
Abstract
Background and aims
To assess how plant genotype and rhizosphere bacterial communities may interact, the genetic structure and diversity of bacterial communities in the rhizosphere soil of different Medicago truncatula genotypes were studied in relation to the plant carbon and nitrogen nutrition at the whole plant level.
Methods
The genetic structure and diversity of plant-associated rhizosphere bacterial communities was analysed by Automated Ribosomal Intergenic Spacer Analysis and 454-pyrosequencing. In parallel, the carbon and nitrogen nutrition of the plants was estimated by a phenotypic description at both structural level (growth) and functional level (using carbon and nitrogen isotope labeling and an ecophysiological framework).
Results
An early effect of the plant genotype was observed on the rhizosphere bacterial communities, while few significant differences were detected at the plant structural phenotypic level. However, at a functional level, the different Medicago truncatula genotypes could be distinguished by their different nutritional strategies. Moreover, a comparison analysis showed that ecophysiological profiles of the different Medicago truncatula genotypes were correlated to the genetic structure and the diversity of the rhizosphere bacterial communities.
Conclusions
The exploration of the genetic structure and diversity of rhizosphere bacterial communities combined with an ecophysiological approach is an innovative way to progress in our knowledge of plant-microbe interactions in the rhizosphere.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Rhizosphere microbial communities are able to interact with plants and, for some kind of interactions, play beneficial roles in plant health and growth (Buee et al. 2009; Franche et al. 2009; Harrison 1999; Raaijmakers et al. 2009; Richardson et al. 2009). Interactions between plants and microbes have been well documented (for reviews, see Buee et al. (2009); Harrison (1999); Hartmann et al. (2009); Jones et al. (2009a); Lambers et al. (2009)). As an example it has been demonstrated that plant genotype is one of the most important factors in the evolution of microbial communities (Buee et al. 2009). The influence of plant genotype is likely to occur through the release of rhizodeposits; significantly stronger rhizosphere effect towards bacterial communities was observed during the plant vegetative period than at later growth stages and can be ascribed to differential root exudation along the plant growth cycle (Mougel et al. 2006).
However, studies to date have avoided considering the bidirectional links between plants and microbial communities and focused either on the microbial or on the plant “side”. Consequently, a major objective is now to study the effect of the plant genotype on the shaping of the soil microbial communities, which could in return improve plant nutrient uptake. This challenge can now be attained by using novel and multidisciplinary approaches (Hartmann et al. 2009).
Recently developed microbiology techniques permit the characterization of microbial communities in complex soil environments (Ranjard et al. 2000; Singh et al. 2004). ARISA (for Automated Ribosomal Intergenic Spacer Analysis) was developed (Fisher and Triplett 1999), rapidly optimized and then validated as a sensitive and robust analysis method for studying soil microbial communities in both soil (Ranjard et al. 2003; Ranjard et al. 2001) and the plant rhizosphere (Mougel et al. 2006). In addition, pyrosequencing, a high-throughput technique allowing the analysis of microbial diversity, was used to examine bacterial communities in soil samples (Roesch et al. 2007). However, as stated above, microbiological studies generally did not assess nutritional variations of the associated plants.
While most plants acquire soil mineral nitrogen (N), predominantly nitrate and to lesser extent ammonium, legumes also possess the capacity to acquire atmospheric dinitrogen (N2), thanks to a symbiosis with bacteria belonging to the Rhizobiaceae family. Legumes contribute to diversifying cropping systems and decrease the need for external inputs (Jensen and Hauggaard-Nielsen 2003; Munier-Jolain and Carrouée 2003; Nemecek et al. 2008). Despite these advantages, legumes are still under exploited in European cropping systems mostly because of fluctuating yields resulting from abiotic and biotic constraints often affecting plant N nutrition. Plant N nutrition is known to interact closely with plant C nutrition (Marschner 1995; Minchin et al. 1981). Ecophysiological tools have been developed to understand the physiological bases involved in legume plant C and N nutrition (Moreau 2007; Moreau et al. 2012). However in these studies plants were grown in inert substrate inoculated with Rhizobium strain, the contribution of the entire rhizosphere microbial communities to plant nutrition was also not assessed.
The aim of our work was to assess plant genotype influence on the genetic structure of the rhizosphere bacterial communities in relation to the plant C and N nutritional strategies. The objective is to propose a global and multidisciplinary approach combining ecophysiology and microbial ecology to study plant-microbe interactions. A greenhouse experiment was conducted on seven genotypes of the legume plant model Medicago truncatula, which were contrasted in their genetic diversity (Ronfort et al. 2006). This study was confined to the vegetative period of Medicago truncatula during which roots and nodules are established and the rhizosphere effect towards bacterial communities is suspected to be stronger. On the one hand, microbiological tools were used to study the influence of the Medicago truncatula genotype on the genetic structure and diversity of the rhizosphere bacterial communities. On the other hand, the different nutritional strategies depending on the plant genotype were studied using an ecophysiological approach with three complementary approaches: a phenotypic growth description, C and N flux measurements using isotope labeling and an ecophysiological framework. Finally, the abundance of the rhizosphere bacterial communities was compared with the plant functional traits to bring some clues in the biological link between some phyla abundances and the plant nutritional strategies.
Materials and methods
Plant growth conditions and experimental design
The seven contrasted Medicago truncatula genotypes (DZA 045-6, DZA 315-16, DZA 315-26, F 83005-5, Jemalong A17, Jemalong J6 and SA 028064) used in this study have been chosen for their different geographic origins and genetic diversity (Ronfort et al. 2006). Seeds of Medicago truncatula genotypes were scarified, surface-sterilized (Mougel et al. 2006), vernalized at 4 °C during 48 h and germinated on 0.7 % (w/v) water agar plates at 25 °C in the dark. One germinated seed of each Medicago truncatula genotype was sown in pot containing 900 g of a silt-clay loam soil (Mas d’Imbert, France) which is poor in mineral N and rich in organic matter and hosts symbiotic microbes (Offre et al. 2007). This soil is characterized by low mineral nitrogen (N) content: 0.018 g.kg−1 of NO3-N and 0.002 g.kg−1 of NH4-N (NF ISO 14256-2). Physicochemical characteristics of this soil were determined as: 11 % of sand, 51 % of silt, 38 % of clay, pH = 8, 1.45 % of organic carbon (NF ISO 10694), 0.1 % of N (NF ISO 13878) and 0.1 % of phosphorus (NF ISO 11885; inductively coupled plasma atomic emission spectroscopy (ICP-AES)).
Plants were cultivated up to the end of vegetative period at a density of 45 plants. m−2 in a greenhouse, under a photoperiod of 15 h, a temperature of 26°/21 °C (day/night). Supplementary artificial light was supplied with sodium lamps (MACS 400 W; Mazda, Dijon, France) to complement photosynthetically active radiation. In order to characterize the interactions between Medicago truncatula and the soil microbial communities in conditions where the plant can interact with N2-fixing bacteria, plants were watered with a N-free solution (Moreau et al. 2008). An automatic watering scheme considering the weight of two control pots was used to maintain soil moisture at 45 % of its maximum soil water-holding capacity and a manual control watering was also done each weeks.
