanisms, which together lower the inoculum potential of Ggt. Hence, no specific microorganism or a selected group of microorganisms is solely responsible for the effect (Cook 2003; Janvier et al. 2007). This form of suppression has been reported to be non-transferrable between soils (Cook and Rovira 1976; Rovira and Wildermuth 1981; Kwak and Weller 2013). In contrast, specific suppression is attributed to particular antagonistic microorganisms that limit host infection and the secondary spread of the pathogen, using similar mechanisms as in general suppression (Graham and Mitchell 1999; Cook 2003). Since this suppression is mediated by specific microorganisms, its characteristic is transferable between soils (Cook 2003).
Many soil microorganisms, representing different taxonomic groups, have been shown to exhibit antagonistic activity against Ggt. The reported mechanisms of activity include cross-protection of the roots by Phialophora graminicola (Deacon 1976; Wong et al. 1996; Zriba et al. 1999), hyphal lysis of Ggt by a sterile red fungus (Limonomyces roseipellis) (Shankar et al. 1994; Aberra et al. 1998; Andjic et al. 2005), antibiosis by a Trichoderma sp. (Simon 1989; Duffy et al. 1996), a Bacillus sp. (Kim et al. 1997), a Pseudomonas sp. (Weller 1983; Raaijmakers and Weller 2001) and a Penicillium sp. (Hornby et al. 1998), as well as competition by Microdochium bolleyi (Kirk and Deacon 1987a, b). Some actinobacteria, in particular Streptomyces spp., have also been reported to be associated with TAD soils (Sivasithamparam and Parker 1978; Andrade et al. 1994a). Since so many different species of microorganisms have been reported to exhibit activity against Ggt, it seems likely that different microorganisms are involved interactively in take-all suppression.
To date, take-all suppressive soils have not been reported in New Zealand, although previous research on some soils (Cromey et al. 2004) suggested that the phenomenon has occurred. The present study aimed to compare the ability of New Zealand soils with different cropping histories to suppress take-all, determine the potential suppressive mechanisms associated with this effect, and to characterise the microbial communities associated with the suppressive mechanisms using denaturing gradient gel electrophoresis (DGGE).
Materials and methods
Soil sampling and processing
Soils from 13 cropping fields in the South Island, New Zealand, were used in this study. Information on previous cropping histories was obtained from growers, and each soil was assigned a specific field code (Table 1). Soil H15, which had been cropped with 5 years of perennial ryegrass (Lolium perenne L.), was included as a non-wheat, putative non-suppressive control. Soil P7, from a field cropped with wheat for 9 consecutive years and, therefore, potentially a take-all suppressive soil, was also included. The remaining 11 soils were identified in an earlier survey that investigated Ggt DNA concentrations in field soils previously cropped with wheat (Cromey et al. 2004). These 11 soils were from fields grown with wheat for 3 or 4 years, which potentially could express TAD, or from fields with high pre-sowing Ggt DNA concentrations (greater than 325 pg Ggt DNA g−1 soil), but low severity of take-all in the field [take-all index < 25, derived from the formula described by Cook (2003), Table 1] in the subsequent wheat crop. All soils, except for soil P7, were collected after crop harvest in March 2004, dried, sieved and stored at 4 °C until use. Soil P7 was collected from within a crop when the flag leaves had fully emerged in Dec 2004 and treated in a similar manner. Storage of soils at 4 °C was considered a suitable method as the soil microbial biomass C and enzyme activities have been reported to be least affected during storage (Lee et al. 2007).
Soils were collected by cutting 10 soil blocks (each ~15 × 15 × 15 cm) from the surface of each field along a ‘W’ pattern within a 1-ha zone (van Elsas et al. 2002). Locations (latitudes and longitudes) where soils were taken were recorded using a global positioning system (Table 1). The blocks were partially dried overnight at 25 °C and then passed through a 4 mm sieve. Stones were discarded but crop debris was retained.
A sample of each soil (300 g) was sent to South Australia Research & Development Institute (SARDI) for quantification of post-harvest Ggt DNA concentrations (Table 1) ( Ophel-Keller et al. 2008; Riley et al. 2010; Bithell et al. 2012). At the time of this study, specific primers for Ggt were not yet available, hence, the extracted DNA was analysed by qPCR using two sets of primers designed to quantify: (a) the combined DNA concentrations of both Ggt and Gaeumannomyces graminis var. Avenae (Gga, which causes take-all in oats, wheat and barley) in the soil, and (b) the DNA concentration of Gga in the same soil. The concentrations of Ggt DNA in each soil was then obtained from the difference between (a) combined Ggt and Gga DNA concentrations and (b) Gga DNA concentration [i.e., Ggt DNA concentration = (a)–(b)] of that soil. Specific primers targeting Ggt in wheat roots have recently been developed and were used for quantifying Ggt DNA concentrations on roots (van Toor et al. 2014). Quantitative PCR was performed using TaqMan® MGB probes and Quanti Tect Probe PCR kits Master Mix (Qiagen) in 10 μL reactions on an ABI PRISM® 7900HT Sequence Detection System. The primers and probes used in the analysis are shown in Table 2. Thermal cycling conditions comprised an initial step at 95 °C for 15 min, followed by a melting step of 95 °C for 15 s and an annealing/extension step (combined) of 60 °C for 1 min for 40 cycles. Cycle threshold (Ct) values (the number of cycles required for the fluorescent signal to cross the threshold value, i.e., exceeds background level) were converted to picograms of DNA per gram of soil using a reference standard curve made of 10-fold dilutions for DNA at 200 pg to 2 fg μl−1 of DNA from Gga isolate 137 T (Bithell et al. 2012).
Soil physico-chemical properties, including pH, available nitrogen (N), Olsen phosphorus (P), potassium (K), calcium (Ca), magnesium (Mg), sodium (Na), cation exchange capacity (CEC) and base saturation were measured (R. J. Hill Laboratories Ltd., Hamilton, New Zealand, Table 3). Overall, Olsen P and K concentrations in the soils were below the recommended range for cereal crops and N concentrations were at the lower end of the range (Morton et al. 2000). Other nutrients were not limiting to plant growth in any of the soils. The soils had CEC values within the normal range, indicating that they were capable of storing nutrients (Morton et al. 2000). Additional nutrients were therefore added to the pots (in solution form) to bring N, P and K levels up to the recommended range for cereal crops in preparation for the pot experiments.
Experiment 1: screening of soils for take-all suppression
The pot experiment was set up in a randomised block design consisting of 13 soils, sterilised or non-sterilised, with or without added Ggt inoculum. Each treatment combination was replicated four times, giving 208 pots in total. Each pot (400 ml capacity, not free-draining) contained 300 g of test soil (sterilised or non-sterilised), which was amended with 4 % (w/w) of a sterile sand/maize-meal mixture inoculated or not inoculated with Ggt (Chng et al. 2013). Soils were sterilised by autoclaving twice at 121 °C for 1 h (Alef 1995), with sterility verified by plating soil dilutions onto nutrient agar and potato dextrose agar. The Ggt inoculum consisted of four New Zealand isolates, H11T3 R1/3, A3SL4, H9T3 R1/1.2 and Biomill1SC3 (Plant & Food Research Culture collection) obtained from the rhizomes of Elytrigia repens and roots of wheat. All four Ggt isolates were found to be pathogenic to wheat but not Avena sativa L., which Ggt does not infect (Deacon 1997), in a separate study using methods described by Chng et al. (2005). They were grown separately in sand/maize-meal mixtures, as described by Hollins et al. (1986), then pooled and mixed thoroughly into the soil.
Each pot of soil was planted with four pre-germinated wheat seedlings, and 20 ml of a nutrient solution were added to give final concentrations of N, P, K and S of 150, 50, 100 and 20 μg g−1 soil, respectively. The pots were then watered to field capacity (FC) at −5kPa by weight. The experiment was set up in growth chambers (Conviron, Controlled Environments Ltd, Canada). Chambers were maintained at 16 °C with 12 h light/12 h dark cycles and 80 % relative humidity for 4 weeks. Throughout this period, all pots were watered to FC by weight twice each week. Preliminary experiments (results not presented) showed that these conditions were optimal for pot bioassays investigating take-all suppression.
Disease assessment
After 4 weeks, roots were recovered, washed and assessed for take-all lesions by immersion in water and examination against a white background. Disease severity was scored as the percentage area of each root system covered with take-all lesions, being categorised as ‘slight’ (<25 % of root area covered with lesions), ‘moderate’ (25 − 75 % of root area covered with lesions) and ‘severe’ (>75 % of root area covered with lesions) (Hornby et al. 1998). The take-all rating (TAR) for each pot was then calculated using the formula, TAR = 1(a) + 2(b) +3(c), where a, b and c are the percentages of plants with slight, moderate or severe infection, respectively (Dyke and Slope 1978). This calculation leads to a maximum TAR of 300.