At three sampling dates (340, 634 and 934 degree-days after sowing), representing three vegetative developmental stages (4 leaves, first ramification and second ramification) (Moreau et al. 2006; Mougel et al. 2006), four plants per genotype were harvested and the leaf area was measured (LI-3100 Area Meter, Li-Cor Inc., Lincoln, NE, USA); shoot and root biomass was determined after drying at 80 °C during 48 h. Data obtained for these three plant harvests were used to calculate integrative parameters. Only the third harvest data point (934 degree-days sowing) was used for both plant phenotypic and bacterial description, which with 33 ± 10 leaves per plant was considered as an early stage of plant development. Thus, at 934 degree-days after sowing, six plants per genotype were used for estimating C and N fluxes in plant. Among these 6 plants, three were used for 13C and 15N labeling while three other plants were taken as controls for non-labeled plants. Additionally, four of these six plants were randomly selected for plant growth description (leaf area and dry shoot and below-ground biomass), calculation of integrative ecophysiological framework parameters and microbial description (Automated Ribosomal Intergenic Spacer Analysis and pyrosequencing analysis).
13C-labelling
Photosynthetic carbon (C) uptake was quantified with 13CO2 (Jeudy et al. 2010; Voisin et al. 2003) at 934 degree-days after sowing. The labeling experiment was conducted in a gas-proof chamber as in Jeudy et al. (2010). The environmental parameters of this chamber were: a 14/10 h day/night photoperiod, a temperature of 22 °C, 60 % relative-humidity, a photosynthetically active radiation of 650 μmol photons.m−2.s−1 (108 DULUX L 55 W lamps, APPRO5-21850, Saint Appolinaire, France). The atmospheric CO2 concentration within the labeling chamber was measured and maintained at 380 μL L−1 CO2 (Jeudy et al. 2010). Plants were exposed for 10 h to a 13C-enriched atmosphere (63.8 % 13C/12C). Isotopic compositions of gases in the labeling chamber were controlled by a modular gas analyser (SIC MAHIAC S710, Hamburg, Germany) using non dispersive infrared.
After 2 days of chase, plants were separated into shoots and nodulated roots whose dry biomass was determined after oven drying at 80 °C for 48 h. Plant 13C content was analysed using a continous-flow isotope ratio mass spectrometer (Sercon, Crewe, UK) coupled to a C-N elemental analyser (Thermo Electron NC2500, Courtaboeuf, France). The quantities of carbon (QC) in plant shoots and nodulated roots acquired during the labeling period were calculated as follows:
where DW is the dry biomass, %C the percentage of carbon in dry biomass (w/w), ECplant and ECcontrol are respectively the 13C abundance of the labeled plant or the non-labeled control plant and ECsource is the 13C enrichment of the atmosphere.
The rhizodeposition was quantified in the same way by analysing the quantity of 13C content in the rhizosphere soil. The labeling measurement was performed on three plants per genotype.
15N-labelling
Nitrogen (N) entering plants through symbiotic N2 fixation was estimated using 15NO −3 labeling and isotope dilution method (Salon et al. 2010). At sowing, 0.23 mg of K15NO3 (10 % 15N atom excess enrichment) was added in three pots without plant and in three pots for each sown genotypes. As control, 0.23 mg of K14NO3 was supplied to three control pots without plant and for each genotype. In order to estimate mineralization, the decrease of 15N of 15NO −3 was measured at 934 days after sowing using a diffusion method (Brooks et al. 1989; Mathieu 2005). The symbiotic N2 fixation (%Ndfa) was then estimated as follows:
where Eref* and Eref are the 15N abundance of respectively labeled soil and nonlabeled control soil; DWleg* and DWleg are the dry biomass of respectively labeled plant and nonlabeled control plant; Eleg* and Eleg are the 15N abundance of labeled plant and nonlabeled control plants respectively; ε fix = −1δ‰
Respiration
Respiration of root and microbial communities was measured at 934 degree-days in a growth chamber with similar environmental conditions as those used for the labeling experiment. The atmospheres of the root systems of each genotype (the three non-labeled plants per genotype), were individually separated from the shoot compartment using an air-tight plexiglass container. The respiration was then estimated by measuring the rate at which the CO2 concentration within the container increased using an infrared gas analyzer (IRGA; Qubitac; Qubit Systems Inc., Kingston, Canada).
C and N nutritional strategies via an ecophysiological framework
The various plant genotypes could employ contrasted carbon (C) and nitrogen (N) nutritional strategies to reach similar growth. An ecophysiological framework (Moreau 2007; Moreau et al. 2012) was used to study these different C and N nutritional strategies. This framework linked four integrative variables (leaf area, total dry biomass, below-ground dry biomass and total amount of N in the plant) with four intermediate parameters:
-
i)
RUE, which represents the Radiation Use Efficiency for biomass production. RUE was calculated as the slope of the linear function between total dry biomass (TDW) and the sum of intercepted PAR (Photosynthetically Active Radiation) (ΣPARintercepted)
$$ TDW = \alpha + RUE \times \Sigma PA{R_{{intercepted}}} $$(3)$$ \Sigma PA{R_{{intercepted}}} = \sum\nolimits_{{i = 1}}^D {L{A_i} \times PA{R_i}} $$(4)where TDW is the total dry biomass (g), α is the y-intercept, ΣPAR intercepted is the sum of PAR intercepted (MJ.plant−1), D is the day (day), LA i is the leaf area at the day i (cm².plant-1) and was calculated as reported by Chenu (2004), PAR i is the incident PAR at the day i (MJ.m-2.day-1).
-
ii)
RTR, which represents the Root: Total biomass Ratio. RTR was calculated as the slope of the linear function between below-ground dry biomass (RDW) and total dry biomass (TDW)
$$ RDW = \alpha + RTR \times TDW $$(5)where RDW is the below-ground dry biomass (g) and TDW is the total dry biomass (g).
-
iii)
SNU, which represents plant-Specific N Uptake. SNU was calculated as the slope of the linear function between total amount of N (Ntotal) and below-ground dry biomass (RDW)
$$ {N_{{total}}} = \alpha + SNU \times RDW $$(6)where N total is the total amount of N (g.plant−1) and RDW is the below-ground dry biomass (g).
-
iv)
NLA, which represents the conversion factor of N to Leaf Area. NLA was calculated as the slope of the linear function between leaf area (LA) and total amount of N (Ntotal).
$$ LA = \alpha + NLA \times {N_{{total}}} $$(7)where LA is the leaf area (cm2.plant−1) and N total is the total amount of N (g.plant−1).
Extraction and purification of total DNA from soil sample
DNA was extracted from two different compartments: rhizosphere soil and root tissue as previously described by Mougel et al. (2006). This sampling design enables to distinguish bacterial communities as a function of their proximity to plant influence (rhizosphere effect). The rhizosphere soil was obtained after manually separating the root system with adhering soil from the container and by washing the root system under agitation (vortex at 30 Hz during 1 min) in 50 ml of sterile 0.025 M K2SO4 solution. The root system was discarded and the soil was collected after centrifugation at 9,000 g for 10 min. The root tissue compartment included both bacterial communities in roots and on the root surface (rhizoplane). For the rhizosphere soil compartment and the root tissue compartment four replicates per genotype were used. Each sample was weighed, frozen in liquid nitrogen and stored at −80 °C for further use in DNA extraction. The DNA extraction procedure was based on chemical and mechanical extraction as described previously by Ranjard et al. (2003) and by Mougel et al. (2006). DNA preparations were quantified as described previously (Mougel et al. 2006).
Automated RISA fingerprinting
The bacterial ribosomal IGS were amplified with primers S-D-Bact-1522-b-S-20 (3′ end of 16S genes) and L-D-Bact-132-a-A-18 (5′ end of 23S genes) (Normand et al. 1996) for bacterial automated ribosomal intergenic spacer analysis (B-ARISA). PCR conditions, PCR template preparation for DNA sequencer loading and electrophoresis conditions were described by Ranjard et al. (2003).
454 sequencing and bioinformatics analysis
Rhizosphere bacterial diversity of the four Medicago truncatula genotypes was analysed using 454 sequencing. Genotype selection was based on results obtained on genetic structure of rhizosphere bacterial communities, where three different genotype groups were identified. One representative genotype of each group (or two representative genotypes for the biggest group) was chosen (DZA 045-6, DZA 315-16, F 83005-5 and Jemalong A17).
A 16S rRNA gene fragment of the appropriate size and sequence variability for 454 pyrosequencing was amplified using the primers 530F (5′- ACTCCTACGGGAGGCAGCAG) (Acosta-Martinez et al. 2008) and 803R (5′-CTACCNGGGTATCTAAT-3′). Ten base pair tags (“Multiplex Identifier”, MID) at 5′ position were added to the primers to specifically identify each sample as recommended by the manufacturer (Beckman Coulter Genomics). For each rhizosphere soil analyzed, the DNA extracted from the four replicates was pooled to perform the PCR. DNA aliquots (2 ng) from each pool were used for a 50 μl PCR reaction under these conditions: 95 °C for 3 min, 30 cycles of 1 min at 95 °C (denaturation), 55 °C for 30 s (annealing) and 72 °C for 30 s (extension), followed by 5 min at 72 °C. Adapter sequences were added to 300 ng per sample as recommended by the manufacturer and pyrosequencing was carried out on a Genome Sequencer FLX 454 (Beckman Coulter Genomics).
The bioinformatics analysis followed those developed by Terrat et al. (2011) corresponding to (a) the orientation for each kept read was checked, reverse-complemented where necessary (average read length: 400) and sorted according to the MID sequences, (b) the reads were then filtered and eliminated if the exact primer sequences were not found, or if the sequences contained any ambiguity (Ns) or if its length was below 300 bases, (c) a Python program was applied to obtain strict dereplication (i.e. clustering of strictly identical sequences). This led to a 30 to 40 % reduction in the number of sequences. The dereplicated reads were then aligned and clustered into Operational Taxonomic Units (OTUs) defined by a 3 % distance level using available tools from the Ribosomal Database Project (RDP) pyrosequencing pipeline (Cole et al. 2009). Then, another homemade filtering step was applied to potentially eliminate sources of errors (PCR chimeras and sequencing errors) based on OTUs definition. Briefly, for each sample, single-singletons (reads detected only once and not clustered) were deleted in order to eliminate these potential errors and to avoid an overestimated number of OTUs (deleted single-singletons represented only 7 % of all reads but more than 30 % of detected OTUs) (Behnke et al. 2011; Pawlowski et al. 2011). In fact, a majority of the single-singletons might be artifacts (PCR chimeras and large sequencing errors) resulting from noise in high-throughput pyrosequencing of the most abundant OTUs (Kunin et al. 2010; Reeder and Knight 2009). Subsequently, all high-quality sequence sets were another time aligned and clustered (at a 3 % genetic distance) using tools from the RDP pyrosequencing pipeline (Cole et al. 2009). Rarefaction curves and diversity indices were finally determined as described previously by Terrat et al. (2011).
For taxonomy-based analysis, the Naïve Bayesian rRNA classifier of the RDP project was used (Cole et al. 2009) (the bootstrap value was set to 80 %).
The sequences collected in this study have been deposited under accession number ERP000919 in the EMBL nucleotide sequence database.
Statistical analysis
All statistical analyses were performed using the statistical software package R 2.9.2. Only differences significant at P < 0.05 were considered.
Biomass, leaf area, photosynthesis rates, rhizodeposition, respiration, total amount of nitrogen (N) and symbiotic N2 fixation were analysed using ANOVA tests. The means were compared using a Tukey-HSD post-hoc test.
Radiation use efficiency, root:total biomass ratio, specific N uptake and N to leaf area ratio were analysed using ANCOVA test, after a Bonferroni correction.
Two principal component analyses (PCA) were performed. The first one, which described the genetic structure of bacterial communities of the different Medicago truncatula genotypes (using ARISA data), was performed as reported by Ranjard et al. (2003) The second one described phenotypic characterization of the different genotypes of Medicago truncatula. A co-inertia analysis was used to correlate the genetic structure of the rhizosphere bacterial communities with the plant phenotypic characteristics. Co-inertia analysis is a standard multivariate analysis technique that describes the relationship between two data tables and serves as a general method to perform simultaneous analysis of two tables for studying species-environment relationship when the number of variables is higher than that of samples (Doledec and Chessel 1994; Dray et al. 2003). Monte-Carlo tests were used to check the significance of the PCA and the co-inertia. The two PCA, the co-inertia analysis and the Monte Carlo test were performed using the Ade4TkGUI software (Thioulouse and Dray 2007).
Co-inertia analysis allowed identifying the major ecophysiological variables and microbial markers that discriminate Medicago truncatula genotypes.
Pearson correlation coefficients were also calculated between the rhizosphere bacterial diversity (454 pyrosequencing data) of the four Medicago truncatula genotypes (DZA 045-6, DZA 315-16, F 83005-5 and Jemalong A17) and the ecophysiological variables.
Two dendrograms were drawn using the “ward” method of R statistical software package after calculating Euclidean distances. The first dendrogram of simple sequence repeat marker diversity was based on microsatellite data published by Ronfort et al. (2006). The second dendrogram, representing the genetic structure of rhizosphere bacterial communities of the different Medicago truncatula genotypes, was done by means of ARISA band intensity of the four replicates per genotype. A Mantel test was performed to test correlation between the two dendrograms obtained.
Results
At an early stage of plant development, the Medicago truncatula genotype modifies the genetic structure of the bacterial communities in the rhizosphere soil but not in the root tissue
The genetic structure of bacterial communities in the rhizosphere soil compartment differed according to Medicago truncatula genotype (Fig. 1a). The first two components accounted for approximately 70 % of the total variance. DZA 315-16 and DZA 315-26 were separated from the other genotypes on the first axis, which explained 43 % of the total variance (Fig. 1a). DZA 045-6 was separated from the others on the second axis, which explained 27 % of the total variance (Fig. 1a). In contrast, the genetic structure of the bacterial communities in the root tissue compartment (Fig. 1b) was not affected by the plant genotype.