Statistical analyses and other calculations
Wheat plants grown in sterilised soils with no Ggt added (i.e., Ster + Ggt−) had no take-all lesions, indicating that there was no contamination and so they were excluded from further statistical analyses. All other TAR data were analysed with analysis of variance which included two sets of contrasts. One compared Ggt addition for the non-sterilised soils (i.e., ‘Ster − Ggt+’ against ‘Ster − Ggt−’), and the other compared the sterilisation treatments for soils amended with Ggt (i.e., ‘Ster − Ggt+’ against ‘Ster + Ggt+’).
The increases or changes in TAR in response to Ggt addition (i.e., ΔTAR Ggt ) and soil sterilisation (i.e., ΔTAR ster ), which would allow differentiation of their effects (Andrade et al. 1994b; Gilligan et al. 1994; Augustin et al. 1997), were calculated using the following formulae:
When ΔTAR Ggt, in response to the addition of Ggt inoculum, was less than the least significant difference (LSD) at the 5 % level (i.e., ΔTAR Ggt < LSD Ggt value), soils were categorised as suppressive to take-all. When ΔTAR ster, in response to soil sterilisation prior to adding Ggt, was greater than the LSD (i.e., ΔTAR ster > LSD ster value), the suppression was categorised as being biological in nature or ‘specific form of suppression’. Alternatively, when ΔTAR ster, in response to soil sterilisation prior to adding Ggt, was less than the LSD (i.e., ΔTAR ster > LSD ster value), the suppression was categorised as a ‘general form of suppression’. Thus, the soils could be separated into the following four categories:
-
1.
Non-suppressive with low levels of biological involvement (ΔTAR Ggt > LSD Ggt and ΔTAR Ster < LSD Ster );
-
2.
Non-suppressive with high levels of biological involvement (ΔTAR Ggt > LSD Ggt and ΔTAR Ster > LSD Ster );
-
3.
Suppressive with low levels of biological involvement (ΔTAR Ggt < LSD Ggt and ΔTAR Ster < LSD Ster );
-
4.
Suppressive with high levels of biological involvement (ΔTAR Ggt < LSD Ggt and ΔTAR Ster > LSD Ster ).
The pot bioassay was repeated for five of the same soils, comprising one ex-ryegrass soil of Category 2 (H15), and four wheat soils, two of Category 4 (H1 and H3), one of Category 3 (M2) and one of Category 2 (P7). The ex-ryegrass soil (H15) was used as a non-suppressive soil control. Therefore, no soil from Category 1 was used in the repeated bioassay. In this bioassay, six pre-germinated seedlings were planted, so that two of the seedlings could be used for DNA extraction and DGGE analysis, which investigated the microbial communities associated with the suppression characteristics (see Section on DNA extraction from rhizosphere samples).
Experiment 2: transferability of suppressive properties between soils
A pot experiment to test the transferability of suppressive properties between soils was set up using methods modified from those described by Shipton et al. (1973). The experiment consisted of 10 treatment combinations made up of five previously used test soils (H15, H1, H3, M2 and P7), which were each added at 1 % w/w (non-sterilised) to a sterilised base soil with or without added Ggt inoculum.
The base soil, collected from a field cropped with wheat, was air-dried, sieved and stored as for the test soils. Before use, the base soil was gamma (γ) irradiated at 2.5 Mrad (Schering Plough Animal Health Ltd., Upper Hutt, New Zealand) (Alef 1995). Sterility of the soil was confirmed by plating as previously described for Experiment 1.
The Ggt inoculum comprised the same four isolates as used in Experiment 1. They were separately grown in oat kernels as described by Weller and Cook (1983). Prior to use, the oat kernels were air-dried at 20 °C for 4 days and ground to about 1 mm diameter particles with a grinder (model FR15, Girmi, Italy) run for four 5 s durations. The four ground cultures were thoroughly mixed, and then added at 0.1 % (w/w) into the respective combined base and test soils. The non-inoculated soils were amended with sterile dried and ground oat kernels.
Subsequent planting, nutrient treatment, incubation conditions and disease assessments were as described in Experiment 1. The pots were laid out in a randomised complete block design, with each treatment replicated four times, giving 40 pots in total. The TAR data were analysed with analysis of variance, and the increases in mean TARs for each soil, in response to the Ggt treatments (±), were calculated as [Mean TAR of (Ggt+)] − [Mean TAR of (Ggt−)].
Microbial communities
DNA extraction from rhizosphere samples
Material from the repeated pot trial of Experiment 1, in which soils were screened for suppression to Ggt, was used in a DGGE study. In order to focus on relevant microbial populations associated with suppression, only material from pots of that experiment that contained non-sterilised soil and also Ggt inoculum (20 of the 80 pots) was used. From each of the 20 pots, two plants per pot were selected, and roots from these that had tightly adhering soil were pooled. Thus, in the DGGE study, there were a total of 20 root samples, comprising four replicates for each of the five soils, all non-sterilised and with Ggt inoculum added.
DNA extraction was performed on unwashed roots, that had loosely adhering soil shaken off, using methods modified from those described by Russell and Bulman (2005). The roots from each pot were cut into 1 cm pieces, pooled and mixed, and then a 100 mg sub-sample was taken and placed in a 2 mL screw-cap microfuge tube containing 1 g of 2.3 mm stainless steel beads. To each tube, 1 mL of CTAB buffer (20 mM Tris pH 8, 20 mM EDTA, 0.8 M NaCl and 2 % cetyltrimethylammonium bromide) and 200 μL of 5 % sarcosyl were added (Russell and Bulman 2005). The root samples were homogenised with a MiniBeadbeater8TM cell disrupter (Biospec Products, BioLab Scientific NZ) twice at 1 min each time, with a 1 min cooling in ice between the two disruptions. After incubating at 65 °C for 1 h, impurities from the solution were extracted by adding 800 μL of chloroform:iso-amyl alcohol (24:1), mixing and centrifuging at 17, 900 × g (Relative Centrifugal Force) for 1 min. The supernatant (consisting of genomic DNA) was transferred into a clean microfuge tube and precipitated with an equal volume of 100 % isopropanol and centrifuged at 17, 900 × g for 5 min. The resultant pellet was recovered and washed with 500 μL of 70 % ethanol, air-dried at room temperature for 30 min, and re-suspended in 50 μL of 10 mM TE buffer (10 mM Tris, 1 mM EDTA, pH 8). The quantity and the quality of the extracted genomic DNA were then determined by electrophoresis, running 2 μL aliquots of the DNA samples in a 1 % agarose gel and comparing them to bands of a high DNA mass ladder (Invitrogen New Zealand Ltd., Auckland). Each root sample yielded 10 to 30 ng μL−1 of genomic DNA.
PCR amplification for DGGE analysis
PCR reactions were performed with the extracted gDNA of each root sample using universal primers targeting 16S and the internal transcribed spacer (ITS) rDNA regions of the generic rhizosphere bacterial and fungal populations, respectively. More specific primers targeting the respective sub-groups, actinobacteria and ascomycota, were also used to amplify from the total extracted gDNA samples (Table 4).
As part of the DGGE method, one primer of each set had a G + C-rich sequence attached (Table 4) to prevent complete disassociation of the double stranded DNA product as they ran through the denaturing gradient in the gel (Heuer et al. 2001). Amplifications were performed in 25 μL reaction mixtures containing: 10–50 ng of template DNA, 200 μM each of dATP, dTTP, dGTP and dCTP (Roche diagnostics NZ Ltd., Auckland, New Zealand); 0.2 μM of each primer (Invitrogen New Zealand Ltd, Auckland); one unit of Taq DNA polymerase (Roche diagnostics NZ Ltd., Auckland, New Zealand); 10 × buffer (Roche diagnostics NZ Ltd., Auckland, New Zealand) and water (UltraPure™, Invitrogen New Zealand Ltd, Auckland). A reaction without added template DNA was used as a negative control. PCR reactions were performed in a GeneAmp® PCR System 9700 (Applied Biosystems, Foster City, U.S.A.) with an initial denaturation at 94 °C for 2 min, followed by 35 cycles of denaturation at 94 °C for 30 s, annealing at the respective temperatures for each primer set (Table 4) for 60 s and extension at 72 °C for 1 min, followed by a final extension at 72 °C for 10 min. The quality and quantity of the resulting amplicons were assessed by electrophoresis in a 1 % agarose gel and compared to bands of a low DNA mass ladder (Invitrogen New Zealand Ltd., Auckland).
Denaturing gradient gel electrophoresis (DGGE)
DGGE analysis was performed using a DCode Mutation Detection System (BioRad, Life Science Research, Hercules, California). To separate the amplified rDNA products, polyacrylamide gels (8 % w/v, using 40 % acrylamide:bisacrylamide 37.5:1) bound to a GelbondTM sheet (Life Science Research, Hercules, California) were used with the denaturing gradients specific for the four microbial groups as outlined in Table 5. These gradients were modified from those referenced in Table 4 according to the manufacturer’s protocol to optimise band resolution. Each gel lane was loaded with 8 μL of amplicon and the electrophoresis was performed in 1 × TAE buffer at 58 °C and 85 V for 16 h. Bands were visualised by silver staining according to the protocols of Heuer et al. (2001). For long term preservation, each DGGE gel, sandwiched between a GelbondTM sheet (BioRad, Life Science Research, Hercules, California) and a sheet of cellophane, was dried overnight at 60 °C.
DGGE band scoring and statistical analyses
DGGE band profiles were analysed using GelCompar II (Applied Maths, Sint-Martens-Latem, Belgium). For each root replicate, bands at a normalised migration distance on the gel were scored as either present or absent by the software. To ensure scoring accuracy, the bands were also visually inspected on a light box to confirm their presence or absence. The resulting binomial data was examined with cluster analysis using the Jaccard similarity coefficient, and the un-weighted paired-group method with arithmetic mean (UPGMA) (Manly 2005). For each microbial group, the bands that differentiated the soils from one another were assessed with principal component analyses (PCA). In this comparison, a band was considered present only when it was in the same position in at least three of the four replicates of a soil type, but treated as absent if in fewer than three replicates. All analyses were carried out with GenStat Release 9.2 (VSN International Ltd. UK).
Recovery, re-amplification, cloning and sequencing of DGGE bands
Distinguishing DGGE bands, as determined from PCA, were excised from the dried gels, re-amplified, cloned and sequenced. Recovery of DNA from the bands was achieved by transferring each excised band into a 1.5 mL microfuge tube containing 50 μL of 1 × TAE buffer (pH 8), and incubating at 50 °C for 2 h with occasional vortexing to release the DNA into the buffer. Re-amplification was performed using 1 μL of this mixture as a template following the PCR conditions described above for DGGE analysis with the exception of the G + C-rich sequence no longer being attached to one of the primers. All amplified products were cloned using a TOPO TA Cloning® kit (Invitrogen, Auckland, New Zealand) following the methods of the manufacturer. Five clones were randomly selected from each amplified product (representing one DGGE band), purified using a QIAquick PCR purification kit (QIAGEN, Biolab Ltd., Auckland, New Zealand), and sequenced (Macrogen sequencing services, Macrogen Ltd. Seoul, Korea) with either the forward or reversed primers without GC nucleotides. The resulting nucleotide sequences (i.e., chromatograms) from the clones of the same band were first visually inspected for their quality using Sequencher® version 4.1 (Gene Codes Corporation, US). Sequences of good quality were then edited and aligned to generate a consensus sequence, which was then subjected to a Genbank Blastn search (National Center for Biotechology Information, US) to identify the microorganism, with the closest sequence similarity to the band in question. The microorganism, which represented the majority of the five clones (i.e., greater than three) from each band, was considered the dominant species that made up that band.
Results
Ggt DNA concentration
Results of pre-sowing, post-crop harvest and changes in the soil Ggt DNA concentrations for the growing season of all the soils tested are summarised in Table 1. Natural Ggt inoculum concentrations decreased in nearly all the soils (except H9) between pre-sowing and post-harvest, generally reducing take-all risk levels from high to medium or low for most of the soils. Soils C12, H1, H3, H10, L9, and M2 showed the greatest reductions in Ggt DNA concentrations (>1000 pg g−1 soil).
Experiment 1: occurrence of take-all suppressive soil
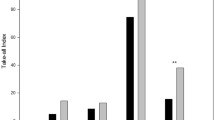
Disease severity (mean TARs) in roots differed substantially among the tested soils (P = 0.014), with plants in soils B6, G2 and M2 having less disease (TARs of 110, 102 and 100, respectively) than those from the other soils (TARs 119 to 135). Overall, the mean TARs were greatest in soils which were sterilised prior to the addition of Ggt inoculum (Ster + Ggt+), followed by the non-sterilised soils with and without added Ggt (i.e., Ster − Ggt + and Ster − Ggt−) (P < 0.001; mean TARs of 160, 112 and 94, respectively).
There was a significant interaction (P = 0.01) between soils, soil sterilisation and the addition of Ggt. Plants from Ggt-amended soils, H15, C12, H1, H3, L9 and P7, had greater TARs in sterilised than non-sterilised soils (Fig. 1). However, addition of Ggt did not increase TARs in most non-sterilised soils (P = 0.377), indicating biological involvement in disease suppression. The exception was in soils P7, H2 and H15, where TARs increased (by 56, 56 and 52, respectively) following the addition of Ggt inoculum (Fig. 1) to the non-sterilised soils.

Mean take-all ratings (TARs) of plants grown in soils with or without sterilisation and addition of Ggt inoculum. Error bar represents LSD0.05 (df = 114)
Changes in mean TARs (i.e., ΔTARs) of plants after Ggt addition, relative to ΔTARs of plants in response to soil sterilisation, and the resulting four suppression categories for tested soils are shown in Fig. 2. Category 1 contained a non-suppressive soil (H2) with low level of biological involvement, and Category 2 contained non-suppressive soils [P7 and H15 (non-wheat)], with indications of moderate to high levels of biological involvement. In Category 3, soils (G2, M2, B6, H7, H9 and H10) suppressed take-all, but had low level of biological involvement, while the soils in Category 4 (C12, L9, H1 and H3) were suppressive and had moderate to high levels of biological involvement. Amongst these four soils, H1 and H3 were most suppressive to take-all and had the highest degree of biological involvement.

Change in mean take-all ratings (ΔTARs) of plants grown in non-sterilised versus sterilised soils amended with Ggt before planting using the formula ΔTAR ster = [Mean TAR of (Ster + Ggt+)] − [Mean TAR of (Ster − Ggt+)] (x-axis), relative to ΔTARs of plants grown in non-sterilised soils with and without added Ggt inoculum using the formula ΔTAR Ggt = [Mean TAR of (Ster − Ggt+)] − [Mean TAR of (Ster − Ggt−)] (y-axis). The dotted lines are LSDs0.05 for each axis to compare each mean ΔTAR with ‘0’. Non-significant ΔTARs lie to the left of the vertical or below the horizontal lines. Soil categories are defined as 1 – non-suppressive with low involvement of biological factors, 2 – non-suppressive with high involvement of biological factors, 3 – suppressive with low involvement of biological factors and 4 – suppressive with high involvement of biological factors
When the experiment was repeated with five of the soils, H1, H3, M2, P7 and H15, there were consistent increases in mean TARs of the five non-sterilised test soils in response to the addition of Ggt inoculum (Fig. 3). The effects of soil sterilisation showed similar trends to those obtained previously, with the roots of Ster + Ggt + plants being the most severely infected, followed by the Ster − Ggt + and the Ster − Ggt − plants (P < 0.001 and mean TARs of 173, 133 and 95, respectively, Fig. 3). There were significant interactions between soils and sterilisation for both Ggt − and Ggt + soils (P = 0.013 and <0.001, respectively). The ΔTARs of plants after Ggt addition were similar to those in the previous results (Figs. 1 and 2). However, the levels of biological involvement (as indicated by soil sterilisation) differed for two of the soils, M2 and P7. Soil M2 had an increased mean TAR (+56) after sterilisation, causing it to be reclassified from Category 3 to 4 and P7 had a decreased mean TAR (−42), causing it to be reclassified from Category 2 to 1.

Change in mean take-all ratings (ΔTARs) of plants grown in non-sterilised versus sterilised soils amended with Ggt before planting (x-axis), relative to ΔTARs of plants grown in non-sterilised soils with and without added Ggt inoculum (y-axis) in the repeated pot experiment. The formulae used for calculating changes were ΔTAR ster = [Mean TAR of (Ster + Ggt+)] − [Mean TAR of (Ster − Ggt+)] and ΔTAR Ggt = [Mean TAR of (Ster − Ggt+)] − [Mean TAR of (Ster − Ggt−)]. The dotted lines are LSDs0.05 for each axis to compare each mean ΔTAR with ‘0’. Non-significant ΔTARs lie to the left of the vertical or below the horizontal lines. Soil categories are defined as 1 – non-suppressive with low involvement of biological factors, 2 – non-suppressive with high involvement of biological factors, 3 – suppressive with low involvement of biological factors and 4 – suppressive with high involvement of biological factors
Experiment 2: transferability of suppressive properties between soils
The mean TARs differed between test soils when 1 % of each soil (non- γ-irradiated) was added to the γ-irradiated base soil (P = 0.05), with the greatest difference being for soil M2 (mean TAR = 131, Fig. 4), and similar differences for the other four soils (mean TARs of 94–109). Soils amended with Ggt had more disease (mean TAR = 125) than those without added Ggt (mean TAR = 86, P < 0.001). There was also a significant interaction between test soils and the presence and absence of Ggt (P = 0.014). Addition of Ggt inoculum significantly increased mean TARs of soils amended with H15 or M2 (by 75 and 63, respectively), whereas the TARs for soils amended with H1, H3 or P7 did not change significantly (Fig. 4). This indicated that the characteristics of soils H1, H3 and P7 were transferred into the base soil, but those of soil M2 did not. As expected, soil H15 (ex-ryegrass, non-suppressive soil control) when added into the base soil, did not help reduce the disease.