Comparison of the genetic structure of bacterial communities by Principal Component Analysis (PCA) of bacterial automated ribosomal intergenic spacer analysis (B-ARISA) profiles from different compartments: a. rhizosphere soil and b. root tissue of different genotypes of Medicago truncatula at 934 degree-days. For the rhizosphere soil compartment (P = 0.002), a. represented the first two components combined account for approximately 70 % of the total variance; For the root tissue compartment (P = 0.06), b. represented the first two components combined account for approximately 71 % of the total variance
At an early stage of plant development, rhizosphere bacterial diversity is contrasted among Medicago truncatula genotypes
To precise bacterial community description, the rhizosphere bacterial diversity of four of the most contrasted Medicago truncatula genotypes (DZA 045-6, DZA 315-16, F 83005-5 and Jemalong A17) based on their rhizosphere bacterial genetic structures were analysed using 454 sequencing (Fig. 2). Diversity of the bacterial communities in the root tissue compartment was not analysed because no significant effect of the Medicago truncatula was observed. Between 93 and 94 % of OTU were assigned to major bacterial phyla using our bioinformatic pipeline.

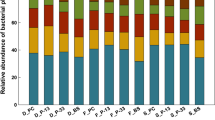
Rhizosphere bacterial phyla of four Medicago truncatula genotypes (DZA 045-6, DZA 315-16, F 83005-5 and Jemalong A17) identified from 454 pyrosequencing
The rhizosphere bacterial diversity was contrasted among Medicago truncatula genotypes specifically between the proportion of Proteobacteria, Acidobacteria and Bacteroidetes phylum which represent between 81 and 83 % of the total OTUs (Fig. 2). The proportion of Proteobacteria varied among genotypes from 22 % of the total sequenced bacteria for DZA 315-16 to 51 % for Jemalong A17 (Fig. 2). Inside Proteobacteria, α-Proteobacteria that include the Medicago truncatula nitrogen fixing symbiots which are preferentially reproducing in association with Medicago truncatula host represented 55 % of sequences among Proteobacteria (data not shown). The proportion of Acidobacteria varied among genotypes from 19 % of the total sequenced bacteria for Jemalong A17 to 56 % for DZA 315-16 (Fig. 2). The proportion of Bacteroidetes varied among genotypes from 3 % of the total sequenced bacteria for DZA 315-16 to 11 % for Jemalong A17 (Fig. 2).
Therefore, the modifications of the genetic structure of the rhizosphere bacterial communities among the different genotypes of Medicago truncatula were representative of a contrasted rhizosphere bacterial diversity. As such, the Medicago truncatula genotype significantly affected the genetic structure and diversity of rhizosphere bacterial communities at an early stage of its development.
At an early stage of plant development, functional descriptors better discriminate Medicago truncatula genotypes than phenotypic structural descriptors
Plant growth was characterised with standard phenotypic structural descriptors such as total dry biomass (DW) and leaf area (Table 1). Responses ranged between 0.63 and 1.31 g for total DW and between 44 and 112 cm² for leaf area (Table 1). At 934 degree-days, the only difference in plant growth was observed for DZA 045-6, which was significantly smaller than DZA 315-16, F 83005-5 and Jemalong A17 (Table 1). These growth descriptors did not permit a distinction among the six other genotypes.
To go further than in previous microbiological studies, some plant functions involved in plant carbon (C) and nitrogen (N) nutrition were assessed using both an isotope labeling experiment and an ecophysiological framework (Moreau 2007). The isotope labeling enabled quantification of photosynthesis, rhizodeposition and symbiotic N2 fixation (Table 1). The ecophysiological framework allowed the calculation of four intermediate parameters: the radiation use efficiency (RUE); the root:total biomass ratio (RTR); the specific nitrogen uptake (SNU) and the conversion factor of nitrogen to leaf area (NLA). The different genotypes were contrasted in their C and N nutrition. Jemalong A17 had higher RUE than DZA 315-16, DZA 315-26 and F 83005-5 (Table 1). DZA 045-6 had a higher photosynthesis than SA 028064 (Table 1). Rhizodeposition did not significantly vary with Medicago truncatula genotype, but DZA 315-16 and its microbial communities had a higher respiration than Jemalong A17 and Jemalong J6 (Table 1). The SNU and the root:total biomass ratio of DZA 315-16 were respectively higher and lower than for Jemalong J6 (Table 1). Symbiotic N2 fixation and NLA did not differ among plant genotypes (Table 1).
Thus, phenotypic structural descriptors (dry biomass and leaf area) significantly differentiated DZA 045-6 from DZA 315-16, F 83005-5 and Jemalong A17. A detailed level of discrimination was achieved with functional descriptors (RUE, SNU and respiration) that significantly differentiated DZA 315-16 from Jemalong A17 and Jemalong J6.
Microbiological and ecophysiological data are correlated
In order to correlate microbiological and ecophysiological data, a co-inertia analysis was carried out by comparing the PCA of the genetic structure of the rhizosphere microbial communities (ARISA data) with a PCA analysis of the ecophysiological data (Online Resource 1). The co-inertia indicated that the differences observed between plant genotypes in both genetic structure of associated bacterial communities and ecophysiological profiles were correlated (P = 0.026) (Fig. 3). The co-inertia represented the first two components which accounted for approximately 71 % of the total variance. The co-inertia analyses allowed to distinguish three different groups of genotypes: a) DZA 045-6; b) DZA 315-16, and c) Jemalong A17 and J6 (Fig. 3a and b).

Comparison of the ecophysiological data with the genetic structure of rhizosphere bacterial communities (B-ARISA) by co-inertia for different genotypes of Medicago truncatula at 934 degree-days (P = 0.026). a. Factorial plane of the genotypes discrimination in the co-inertia as a function of their B-ARISA profile ; b. Factorial plane of the genotypes discrimination in the co-inertia as a function of their ecophysiological profile ; c. Factorial plane of B-ARISA bands contribution in the co-inertia ; each number represented size of B-ARISA band (or DNA fragment) (e.g. X286 represent 286–287 base pair DNA fragments) ; d. Factorial plane of ecophysiological characteristics contribution in the co-inertia. The first two components of PCA represented account for approximately 71 % of the total variance
Microbial molecular markers that mostly discriminated the three different groups of genotypes were ranged from 286 base pairs (pb) to 712 bp (Fig. 3c): X692, X640, X670, X398, X712 and X362 on the first principal component; and X286, X640, X692, X670, X562, X392 and X394 on the second principal component. Ecophysiological variables that mostly discriminated the three different groups of genotypes were respiration, N fixation, leaf area, RUE and NLA on the first principal component and total biomass, leaf area, N total content and RUE on the second principal component (Fig. 3d).