Change in the mean take-all ratings (ΔTARs) of plants due to the addition of Ggt inoculum to γ-irradiated base soil, to which 1 % w/w of five different test soils was added. The dotted line represents LSD0.05 to compare a mean with ‘0’. Non-significant ΔTARs after the addition of Ggt inoculum lie below this line. The formula used was ΔTARs = [Mean TAR of (Ggt+)] − [Mean TAR of (Ggt−)]
Microbial communities
The banding profiles generated on the DGGE gels revealed 40, 44, 28 and 40 individual bands for the respective four microbial groups: bacteria, fungi, actinobacteria and ascomycota across the five soils, H1, H3, M2, P7 and H15. With 28 bands, the actinobacteria population appeared to be less diverse in these soils compared to the other groups. Table 6 shows the number of bands which were present in at least three of the four replicates for each of the soils. In general, there were noticeable differences among the soils, with the bacterial group being the most diverse, generating 19–31 bands across all soils. The greatest range difference for the bacterial group was observed between soils H1 (19 bands) and H3 (31 bands). The remaining soils, H15, M2 and P7 (9 year wheat soil) showed similar diversities in their bacterial populations (23–24 bands, Table 6). For the fungal group, the highest number of bands was generated by soil H3. In contrast, soils P7 and M2 generated the lowest number of fungal bands (18 and 19, respectively) while H1 and H15 produced similar numbers of fungal bands (25 and 24, respectively). For the actinobacterial group, soil H3 produced the least bands (16 bands), while the rest of the soils had bands in the range of 25–30. The ascomycota appeared to be the least diverse group. Among the soils, M2 had the least diverse asccomycota communities. (19 bands, Table 6).
Similarity analysis
Similarity analysis of the total 152 DGGE bands of all the soils and their replicates for all four microorganism groups showed greater variation between the band profiles for different soils, than between profiles for replicate samples of the same soil. Similarities between replicate samples of the same soil varied from 0.84 to 0.95, whereas similarities between samples of different soils were noticeably smaller, varying from 0.55 to 0.76. The resulting UPGMA dendogram derived from the calculated mean similarities clearly groups the samples by their soil origins (Fig. 5). Cluster analysis of the soils showed that the ex-ryegrass soil (H15) and the 9 year wheat soil (P7) were most similar in their band profiles, followed by soils H1 and H3. In contrast, the band profile of soil M2 appeared to be most different from the other four soils (Fig. 5).

A dendogram showing relationships among the five soils (H1, H3, H15, P7 and M2) from cluster analysis of the 152 bands generated from all the DGGE profiles. Similarity is expressed as a value of the Jaccard correlation coefficient with a value of ‘0’ indicating the soils had no bands in common (i.e., completely different), whereas a value of ‘1’ indicated the soils had the same bands present (i.e., completely identical). The numbers next to the soil codes are the soil replicate numbersy
Identification of bands that differentiated the soils
Principal component analyses (PCA) of all the bands present in all four microbial groups showed that among the five soils, 88 % of the variation was accounted for by the first three principal components (36, 34 and 18 %, respectively). A scatter plot matrix representing the first three principal components is shown in Fig. 6. The first principal component (Face a) differentiated the soils into two groups with the first group comprising of H1, H3 and M2, and second group comprising P7 and H15. The second component (Face b), further separated soil H3 from H15 and P7, while M2 was further separated from soils H1 and H3. In the third component (Face c), soils H1 and H3 were separated further from each other, and H15 (ryegrass soil) and P7 (9 year wheat soil) were separated slightly from each other as well (Fig. 6).

A scatter plot matrix of the first three principal components showing separation of the five soils (H1, H3, M2, P7 and H15) from the analysis of bands separated on the four DGGE gels specific for generic bacteria, generic fungi, actinobacteria and ascomycota. The plots represent three side views (faces a, b and c) of a single cube formed by plotting the data using the three axes (components) in three dimensions. The component coefficient refers to the length of the vector in relation to the principal component of interest
As the aim of this study included identification of key microorganisms that differentiated suppressive from the non-suppressive soils, only bands responsible for separating the five soils analysed in PCA were excised from the gels and sequenced. The number of distinguishing bands selected from the DGGE gels of bacteria, fungi, actinobacteria and ascomycota were, six, seven, six and four, respectively (see supplementary figure).
Sequence analysis of the distinguishing DGGE bands from soils of different suppression characteristics
The microorganisms found in Genbank whose corresponding sequences most closely matched those of the distinguishing DGGE bands are shown in Table 7. Within the rhizosphere of wheat plants grown in the five test soils, there were five, five, five and three distinguishing genera for the respective microbial groups: bacteria, fungi, actinobacteria and ascomycota (Table 7). In general, the dominating distinguishing bacterial genus was Pseudomonas, which represented two of the six distinguishing bands, and was most common in soil H3. The genus, Penicillium, dominated among the distinguishing bands in both the fungal (three out of seven bands) and ascomycota (two out of four bands) DGGE profiles. While in the actinobacterial DGGE profile, the most common distinguishing bands belonged to the genus, Streptomyces (two out of six bands). The remaining distinguishing bands from the bacterial, fungal, actinobacterial and ascomycota DGGE profiles were made up of various genera (Table 7). Only one species, Penicillium echinulatum, was identified in more than one distinguishing band, two from the fungal (bands 6 and 33) and one from the ascomycota (band 1) DGGE profiles.
Six distinguishing bands were common in soils H1, H3 and M2. Their sequences matched Streptomyces bingchengensis, Terrabacter sp., Nocardioides sp., Fusarium lateritium, Microdochium bolleyi and an uncultured fungal clone, whose next closest matched relative was Mortierella elongata.
Soils H1 and H3 shared two distinguishing microorganisms, Nocardioides oleivorans and Gibberella zeae. The sequences from the four distinguishing bands generated only by soil H3 were identified as Pseudomonas putida, P. fluorescens, Penicillium echinulatum and Penicillium allii. However, the single distinguishing band produced by M2, also matched Penicillium echinulatum.
The similarity between the band profiles of soils H15 and P7 (second group in PC analyses) was largely due to five bands of the actinobacterial group. The nucleotide sequences of these bands, which were found only in these two soils, matched those of Glycomyces sp. Streptomyces sp., Actinosynnema violaceoruber, Hongia sp. and Actinokineospora diospyrosa.
Discussion
Take-all suppression and mechanisms
Monoculture of a susceptible host, the presence of Ggt, and at least one severe outbreak of take-all are the three components required for take-all decline (TAD) to develop (Weller et al. 2002). The soils used in this study had either low disease severity coupled with high Ggt DNA concentrations in the field prior to sowing, or were in their third, fourth or ninth year of continuous wheat cropping (Table 1). Although the timing of any previous take-all outbreaks in the fields from which test soils were collected was unknown, their histories met two of the three components for developing TAD. Whipps (1997a) considered that soils in which diseases should have occurred but failed to materialise were ideal for the initial selection of suppressing microorganisms because those soils were already likely to have a range of indigenous potential antagonists present. Therefore, the 12 wheat soils selected for this study were considered to have potential to contain microorganisms capable of suppressing take-all.
Although the test soils used contained natural Ggt inoculum, the concentrations of the pathogen, as indicated by Ggt DNA, varied between soils (Table 1). These uneven amounts of natural Ggt inocula make it difficult to determine the different suppressive capacities of the soils, as it is not possible to tell whether low disease severity was due to low inoculum pressure or to high suppressive capacity caused by the presence of potential antagonists (Rovira and Cook 1981). The addition of a uniform mixture of virulent Ggt inocula to the soils would thus ensure a consistent level of disease pressure and allow differentiation of the suppressive activity among non-sterilised soils. Results showed that non-sterilised soils differed in their ability to suppress take-all, as indicated by the ΔTARs following the addition of Ggt inoculum to the non-sterilised soils. The pot bioassay used in Experiment 1 was therefore, capable of effectively distinguishing the levels of suppression of the soils in question, and confirmed the existence of TAD in New Zealand wheat soils (i.e., those with increased TARs below dotted line parallel to x-axis in Fig. 2). The reliability of the method was shown by the repeated pot bioassay, which demonstrated consistent increases in the mean TARs of the five test soils in response to the addition of Ggt inoculum (i.e., consistent changes in TARs and soils remained below the dotted line parallel to x-axis, Fig. 3).
In this study (Experiment 1), biological involvement (investigated by autoclaving the soils before the addition of Ggt inoculum), was indicated to be associated with take-all suppression in most of the soils (except G2 and M2), since the autoclaving of soils eliminated this property and caused TARs to increase after Ggt inoculum was added. In a similar study, Cook et al. (1986) found that the infection efficiency of added Ggt was greater in a pasteurised (60 °C) suppressive soil, and caused more lesions on the infected roots than on roots in a pasteurised conducive soil. A similar result was also reported by Shipton et al. (1973), who used three soils from fields that had been cropped with wheat and barley for 4–22 years. When those soils were heat-treated at 60, 70, 80 or 121 °C for 30 min, suppression was eliminated in all soils at all four temperatures. Hence, the conditions used to sterilise soils in the present study were likely to have removed any biological components involved in disease suppression.
Variation in the degree of take-all suppression exhibited by the soils in this study could be attributed, in part, to the impact of their different wheat cropping histories on the soil microbiota. Repeated monoculture of a particular crop has been known to favour particular microbial species. For wheat, this can lead to the establishment of microbiota involved in TAD development (Baker and Cook 1974). However, biological buffering in relation to the Ggt inoculum in the soil can be achieved by introducing different host crops to the rotation sequence, since each crop results in different microorganisms dominating in the rhizosphere (Baker and Cook 1974). In contrast, repeated monoculture will favour perpetuation of dominating microbial species in the rhizosphere (Baker and Cook 1974). Hence, the dominating microbial species, or the antagonists associated with TAD, are likely to differ between soils with different durations of wheat monoculture, different rotation crops and soil types. This is likely to result in the differing capability of soils, to suppress take-all observed in this study. However, it is important to note that other factors, such as soil types and local environmental conditions are just as important in contributing to TAD development. For instance, physico-chemical characteristics (such as pH) of a soil have often been shown to influence take-all suppression (Whipps 1997b).
Another explanation for the TAD mechanism during wheat monoculture is that there may be a gradual reduction in populations of aggressive Ggt strains over time as they are replaced by less aggressive strains that allow other soil antagonists to compete for colonisation sites on plant roots (Shipton 1977). Lebreton et al. (2004) found that populations of aggressive Ggt strains in the soil peaked after three or four consecutive wheat crops and then decreased to the same level as less aggressive Ggt strains by the sixth crop. The shift in Ggt population structure was not discernible in the present study as virulent Ggt inocula had been added to the soils to ensure a high disease pressure.
Both general and specific suppression were indicated in the different soils tested in this study. In Experiment 1, soils G2, M2, H7, H9, B6 and H10 demonstrated no or very little increase in TARs following autoclaving prior to the addition of Ggt inoculum (i.e., increases in TARs beyond the dotted line parallel to the y-axis, Fig. 2). This indicated that the apparent suppressive activity may be the result of ‘general suppression’. However, in the repeated pot bioassay, the TAR of soil M2 was significantly greater in the autoclaved soil than in the non-sterilised soil, suggesting the possibility of a specific biological basis for suppression. In Experiment 2, the ability of soil M2 to suppress take-all was not transferred to the γ-irradiated base soil, again indicating a general form of suppression. In a series of studies to demonstrate TAD with wheat monoculture in Dutch polder soils, Gerlagh (1968) found that although TAD could be due to specific suppression of the pathogen, general suppression usually occurred before the onset of specific suppression. This may be the case for soil M2, but more work is required to validate whether general forms of suppression will become specific over time.
Experiment 2 (Fig. 4) showed that soils H1 and H3 had specific suppression, or suppression caused by specific microbial populations, because when small amounts of these soils were transferred to the γ-irradiated inoculated base soil, similar degrees of suppression occurred to those found in non-sterilised soils in Experiment 1. Similar studies by Shipton et al. (1973) showed that the addition of 1 % (w/w) of a non-treated take-all suppressing soil into a methyl bromide fumigated soil amended with G. graminis, re-instated the suppressive capacity of the fumigated soil.
Three years of successive wheat cropping in the H1 and H3 fields would be considered sufficient for the development of specific suppression or TAD in soils (Baker and Cook 1974). The Ggt DNA concentrations of the two soils reduced during the 2003/04 season to 12 and 10 % of pre-sowing levels, respectively (Table 1), are consistent with reports of the development of specific suppression. A decline in the levels of the natural Ggt inoculum in a soil from beginning to the end of a growing season is considered an indication that the pathogen has been suppressed, probably by inhibition of saprophytic growth and survival of Ggt in the plant debris or soil (Hornby 1983). However, this phenomenon alone is not a conclusive indication for the development of a specific suppressive soil. Some studies have shown that soil Ggt continually reduced through degradation of the crop debris (Garrett 1970, 1975, 1981; Shipton 1981; Skou 1981). These studies, however, did not specify the exact amount of Ggt inoculum being reduced. Future studies should investigate the relationship between the Ggt DNA concentrations in/on the roots of host plants and disease severity, during the crop cycle of successive wheat monoculture. Such a study would improve understanding of suppression processes/mechanisms on the parasitic and saprotrophic growth of Ggt in the soil.
The soil expected to have the greatest suppressive capacity was P7, which had 9 years of continuous wheat cropping and low concentration of Ggt DNA at the start of the experiments (Table 1). However, in Experiment 1 (both main and repeated pot bioassays), the addition of Ggt inoculum to this soil consistently caused large increases in disease severity (Figs. 2 and 3, above the dotted line parallel to the x-axis), indicating non-suppression, whereas the addition of 1 % P7 soil to the γ-irradiated base soil in Experiment 2 resulted in apparent suppression of the disease (Fig. 4). This suggests that the soil may have possessed a specific form of suppression. Since the soil categorisation indicated variable levels of biological involvement with low ability to suppress take-all development (Figs. 2 and 3), the putative antagonistic effects of the microorganisms in P7 might have been impaired, repressing their ability to suppress the disease in bulk soil. A similar result was reported by Andrade et al. (1994b), who found that it was possible to transfer disease suppression to a chemically and physically different soil from a soil with an initially low level of suppressive activity, resulting in reductions in take-all and increases in shoot dry weights. In addition, at the time of soil sampling, P7 had Ggt DNA concentration of 168 pg g−1 soil (predicted to be of medium risk to take-all), but the crop had a rather high field take-all index of 44.1 (Table 1), suggesting that the natural Ggt inoculum in the field could either be highly pathogenic or, the existing natural antagonists against Ggt were at insufficient levels to control the pathogen. Additionally, the onset of disease outbreak and development of TAD vary with cropping histories, locations and local environmental factors (Rovira and Cook 1981; Mazzola 2002; Weller et al. 2002), hence the soils, which originated from different regions in this study, could also be at different stages of TAD development. For these reasons, soil P7 was classified as a non-suppressive soil in the current study.
Microbial communities associated with take-all suppression
In the DGGE analysis of the five soils, H1, H3 and M2 were clustered according to their putative suppressive soil characteristics: specific, specific and general forms of suppression, respectively (Figs. 5 and 6). Given that the band profiles of P7 and the ex-ryegrass non-suppressive soil control, H15, were similar, and that the natural Ggt DNA concentration and take-all severity (measured as take-all index, TAI) in the field crop were both high (Table 1), it is likely that the potential suppression mechanisms in P7 had not yet developed at the time of soil sampling.
The total number of DGGE band migratory positions, which represented the numbers of different microorganism species, varied among the five soils but not between soils with similar forms of suppression (Table 6), suggesting that the suppression characteristics in these soils might not be associated with the diversity of the microbial community structures. This agrees with the findings of Vojinović (1972) in: Vojinović 1973; Baker and Cook 1974) who reported similar numbers of microorganisms from the bulk TAD and non-suppressive soils using conventional isolation techniques. A study by Hiddink et al. (2005) using DGGE to investigate the effects of single and mixed cropping systems on bacterial and fungal communities in rhizosphere soils, also showed no differences in the microbial diversities among the soils. However, after conducting principal component analyses on the numbers of bands and intensity of band data, they were able to cluster the soils according to their locations, suggesting that the abundance of individual microorganism species inhabiting the soils were specific to the soil/cropping systems. As not all DNA templates amplify equally due to occurrence of anomalies with PCR, results derived from analysing band intensity data should be considered with caution (Garbeva et al. 2004) and therefore, were not used in this study.