DZA 315-16 was separated from DZA 045-6, Jemalong A17 and J6 on the first axis, which explained 38 % of the total variability (Fig. 3a and b). This difference could be explained, at the bacterial level, by two molecular markers of 692–693 bp and 712–713 bp, which were present for DZA 315-16 but not for DZA 045-6, Jemalong A17 and Jemalong J6 and by two molecular markers of 362–363 bp, 398–399 bp, which had a lower intensity for DZA 315-16 than for DZA 045-6, Jemalong A17 and J6 (Fig. 3c). At the plant ecophysiological level, DZA 315-16 was related to a high leaf area, a high respiration and a high NLA contrary to DZA 045-6, Jemalong A17 and J6 (Fig. 3d).
DZA 045-6 was separated from DZA 315-16, Jemalong A17 and J6 on the second axis, which explained 33 % of the total variability (Fig. 3a and b). This difference could be explained, at the bacterial level, by both two molecular markers of 286–287 bp and 392–395 bp, which had a higher intensity for DZA 045-6 than for DZA 315-16, Jemalong A17 and Jemalong J6, and one molecular marker of 562–563 bp, which had a lower intensity for DZA 045-6 than for DZA 315-16, Jemalong A17 and Jemalong J6 (Fig. 3c). At the plant physiological level, DZA 045-6 was related to a low total biomass, a low leaf area and a low total amount of N, contrary to DZA 315-16, Jemalong A17 and Jemalong J6 (Fig. 3d).
DZA 045-6 and DZA 315-16 were separated from Jemalong A17 and J6 on both the first and second axis (Fig. 3a and b). This difference could be explained, at the bacterial level, by two other molecular markers of 640–641 bp and 670–671 bp, which had a higher intensity for Jemalong A17 and Jemalong J6 than for DZA 045-6 and DZA 315-16 (Fig. 3c). At the plant ecophysiological level, DZA 045-6 and DZA 315-16 were related to a higher SNU and N fixation and a lower RUE than for Jemalong A17 and J6 (Fig. 3d).
The Pearson correlation coefficients between rhizosphere bacterial diversity data (454 pyrosequencing) and ecophysiological variable for the four Medicago truncatula genotypes (DZA 045-6, DZA315-16, F 83005-5 and Jemalong A17) were calculated (Table 2). While Acidobacteria, Verrucomicrobia, Planctomycetes and BCR1 abundances were positively correlated to N fixation Proteobacteria, Bacteroidetes, and Actinobacteria abundances negatively correlated to N fixation (Table 2). Verrucomicrobia abundance was negatively correlated to RTR while Gemmatimonadetes and Nitrospira abundances were positively correlated to RTR (Table 2). Actinobacteria and WS3 abundances were respectively positively and negatively correlated to RUE; WS3 abundance was positively correlated to SNU; TM7 abundance was positively correlated to NLA; Gemmatimonadetes and Fibrobacteres abundances were negatively correlated to respiration (Table 2). TM7 abundance was positively correlated to leaf area and Bacteria incertae sedis and OP10 abundances were negatively correlated to total biomass and N total (Table 2).
Microbiological and genetic data are correlated
A comparison between two dendrograms, one based on bacterial ARISA data and one based on 13 M. truncatula microsatellites showed a highly significant correlation between the genetic structure of the rhizosphere bacterial communities and the genetic diversity of Medicago truncatula genotypes (P < 0.001) (Fig. 4).

Dendrogram comparison between the Medicago truncatula phylogenic structure (a) and the genetic structure of their associated rhizosphere bacterial communities (b). Phylogenetic dendrogram of the seven Medicago truncatula genotypes based on simple sequence repeat marker diversity (modified from Ronfort et al. 2006). Dendrogram of the genetic structure of the Medicago truncatula rhizosphere bacterial communities based on the bacterial ribosomal intergenic spacer data. A Mantel test showed a significant correlation (P < 0.001) between these two dendrograms
Discussion
The interactions between plants and soil microbial communities are bidirectional: on the one side, plant directs the selection of microbes via rhizodeposition (Hartmann et al. 2009) and on the other side, microbes can influence plant health, development and growth, for example, by enhancing nutrient availability and uptake (Richardson et al. 2009). Nevertheless, plant-microbe interactions are usually studied focusing either on the plant or on the microbe side. To our knowledge, our study is the first one assessing the plant genotype’s effect on the genetic structure of the entire rhizospheric and endophytic bacterial community in relation with plant growth and carbon (C) and nitrogen (N) nutrition at the whole plant level.
The plant genotype has an early effect on the rhizosphere bacterial communities
The rhizosphere harbours multiple bacterial communities. Nevertheless, in the literature, plant genotype effects were particularly studied on specific rhizobacterial communities such as plant growth-promoting bacteria (Dalmastri et al. 1999; Mazzola et al. 2004; Paffetti et al. 1996). Our data confirmed the importance of the plant genotype in shaping the genetic structure and diversity of the entire bacterial rhizosphere for the model legume Medicago truncatula. Although Medicago truncatula is able to fix atmospheric N2, this effect was not limited to only rhizobial strains. Besides, no significant difference was observed in nodulation intensity among Medicago truncatula genotypes (Online resource 2). To date, the influence of plant genotype on the entire rhizosphere bacterial communities has only been demonstrated for three genotypes of Solanum tuberosum L. (Andreote et al. 2009; van Overbeek and van Elsas 2008) and for eight accessions of Arabidopsis thaliana (Micallef et al. 2009).
Furthermore, the effect of the Medicago truncatula genotype on bacterial communities is seen early during the growth cycle. This is in agreement with two previous studies, one on canola at the rosette stage (Dunfield and Germida 2003), the other on three different potato genotypes at the first growth stage (Andreote et al. 2009).
Altogether, these studies support the idea of the high sensitivity of bacterial communities to intra-species plant genotype variation. Studies on plant-bacterial community interactions should focus on early stages of plant development.
At an early stage of development, functional descriptors are essential to discriminate plant genotypes
Few microbiological studies have assessed plant phenotypic variations in relation to the modifications of rhizosphere microbial communities. The observed variations were generally based on structural descriptors (Mougel et al. 2006; Parra-Colmenares and Kahn 2005). In our study, structural phenotypic descriptors such as biomass and leaf area only discriminated DZA045-6 from the other genotypes.
To thoroughly characterize the different Medicago truncatula genotypes in relation with their nutritional strategies, an ecophysiological framework with integrative variables (Moreau 2007) was used. Our study enabled us to identify three contrasted nutritional strategies. The first one was illustrated by DZA 045-6, which had a lower biomass than the other genotypes. This can be explained by the low conversion of N to leaf area of DZA 045-6, as leaf area is instrumental to acquire carbon resources and thus biomass. The second nutritional strategy was that of DZA 315-16. This genotype allocated more biomass to its shoots which displayed the least C uptake efficiency among the studied genotypes. The third strategy, illustrated by Jemalong A17 and Jemalong J6, was to allocate more biomass to their below-ground parts, which displayed the least specific N uptake efficiency among the studied genotypes. As such, these differences in biomass allocation by the various genotypes seem to reflect a compensation process, termed the “functional equilibrium” theory (Brouwer 1983; Farrar and Jones 2000). The dry biomass distribution between root and shoot can be regulated by equilibrium between root activity (water and nutrient absorption) and shoot activity (photosynthesis). The PCA of Medicago truncatula ecophysiological profiles (Online Resource 1) illustrate this theory well, as RUE is positively correlated with root:total biomass ratio and is negatively correlated with SNU. Such contrasted nutritional strategies have been identified in several species during their vegetative growth (Lemaire et al. 2008; Moreau 2007) but, to our knowledge, they have not been studied in relation to the rhizosphere microbial communities.