Past studies using conventional isolation techniques to compare the microbial communities in TAD and non-suppressive soils, had revealed that bacteria could be linked to the specific form of suppression, and fungi, to the general form of suppression (Vojinović 1972 in: Pope 1972; Vojinović 1973; Baker and Cook 1974). In the current DGGE analysis, with the exception of Streptomyces sp., there is no reported antagonistic activity of the distinguishing microorganisms detected in the non-suppressive soils (P7 and H15) (Table 7) against Ggt. In fact, some of these distinguishing microorganisms (e.g., Actinosynnema sp. and Hongia sp.) are common in soils not suppressive to take-all (Conn 2005). Hence, these distinguishing microorganisms are unlikely to be associated with TAD. In addition, it might be possible that these indigenous species could have inhibited the activities of microorganisms potentially responsible for take-all suppression, and thus the expression of TAD in soil P7.
In contrast, the distinguishing microorganisms which were only detected in the putative suppressive soils (both specific and general), but not in the non-suppressive soils in the DGGE analysis (Table 7) could interactively play a role in take-all suppression. Streptomyces spp., Nocardiodes spp., and Microdochium bolleyi have all been shown to suppress Ggt in both plate and plant assays (Lascaris and Deacon 1991; Coombs and Franco 2003; Conn and Franco 2004). For example, Nocardiodes sp., Streptomyces argenteolus and S. caviscabies were demonstrated to control Ggt and reduce root infection by up to 25, 37 and 41 %, respectively, in a pot assay using field soils (Coombs et al. 2004). Similarly, Kirk and Deacon (1987a; b) showed that M. bolleyi was able to control Ggt and reduce take-all when used as a seed-applied inoculum. However, there is no recorded evidence of S. bingchengensis, Terrabacter sp., Penicillium echinulatum and Fusarium lateritium, which were also present in the suppressive soils, being effective in antagonising or controlling Ggt. The efficacy of F. lateritium as a biocontrol agent to other soil pathogens however, is widely documented (Sitepu and Wallace 1984; Christen et al. 2005; Ho et al. 2005; John et al. 2005). Since most of the distinguishing microorganisms detected in the suppressive soils (H1, H3 and M2) have been reported to antagonise Ggt or other soilborne pathogens to some degree, it is likely that some of them may have specific functions in disease suppression in general, but they may not be responsible for the more specific form of suppression.
On the other hand, the distinguishing microorganisms, Pseudomonas putida, P. fluorescens, Nocardioides oleivorans, Gibberella zeae and Penicillium allii are more likely to be responsible for the specific form of suppression since these microorganisms were detected only in soils H1 and/or H3 that had specific form of suppression, but not in soil M2 that had general form of suppression. The antagonistic effects of many Pseudomonas spp. on Ggt and the development of TAD through the production of antibiotics (e.g., phenazine-1carboxylic acid and 2,-4-diacetylphloro-glucinol) and other metabolites (e.g., siderophore pseudobactin and hydrogen cyanide), are widely documented (Kloepper et al. 1980; Brisbane and Rovira 1988; Thomashow and Weller 1988; Harrison et al. 1993; Hornby et al. 1998; Raaijmakers and Weller 1998; Coombs et al. 2004). Hence, they are likely to play a part in the specific suppression in the soils. Attempts were made to amplify the 16S regions using primers [S-G-Psmn-028-a-S-20 (PS-for) and S-G-Psmn-1258-a-A-18 (PS rev-GC)] specific for the Pseudomonas genus (Widmer et al. 1998) in the current research, but these primers were not successful (results not presented). The ascomycete, Gibberella zeae was also unique in the specific suppressive soils in the current research. As there has been no reported antagonistic activity of G. zeae against Ggt or any other soilborne pathogens, the role of G. zeae in the specific suppression of TAD soil is unknown. In vitro antagonism to Ggt has been demonstrated by many Penicillium spp. isolated from wheat soils (Sivasithamparam and Parker 1980; Dewan and Sivasithamparam 1988; Hornby et al. 1998), but this has not been reported for the two species, Penicillium echinulatum and P. allii, identified in the current study (Table 7).
According to Kisand and Wikner (2003), it is possible that a single DNA sequence (individual microorganism) can have more than one migration point or multiple melting domains on a DGGE gel. As a result, several bands can be generated from a single sequence. In the current research, two bands (band 6 from soil M2 and band 33 from soil H3) on the same fungal DGGE gel matched P. echinulatum, indicating that two migratory positions are possible for ITS DNA sequences of the same fungus from different sources. Since soils H3 (specific suppression) and M2 (general suppression) shared P. echinulatum as a distinguishing microorganism, and it was the only microbial band unique to the general suppression soil, it would appear there are no specific microbes that distinguish it from the other soils in the current study.
This study is the first report of using DGGE to investigate microbial communities in the rhizosphere of wheat plants grown in soils showing different forms of take-all suppression (i.e., specific, general forms of suppression and non-suppression) and identify the key microorganisms unique to these soils. However, similar to other molecular applications (e.g., restriction fragment length polymorphism and 454 or next generation sequencing) used for profiling microbial communities of environmental samples, there are drawbacks and limitations of DGGE. For instance, one rDNA sequence may produce two or more bands on the DGGE gel due to heterogeneities of some rDNA sequences (Nübel et al. 1996 in: Heuer and Smalla 1997), such as those observed in Penicillium echinulatum in the current study. Alternatively, only one band may be observed for closely related, and even for phylogenetically-unrelated rDNA sequences of different microbial species due to the species having similar electrophoretic mobility (Heuer and Smalla 1997; Kowalchuk et al. 2003). These limitations might also have occurred in the current study. In addition, not all the distinguishing microorganisms present in the rhizosphere samples in the current study were detected by the DGGE method. The fungal and the bacterial primers yielded similar or fewer bands than the actinomycota- and ascomycota-specific primers for most of the five soils (Table 6), suggest that either the ascomycota or actinomycota were the most dominating microbial groups in the rhizosphere of wheat plants in these soils, or that the universal primers were showing a bias to those microorganisms present in higher numbers. To discount the latter possibility, future experiments involving profiling the microbial community in the rhizosphere of plants should include a wider range of group-specific primers. In addition to those employed in the current study, primers specific to oomycetes, basidiomycetes, pseudomonads, and Streptomyces spp. should be used as well. Another factor which might have affected the specificity of the method was the short rDNA fragments (300–550 bp) targeted by the primer sets used in the current study. Although some authors (Vainio and Hantula 2000) have reported high quality band resolution on DGGE using DNA fragments in excess of 1 kb, short fragments are generally known to provide better resolution but they yield less sequence information (Kowalchuk et al. 2003). Nearly all the distinguishing microorganisms identified in the DGGE analysis in the current study have been shown to control Ggt or other soilborne pathogens to some degree, and thus likely to be associated with the different forms of take-all suppression. However, more research is required to show that these distinguishing microbes were indeed responsible for the described results. The antagonistic effects of the identified microbes on Ggt either independently or interactively in plate and plant assays and under field conditions have to be determined.
References
Aberra MB, Seah S, Sivasithamparam K (1998) Suppression of the take-all fungus (Gaeumannomyces graminis var. tritici) by a sterile red fungus through induced resistance in wheat (Triticum aestivum) seedling roots. Soil Biol Biochem 30:1457–1461
Alef K (1995) Sterilisation of soil and inhibition of microbial activity. In: Nannipieri AKP (ed) Methods in applied soil microbiology and biochemistry. Academic, London, pp 52–54
Andjic V, Cole ALJ, Klena JD (2005) Taxonomic identity of the sterile red fungus inferred using nuclear rDNA ITS 1 sequences. Mycol Res 109:200–204
Andrade OA, Mathre DE, Sands DC (1994a) Natural suppression of take-all disease of wheat in Montana soils. Plant Soil 164:9–18
Andrade OA, Mathre DE, Sands DC (1994b) Suppression of Gaeumannomyces graminis var. tritici in Montana soils and its transferability between soils. Soil Biol Biochem 26:397–402
Augustin C, Jacob HJ, Werner A (1997) Effects on growth of wheat plants of isolates of Gaeumannomyces/Phialophora-complex fungi in different conditions of soil moisture, temperature, and photoperiod. Eur J Plant Pathol 103:417–426
Baker KF, Cook RJ (1974) Biological control of plant pathogens. Freeman, San Francisco
Bithell SL, McKay A, Butler RC, Ophel-Keller K, Hartley D, Cromey MG (2012) Predicting take-all severity in second-year wheat using soil DNA concentrations of Gaeumannomyces graminis var. tritici determined with qPCR. Plant Dis 96:443–451
Brisbane PG, Rovira AD (1988) Mechanisms of inhibition of Gaeumannomyces graminis var. tritici by fluorescent pseudomonads. Plant Pathol 37:104–111
Chng SF, Cromey MG, Butler RC (2005) Evaluation of the susceptibility of various grass species to Gaeumannomyces graminis var. tritici. New Zealand Plant Protection, Vol 58. Proceedings of a conference Wellington, New Zealand, pp 261–267
Chng SF, Stewart A, Cromey MG, Dodd SL, Butler RC, Jaspers MV (2013) Effects of different rates of Gaeumannomyces graminis var. tritici inoculum for detecting take-all suppression in soils. Australas Plant Pathol 42:103–109
Christen D, Tharin M, Perrin-Cherioux S, Abou-Mansour E, Tabacchi R, Defago G (2005) Transformation of Eutypa dieback and Esca disease pathogen toxins by antagonistic fungal strains reveals a second detoxification pathway not present in Vitis vinifera. J Agric Food Chem 53:7043–7051
Conn VM (2005) Molecular interactions of endophytic actinobacteria in wheat and arabidopsis. PhD thesis. Flinder University
Conn VM, Franco CMM (2004) Effect of microbial inoculants on the indigenous actinobacterial endophyte population in the roots of wheat as determined by terminal restriction fragment length polymorphism. Appl Environ Microbiol 70:6407–6413
Cook RJ (2003) Take-all of wheat. Physiol Mol Plant Pathol 62:73–86
Cook RJ, Rovira AD (1976) The role of bacteria in the biological control of Gaeumannomyces graminis by suppressive soils. Soil Biol Biochem 8:269–273
Cook RJ, Wilkinson HT, Alldredge JR (1986) Evidence that microorganisms in suppressive soil associated with wheat take-all decline do not limit the number of lesions produced by Gaeumannomyces graminis var. tritici. Phytopathology 76:342–345
Coombs JT, Franco CMM (2003) Isolation and identification of actinobacteria from surface-sterilized wheat roots. Appl Environ Microbiol 69:5603–5608
Coombs JT, Michelsen PP, Franco CMM (2004) Evaluation of endophytic actinobacteria as antagonists of Gaeumannomyces graminis var. tritici in wheat. Biol Control 29:359–366
Cromey MG, Fraser PM, Francis GS, Mckay A, Stewart A, Walter M, Waipara NW, Wakelin S (2004) nfluence of crop management on the incidence of take-all of cereals in New Zealand. Proceedings of the third Australasian soilborne diseases symposium, Adelaide, pp 61–62
Deacon JW (1976) Biological control of the take-all fungus, Gaeumannomyces graminis, by Phialophora radicicola and similar fungi. Soil Biol Biochem 8:275–283
Deacon JW (1997) Modern mycology. Blackwell Science, Scotland
Dewan MM, Sivasithamparam K (1988) Occurrence of species of Aspergillus and Penicillium in roots of wheat and ryegrass and their effects on root rot caused by Gaeumannomyces graminis var. tritici. Aust J Bot 36:701–710
Duffy BK, Simon A, Weller DM (1996) Combination of Trichoderma koningii with fluorescent Pseudomonas for control of take-all on wheat. Phytopathology 86:188–194
Dyke GV, Slope DB (1978) Effects of previous legume and oat crops on grain yield and take-all in spring barley. J Agric Sci UK 91:443–451
Garbeva P, van Veen JA, van Elsas JD (2004) Microbial diversity in soil: selection of microbial populations by plant and soil type and implications for disease suppressiveness. Annu Rev Phytopathol 42:243–270
Gardes M, Bruns TD (1993) ITS primers with enhanced specificity for basidiomycetes - application to the identification of mycorrhizae and rusts. Mol Ecol 2:113–118
Garrett SD (1970) Pathogenic root-infecting fungi. Cambridge University Press, London, p 294
Garrett SD (1975) Cellulolysis rate and competitive saprophytic colonisation of wheat straw by foot-rot fungi. Soil Biol Biochem 7:323–327
Garrett SD (1981) Introduction. In: Asher MJC, Shipton PJ (eds) Biology and control of take-all. Academic Press Inc. (London) Ltd, London, pp 1–11
Gerlagh M (1968) Introduction of Ophiobolus graminis into new polders and its decline. Neth J Plant Pathol 74:1–97
Gilligan CA, Brassett PR, Campbell A (1994) Modeling of early infection of cereal roots by the take-all fungus - a detailed mechanistic simulator. New Phytol 128:515–537
Graham JH, Mitchell DJ (1999) Biological control of soilborne plant pathogens and nematodes. In: Sylvia DM, Fuhrmann JJ, Hartel PG, Zuberer DA (eds) Principles and applications of soil microbiology. Prentice Hall, New Jersey, pp 427–446
Harrison LA, Letendre L, Kovacevich P, Pierson E, Weller D (1993) Purification of an antibiotic effective against Gaeumannomyces graminis var. tritici produced by a biocontrol agent, Pseudomonas aureofaciens. Soil Biol Biochem 25:215–221
Heuer H, Smalla K (1997) Application of denaturing gradient gel electrophoresis and temperature gradient gel electrophoresis for studying soil microbial communities. In: Elsas JDV, Trevors JT, Wellington EMH (eds) Modern soil microbiology. Marcel Dekker, Inc, New York, pp 353–373
Heuer H, Krsek M, Baker P, Smalla K, Wellington EMH (1997) Analysis of actinomycete communities by specific amplification of genes encoding 16S rRNA and gel-electrophoretic separation in denaturing gradients. Appl Environ Microbiol 63:3233–3241
Heuer H, Wieland G, Schonfeld J, Schonwalder A, Gomes NCM, Smalla K (2001) Bacterial community profiling using DGGE or TGGE analysis. Environmental Molecular Microbiology : Protocols and Applications. Horizon Scientific Press, Wymondham, pp 177–190
Hiddink GA, Termorshuizen AJ, Raaijmakers JM, Bruggen AHCV (2005) Effect of mixed and single crops on disease suppressiveness of soils. Phytopathology 95:1325–1332
Ho MA, Squire LM, Sabeh NC, Giles DK, VanderGheynst JS (2005) Design and evaluation of a grapevine pruner for biofungicide application. Bioresour Technol 96:963–968
Hollins TW, Scott PR, Gregory RS (1986) The relative resistance of wheat, rye and triticale to take-all caused by Gaeumannomyces graminis. Plant Pathol 35:93–100
Hornby D (1983) Suppressive soils. Annu Rev Phytopathol 21:65–85
Hornby D, Bateman GL, Gutteridge RJ, Ward E, Yarham DJ (1998) Take-all disease of cereals: a regional perspective. CAB International, UK
Janvier C, Villeneuve F, Alabouvette C, Edel-Hermann V, Mateille T, Steinberg C (2007) Soil health through soil disease suppression: which strategy from descriptors to indicators? Soil Biol Biochem 39:1–23
John S, Wicks TJ, Hunt JS, Lorimer MF, Oakey H, Scott ES (2005) Protection of grapevine pruning wounds from infection by Eutypa lata using Trichoderma harzianum and Fusarium lateritium. Australas Plant Pathol 34:569–575
Kim D, Cook RJ, Weller DM (1997) Bacillus sp. L324-92 for biological control of three root diseases of wheat grown with reduced tillage. Phytopathology 87:551–558
Kirk JJ, Deacon JW (1987a) Control of the take-all fungus by Microdochium bolleyi, and interactions involving M. bolleyi, Phialophora graminicola and Periconia macrospinosa on cereal roots. Plant Soil 98:231–237
Kirk JJ, Deacon JW (1987b) Invasion of naturally senescing root cortices of cereal and grass seedlings by Microdochium bolleyi. Plant Soil 98:239–246
Kisand V, Wikner J (2003) Limited resolution of 16S rDNA DGGE caused by melting properties and closely related DNA sequences. J Microbiol Methods 54:183–191
Kloepper JW, Leong J, Teintze M, Schroth M (1980) Pseudomonas siderophores: a mechanism explaining disease-suppressive soils. Curr Microbiol 4:317–320