Thus, at an early stage of the plant development, while plant structural descriptors did not reveal effects of plant-microbe interactions on the plant, functional descriptors could discriminate plant genotypes.
Rhizosphere bacterial diversity could be controlled by labile carbon availability and rhizosphere CO2 concentration
By traditional culture methods, 95–99 % of the microbial communities present in the soil are not accessible. Thanks to the development of culture-independent approaches, the most dominant bacterial phyla present in the rhizosphere soil could be identified: Proteobacteria, Actinobacteria, Acidobacteria, Verrucomicrobia, Planctomycetes, Bacteriodetes and Firmicutes (da Rocha et al. 2009). In the rhizosphere of Medicago truncatula, the two most dominant bacterial phyla were Proteobacteria and Acidobacteria (Fig. 2). This result is in agreement with a meta-analysis performed by Fierer et al. (2007) showing the high abundance of Proteobacteria (50–60 %) and Acidobacteria (10–15 %) in the rhizosphere. More interestingly, in our study, we found a high Acidobacteria abundance (between 19 to 56 %) and a modified Acidobacteria: Proteobacteria ratio between DZA315-16, DZA045-6, F83005-5 and Jemalong A17 (Fig. 2). If Proteobacteria phylum was successful rhizosphere colonizers, the high abundance of Acidobacteria is more surprising. Two major hypotheses could be used to explain these results in relation to the ecological life strategies of Proteobacteria and Acidobacteria and to the plant ecophysiological parameters.
Acidobacteria is considered as an oligotrophic phylum (K-strategists) whereas Proteobacteria is considered as a copiotrophic phylum (r-strategists). Ecological attributes of copiotrophs were their greater ability to consume labile organic C pools and their high growth rates in non limiting resource conditions. In contrast, oligotrophs exhibit slower growth rate and are suppose to outcompete copiotrophs in low nutrient availability conditions due to a higher substrate affinities (Fierer et al. 2007). Acidobacteria are composed mostly of non-cultivable bacteria, which may have become adapted to elevated concentrations of CO2 and concentrations of O2 lower than the atmospheric O2 concentrations (Stevenson et al. 2004) and their abundance and diversity were strongly positively correlated with pH (Jones et al. 2009b). The rhizosphere environment has generally a lower pH compare to soil, lower oxygen and higher carbon dioxide concentrations and is a rich nutrient environment with a great quantity of labile organic C.
Among ecophysiological variables, radiation use efficiency (RUE) was significantly higher in Jemalong A17. The RUE, which corresponds to the efficiency of conversion of atmospheric CO2 in plant biomass could be directly correlate to the quantity of rhizodeposits. In our study the rhizodeposition was not different among Medicago truncatula genotypes but we did not measured the quantity of labile carbon which could affect Acidobacteria: Proteobacteria ratio in the soil. Indeed, according to Fierer et al. (2007), contrary to Proteobacteria, Acidobacteria abundance is negatively correlated to carbon mineralisation rates. This point could explain the high abundance of the r-strategists Proteobacteria phylum compare to Acidobacteria in the rhizosphere of Jemalong A17. Rhizosphere respiration, corresponding to root and microbial respiration, was four-fold higher in the rhizosphere of DZA 315-16 but was not directly correlated to Acidobacteria phylum (Table 2). The N fixation was positively correlated to Acidobacteria phylum abundance (Table 2). A stronger rhizosphere respiration and nodule activity could explain a more important CO2 concentration in the rhizosphere of DZA 315-16 which would positively select Acidobacteria. Altogether, the abundance and modification of the ratio between these two major phyla could be the mirror of modifications of labile carbon availability, CO2 concentration and pH in the rhizosphere.
Quantitative rhizodeposition analyse is not sufficient to understand the basis of plant-microbe interactions
The total amount of plant C deposited in the rhizosphere is known to greatly vary with the plant ecophysiology, as determined by environment, genetics and physiology (Nguyen 2003). Micallef et al. (2009) explained the different bacterial communities present in the plant rhizosphere by quantitative and qualitative differences in the exudation pattern of the different Arabidopsis thaliana accessions. In our study, no significant difference could be observed for the total amount of C lost by plants via rhizodeposition. Although rhizodeposition was correlated with a microbial marker (X562), the bacterial community diversity represented by this marker is unknown. The dissection of taxonomic structures and the metabolic/ecological functions of the microbial communities could be assessed using metagenomics approaches. However, this result does not exclude quantitative difference in root exudation or qualitative differences in the signalling pathways among plant genotypes as it was shown for the legume–Rhizobia molecular dialogue (Bais et al. 2006; Oldroyd and Downie 2008). To understand further the basis of plant-microbe interactions, qualitative rhizodeposition analysis should be promoted instead of quantitative analysis.
A global and multidisciplinary approach including ecophysiology, microbial ecology and quantitative genetics is a key tool in deciphering plant-microbe interactions
In our study, ecophysiology of the different Medicago truncatula genotypes was assessed using two complementary levels of discrimination: the first based on phenotypic structural descriptors; the second based on functional descriptors. Ecophysiological profiles of the different Medicago truncatula genotypes were correlated to the genetic structure of the rhizosphere bacterial communities. Nevertheless, microbial approach used (ARISA) did not permit to assess diversity and functions of the microbial communities. A direct causal link between the assembly of the rhizosphere bacterial and plant physiological functions could not be ascertained in the present study. Establishing this connection will be a challenge for the future.
Our study highlighted that Medicago tuncatula plant genotypes-associated microbial communities are diverse and potentially under selection by their host plant. In our study few bacterial phyla (Proteobacteria and Acidobacteria) show their abundance modified in relation with plant functional traits. At this stage we need to generalize this result in order to define diversity profile of bacteria associated with root in relation with plant and microbial traits. High-throughput sequencing technology will facilitate surveys of taxonomic loci as well as loci implicated in impacting plant phenotypes (e.g., nitrogen fixation, root architecture, leaf area,…). Our study demonstrated the importance today to have a multidisciplinary approach in plant-microbe interactions. In addition, the relationship between plant functional traits and microbial diversity and fitness needs to be quantified simultaneously. Moreover, the highly significant correlation observed between the genetic structure of the rhizosphere bacterial communities and the genetic diversity of Medicago truncatula genotypes (Fig. 4) should be confirmed with a Medicago truncatula core collection whose genetic diversity is established and that presents a greater number of genotypes. Furthermore, in our study, the environmental effect on Medicago truncatula associated rhizosphere bacterial communities was not evaluated ie one soil-type and one cultural condition. Therefore, in our experiment, the genetic effect is probably overestimated. In fact, according to van Overbeek and van Elsas (2008), potato-associated bacterial communities were affected by both plant growth stage and experimental factors (soil and year) rather than by plant genotype. It should be of great interest to observe the ecophysiological behaviour of a Medicago truncatula core collection in variable environmental conditions to distinguish genetic and environmental effects on rhizosphere bacterial communities. If relevant, a quantitative genetic approach could be implemented to identify the genetic basis of the plant-microbial community interactions. In summary, it will be necessary to combine ecophysiology, microbial ecology and quantitative genetics to increase our understanding of plant-microbe interactions. In the near future, this could be useful for plant breeders, who will be able to select their plant genotype as a function of the associated soil microbial communities so as to obtain better crop yields (biofertilization) and pathogen resistance (biocontrol).
References
Acosta-Martinez V, Dowd S, Sun Y, Allen V (2008) Tag-encoded pyrosequencing analysis of bacterial diversity in a single soil type as affected by management and land use. Soil Biol Biochem 40:2762–2770
Andreote FD, de Araujo WL, de Azevedo JL, van Elsas JD, da Rocha UN, van Overbeek LS (2009) Endophytic colonization of potato (Solanum tuberosum L.) by a novel competent bacterial endophyte, pseudomonas putida strain P9, and its effect on associated bacterial communities. Appl Environ Microbiol 75:3396–3406
Bais HP, Weir TL, Perry LG, Gilroy S, Vivanco JM (2006) The role of root exudates in rhizosphere interations with plants and other organisms. Annu Rev Plant Biol 57:233–266
Behnke A, Engel M, Christen R, Nebel M, Klein RR, Stoeck T (2011) Depicting more accurate pictures of protistan community complexity using pyrosequencing of hypervariable SSU rRNA gene regions. Environ Microbiol 13:340–349
Brooks PD, Stark JM, McInteer BB, Preston T (1989) Diffusion method to prepare soil extracts for automated N-15 analysis. Soil Sci Soc Am J 53:1707–1711
Brouwer R (1983) Functional equilibrium - sense or nonsense. Neth J Agric Sci 31:335–348
Buee M, De Boer W, Martin F, van Overbeek L, Jurkevitch E (2009) The rhizosphere zoo: an overview of plant-associated communities of microorganisms, including phages, bacteria, archaea, and fungi, and of some of their structuring factors. Plant Soil 321:189–212
Chenu K (2004) Variabilité phénotypique de l’architecture de la rosette d’Arabidopsis thaliana en réponse au rayonnement: analyse et modélisation de la réponse de différents génotypes. Université Montpellier 2, Montpellier, France
Cole JR, Wang Q, Cardenas E, Fish J, Chai B, Farris RJ, Kulam-Syed-Mohideen AS, McGarrell DM, Marsh T, Garrity GM, Tiedje JM (2009) The ribosomal database project: improved alignments and new tools for rRNA analysis. Nucleic Acids Res 37:D141–D145
da Rocha UN, van Overbeek L, van Elsas JD (2009) Exploration of hitherto-uncultured bacteria from the rhizosphere. FEMS Microbiol Ecol 69:313–328
Dalmastri C, Chiarini L, Cantale C, Bevivino A, Tabacchioni S (1999) Soil type and maize cultivar affect the genetic diversity of maize root-associated Burkholderia cepacia populations. Microb Ecol 38:273–284
Doledec S, Chessel D (1994) Co-inertia analysis - an alternative method for studying species environment relationships. Freshw Biol 31:277–294
Dray S, Chessel D, Thioulouse J (2003) Co-inertia analysis and the linking of ecological data tables. Ecology 84:3078–3089
Dunfield KE, Germida JJ (2003) Seasonal changes in the rhizosphere microbial communities associated with field-grown genetically modified canola (Brassica napus). Appl Environ Microbiol 69:7310–7318
Farrar JF, Jones DL (2000) The control of carbon acquisition by roots. New Phytol 147:43–53
Fierer N, Bradford MA, Jackson RB (2007) Toward an ecological classification of soil bacteria. Ecology 88:1354–1364
Fisher MM, Triplett EW (1999) Automated approach for ribosomal intergenic spacer analysis of microbial diversity and its application to freshwater bacterial communities. Appl Environ Microbiol 65:4630–4636
Franche C, Lindstrom K, Elmerich C (2009) Nitrogen-fixing bacteria associated with leguminous and non-leguminous plants. Plant Soil 321:35–59
Harrison MJ (1999) Molecular and cellular aspects of the arbuscular mycorrhizal symbiosis. Annu Rev Plant Physiol Plant Mol Biol 50:361–389
Hartmann A, Schmid M, van Tuinen D, Berg G (2009) Plant-driven selection of microbes. Plant Soil 321:235–257
Jensen ES, Hauggaard-Nielsen H (2003) How can increased use of biological N-2 fixation in agriculture benefit the environment? Plant Soil 252:177–186
Jeudy C, Ruffel S, Freixes S, Tillard P, Santoni AL, Morel S, Journet EP, Duc G, Gojon A, Lepetit M, Salon C (2010) Adaptation of Medicago truncatula to nitrogen limitation is modulated via local and systemic nodule developmental responses. New Phytol 185:817–828
Jones DL, Nguyen C, Finlay RD (2009a) Carbon flow in the rhizosphere: carbon trading at the soil-root interface. Plant Soil 321:5–33
Jones RT, Robeson MS, Lauber CL, Hamady M, Knight R, Fierer N (2009b) A comprehensive survey of soil acidobacterial diversity using pyrosequencing and clone library analyses. ISME J 3:442–453
Kunin V, Engelbrektson A, Ochman H, Hugenholtz P (2010) Wrinkles in the rare biosphere: pyrosequencing errors can lead to artificial inflation of diversity estimates. Environ Microbiol 12:118–123
Lambers H, Mougel C, Jaillard B, Hinsinger P (2009) Plant-microbe-soil interactions in the rhizosphere: an evolutionary perspective. Plant Soil 321:83–115
Lemaire G, van Oosterom E, Jeuffroy MH, Gastal F, Massignam A (2008) Crop species present different qualitative types of response to N deficiency during their vegetative growth. Field Crop Res 105:253–265
Marschner H (1995) Mineral nutrition of higher plants. Academic, London
Mathieu O (2005) Application du traçage isotopique 15N à l’étude du protoxyde d’azote (N2O), gaz à effet de serre produit par l’acitivité microbienn des sols. Quantification des flux et approche spaciale au terrain. Université Bourgogne, Dijon, France
Mazzola M, Funnell DL, Raaijmakers JM (2004) Wheat cultivar-specific selection of 2,4-diacetylphloroglucinol-producing fluorescent Pseudomonas species from resident soil populations. Microb Ecol 48:338–348
Micallef SA, Shiaris MP, Colon-Carmona A (2009) Influence of Arabidopsis thaliana accessions on rhizobacterial communities and natural variation in root exudates. J Exp Bot 60:1729–1742
Minchin FR, Summerfield RJ, Hadley P, Roberts EH, Rawsthorne S (1981) Carbon and nitrogen nutrition of nodulated roots of grain legumes. Plant Cell Environ 4:5–26