Kowalchuk GA, Os GJ, Aartrijk J, Veen JA (2003) Microbial community responses to disease management soil treatments used in flower bulb cultivation. Biol Fertil Soils 37:55–63
Kwak Y-S, Weller DM (2013) Take-all of wheat and natural disease suppression: a review. Plant Pathol J 29:125–135. doi:10.5423/PPJ.SI.07.2012.0112
Lascaris D, Deacon JW (1991) Colonization of wheat roots from seed-applied spores of Idriella (Microdochium) bolleyi: a biocontrol agent of take-all. Biocontrol Sci Tech 1:229–240
Lebreton L, Lucas P, Dugas F, Guillerm AY, Schoeny A, Sarniguet A (2004) Changes in population structure of the soilborne fungus Gaeumannomyces graminis var. tritici during continuous wheat cropping. Environ Microbiol 6:1174–1185
Lee YB, Lorenz N, Dick LK, Dick RP (2007) Cold storage and pretreatment incubation effects on soil microbial properties. Soil Sci Soc Am J 71:1299–1305. doi:10.2136/sssaj2006.0245
Manly BFJ (2005) Multivariate statistical methods : a primer, 3rd edn. Chapman and Hall, London
Mazzola M (2002) Mechanisms of natural soil suppressiveness to soilborne diseases. Proceedings of the 9th International Symposium on Microbial Ecology, August 2001, Amsterdam, pp 557–564
Morton J, Craighead M, Stevenson K (2000) Managing soil fertility on cropping farms. New Zealand Fertiliser Manufacturers Research Association and New Zealand Pastoral Agriculture Research Institute Ltd, New Zealand, p 46
Nikolcheva LG, Bärlocher F (2004) Taxon-specific fungal primers reveal unexpectedly high diversity during leaf decomposition in a stream. Mycol Prog 3:41–49
Nübel U, Engelen B, Felske A, Snaidr J, Wieshuber A, Amann RI, Ludwig W, Backhaus H (1996) Sequence heterogeneities of genes encoding 16S rRNAs in Paenibacillus polymyxa detected by temperature gradient gel electrophoresis. J Bacteriol 178:5636–5643
Ophel-Keller K, McKay A, Hartley D, Herdina CJ (2008) Development of a routine DNA-based testing service for soilborne diseases in Australia. Australas Plant Pathol 37:243–253
Pope AMS (1972) The decline phenomenon in take-all disease of wheat. PhD thesis. University of Surrey
Raaijmakers JM, Weller DM (1998) Natural plant protection by 2,4-diacetylphloroglucinol - producing Pseudomonas spp. in take-all decline soils. Mol Plant-Microbe Interact 11:144–152
Raaijmakers JM, Weller DM (2001) Exploiting genotypic diversity of 2,4-diacetylphloroglucinol-producing Pseudomonas spp.: characterization of superior root-colonizing P. fluorescens strain Q8r1-96. Appl Environ Microbiol 67:2545–2554
Riley IT, Wiebkin S, Hartley D, McKay AC (2010) Quantification of roots and seeds in soil with real-time PCR. Plant Soil 331:151–163. doi:10.1007/s11104-009-0241-5
Rovira AD, Cook RJ (1981) The nature and mechanisms of suppression. In: Asher MJC, Shipton PJ (eds) Biology and control of take-all. Academic, London, pp 385–415
Rovira AD, Wildermuth GB (1981) The nature and mechanisms of suppression. In: Asher MJC, Shipton PJ (eds) Biology and control of take-all. Academic Press Inc. (London) Ltd, London, pp 385–415
Russell J, Bulman S (2005) The liverwort Marchantia foliacea forms a specialized symbiosis with arbuscular mycorrhizal fungi in the genus Glomus. New Phytol 165:567–579
Shankar M, Kurtboke DI, Gillespie-Sasse LMJ, Rowland CY, Sivasithamparam K (1994) Possible roles of competition for thiamine, production of inhibitory compounds, and hyphal interactions in suppression of the take-all fungus by a sterile red fungus. Can J Microbiol 40:478–483
Shipton PJ (1977) Monoculture and soilborne plant pathogens. Annu Rev Phytopathol 15:387–407
Shipton PJ (1981) Saprophytic survival between susceptible crops. In: Asher MJC, Shipton PJ (eds) Biology and control of take-all. Academic Press Inc. (London) Ltd, London, pp 295–316
Shipton PJ, Cook RJ, Sitton JW (1973) Occurrence and transfer of a biological factor in soil that suppresses take-all of wheat in Eastern Washington. Phytopathology 63:511–517
Simon A (1989) Biological control of take-all of wheat by Trichoderma koningii under controlled environmental conditions. Soil Biol Biochem 21:323–326
Sitepu D, Wallace HR (1984) Biological-control of Sclerotinia sclerotiorum in luttuce by Fusarium lateritium. Aust J Exp Agric 24:272–276
Sivasithamparam K, Parker CA (1978) Effects of certain isolates of bacteria and actinomycetes on Gaeumannomyces graminis var. tritici and take-all of wheat. Aust J Bot 26:773–782
Sivasithamparam K, Parker CA (1980) Interaction of certain isolates of soil fungi with Gaeumannomyces graminis var. tritici on agar media. Aust J Bot 28:411–419
Skou JP (1981) Morphology and cytology of the infection process. In: Asher MJC, Shipton PJ (eds) Biology and control of take-all. Academic, London, pp 175–197
Thomashow LS, Weller DM (1988) Role of Phenazine antibiotic from Pseudomonas fluorescens in biological control of Gaeumannomyces graminis var. tritici. J Bacteriol 170:3499–3508
Vainio EJ, Hantula J (2000) Direct analysis of wood-inhabiting fungi using denaturing gradient gel electrophoresis of amplified ribosomal DNA. Mycol Res 104:927–936
van Elsas JD, Smalla K, Lilley AK, Bailey MJ (2002) Methods for sampling soil microbes. In: Hurst CJ, Crawford RL, Knudsen GR, McLnerney MJ, Stetzenbach LD (eds) Manual of environmental microbiology, 2nd edn. ASM Press, USA, pp 505–515
van Toor RF, Chng SF, Warren RM, Butler RC, Cromey MG (2014) he influence of growth stage of different cereal species on host susceptibility to Gaeumannomyces graminis var. tritici and on Pseudomonas populations in the rhizosphere. Australas Plant Pathol. doi:10.1007/s13313-014-0324-5
Vojinović ZD (1972) Antagonists from soil and rhizosphere to phytopathogens. Final technical report., Institute of soil science, Beograd
Vojinović ŽD (1973) The influence of micro-organisms following Ophiobolus graminis Sacc. on its further pathogenicity. EPPO Bulletin: 91–101
Weller DM (1983) Colonization of wheat roots by a fluorescent pseudomonad suppressive to take-all. Phytopathology 73:1548–1553
Weller DM (2007) Pseudomonas biocontrol agents of soilborne pathogens: looking back over 30 years. Phytopathology 97:250–256. doi:10.1094/phyto-97-2-0250
Weller DM, Cook RJ (1983) Suppression of take-all of wheat by seed treatments with fluorescent pseudomonads. Phytopathology 73:463–469
Weller DM, Zhang BX, Cook RJ (1985) Application of a rapid screening test for selection of bacteria suppressive to take-all of wheat. Plant Dis 69:710–713
Weller DM, Raaijmakers JM, Gardener BBM, Thomashow LS (2002) Microbial populations responsible for specific soil suppressiveness to plant pathogens. Annu Rev Phytopathol 40:309–348
Whipps JM (1997a) Developments in the biological control of soil-borne plant pathogens. Adv Bot Res 26:1–134
Whipps JM (1997b) Ecological considerations involved in commercial development of bioligical control agents for soil-borne diseases. In: van Elsas JD, Trevors JT, Wellington EMH (eds) Modern soil microbiology. Marcel Dekker, Inc., New York, pp 525–548
White TJ, Bruns T, Lee S, Tayler J (1990) Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. In: Innis MA, Gelfand DH, Sninsky JJ, White TJ (eds) PCR protocols: a guide to methods and applications. Academic, California, pp 315–322
Widmer F, Seidler RJ, Gillevet PM, Watrud LS, Giovanni GDD (1998) A highly selective pcr protocol for detecting 16S rRNA genes of the genus Pseudomonas (Sensu Stricto) in environmental samples. Appl Environ Microbiol 64:2545–2553
Wong PTW, Mead JA, Holley MP (1996) Enhanced field control of wheat take-all using cold tolerant isolates of Gaeumannomyces graminis var. graminis and Phialophora sp. (lobed hyphopodia). Plant Pathol 45:285–293
Yarham DJ (1981) Practical aspects of epidemiology and control. In: Asher MJC, Shipton PJ (eds) Biology and control of take-all. Academic, London, pp 353–384
Zriba N, Sherwood JE, Mathre DE (1999) Characterization and effectiveness of Phialophora spp. isolated from a Montana take-all suppressive soil in controlling take-all disease of wheat. Can J Plant Pathol Rev 21:110–118
Acknowledgments
The authors would like to thank South Australia Research & Development Institute, Adelaide for analysing soil Ggt DNA concentrations and the Ministry of Business, Innovation and Employment (MBIE) (Programme LINX0304) for funding the research.
Author information
Authors and Affiliations
Corresponding author
Additional information
Responsible Editor: Simon Jeffery.
Rights and permissions
About this article

Cite this article
Chng, S., Cromey, M.G., Dodd, S.L. et al. Take-all decline in New Zealand wheat soils and the microorganisms associated with the potential mechanisms of disease suppression. Plant Soil 397, 239–259 (2015). https://doi.org/10.1007/s11104-015-2620-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11104-015-2620-4