Moreau D (2007) Réponse du développement et de la croissance de Medicago truncatula aux facteurs environnementaux: contribution à l’élaboration d’outils de phénotypage pour l’analyse de la variabilité génétique associée à la nutrition azotée. Université Bourgogne, Dijon, France
Moreau D, Salon C, Munier-Jolain N (2006) Using a standard framework for the phenotypic analysis of Medicago truncatula: an effective method for characterizing the plant material used for functional genomics approaches. Plant Cell Environ 29:1087–1098
Moreau D, Voisin AS, Salon C, Munier-Jolain N (2008) The model symbiotic association between Medicago truncatula cv. Jemalong and Rhizobium meliloti strain 2011 leads to N-stressed plants when symbiotic N-2 fixation is the main N source for plant growth. J Exp Bot 59:3509–3522
Moreau D, Burstin J, Aubert G, Huguet T, Ben C, Prosperi JM, Salon C, Munier-Jolain N (2012) Using a physiological framework for improving the detection of quantitative trait loci related to nitrogen nutrition in Medicago truncatula. Theor Appl Genet 124:755–768
Mougel C, Offre P, Ranjard L, Corberand T, Gamalero E, Robin C, Lemanceau P (2006) Dynamic of the genetic structure of bacterial and fungal communities at different developmental stages of Medicago truncatula Gaertn. cv. Jemalong line J5. New Phytol 170:165–175
Munier-Jolain N, Carrouée B (2003) Considering pea in sustainable agriculture: agricultural and environmental arguments. Cahiers Agricultures 12:111–120
Nemecek T, von Richthofen JS, Dubois G, Casta P, Charles R, Pahl H (2008) Environmental impacts of introducing grain legumes into European crop rotations. Eur J Agron 28:380–393
Nguyen C (2003) Rhizodeposition of organic C by plants: mechanisms and controls. Agronomie 23:375–396
Normand P, Ponsonnet C, Nesme X, Neyra M, Simonet P (1996) ITS analysis of prokaryotes. In: Akkermans DL, Van Elsas JD, De Bruijn EI (eds) Molecular microbial ecology manual. Kluwer, Amsterdam, pp 1–12
Offre P, Pivato B, Siblot S, Gamalero E, Corberand T, Lemanceau P, Mougel C (2007) Identification of bacterial groups preferentially associated with mycorrhizal roots of Medicago truncatula. Appl Environ Microbiol 73:913–921
Oldroyd GED, Downie JM (2008) Coordinating nodule morphogenesis with rhizobial infection in legumes. Annu Rev Plant Biol 59:519–546
Paffetti D, Scotti C, Gnocchi S, Fancelli S, Bazzicalupo M (1996) Genetic diversity of an Italian Rhizobium meliloti population from different Medicago sativa varieties. Appl Environ Microbiol 62:2279–2285
Parra-Colmenares A, Kahn ML (2005) Determination of nitrogen fixation effectiveness in selected Medicago truncatula isolates by measuring nitrogen isotope incorporation into pheophytin. Plant Soil 270:159–168
Pawlowski J, Christen R, Lecroq B, Bachar D, Shahbazkia HR, Amaral-Zettler L, Guillou L (2011) Eukaryotic richness in the abyss: insights from pyrotag sequencing. PLoS One 6:e18169
Raaijmakers JM, Paulitz TC, Steinberg C, Alabouvette C, Moenne-Loccoz Y (2009) The rhizosphere: a playground and battlefield for soilborne pathogens and beneficial microorganisms. Plant Soil 321:341–361
Ranjard L, Poly F, Nazaret S (2000) Monitoring complex bacterial communities using culture-independent molecular techniques: application to soil environment. Res Microbiol 151:167–177
Ranjard L, Poly F, Lata JC, Mougel C, Thioulouse J, Nazaret S (2001) Characterization of bacterial and fungal soil communities by automated ribosomal intergenic spacer analysis fingerprints: biological and methodological variability. Appl Environ Microbiol 67:4479–4487
Ranjard L, Lejon DPH, Mougel C, Schehrer L, Merdinoglu D, Chaussod R (2003) Sampling strategy in molecular microbial ecology: influence of soil sample size on DNA fingerprinting analysis of fungal and bacterial communities. Environ Microbiol 5:1111–1120
Reeder J, Knight R (2009) The ‘rare biosphere’: a reality check. Nat Methods 6:636–637
Richardson AE, Barea JM, McNeill AM, Prigent-Combaret C (2009) Acquisition of phosphorus and nitrogen in the rhizosphere and plant growth promotion by microorganisms. Plant Soil 321:305–339
Roesch LF, Fulthorpe RR, Riva A, Casella G, Hadwin AK, Kent AD, Daroub SH, Camargo FA, Farmerie WG, Triplett EW (2007) Pyrosequencing enumerates and contrasts soil microbial diversity. ISME J 1:283–290
Ronfort J, Bataillon T, Santoni S, Delalande M, David JL, Prosperi JM (2006) Microsatellite diversity and broad scale geographic structure in a model legume: building a set of nested core collection for studying naturally occurring variation in Medicago truncatula. Bmc Plant Biology 6
Salon C, Voisin AS, Delfosse O, Mary B (2010) Methodologies for measuring symbiotic nitrogen fixation in the field. In: Munier-Jolain N, Biarnes V, Chaillet I, Lecoeur J, Jeuffroy MH (eds) Physiology of the pea crop. CRC Press, Editions Quae, Versailles, pp 83–87
Singh BK, Millard P, Whiteley AS, Murrell JC (2004) Unravelling rhizosphere-microbial interactions: opportunities and limitations. Trends Microbiol 12:386–393
Stevenson BS, Eichorst SA, Wertz JT, Schmidt TM, Breznak JA (2004) New strategies for cultivation and detection of previously uncultured microbes. Appl Environ Microbiol 70:4748–4755
Terrat S, Christen R, Dequiedt S, Lelievre M, Nowak V, Regnier T, Bachar D, Plassart P, Wincker P, Jolivet C, Bispo A, Lemanceau P, Maron PA, Mougel C, Ranjard L (2011) Molecular biomass and metataxogenomic assessment of soil microbial communities as influenced by soil DNA extraction procedure. Microb Biotechnol
Thioulouse J, Dray S (2007) Interactive multivariate data analysis in R with the ade4 and ade4TkGUI packages. J Stat Softw 22:1–14
van Overbeek L, van Elsas JD (2008) Effects of plant genotype and growth stage on the structure of bacterial communities associated with potato (Solanum tuberosum L.). FEMS Microbiol Ecol 64:283–296
Voisin AS, Salon C, Jeudy C, Warembourg FR (2003) Root and nodule growth in Pisum sativum L. in relation to photosynthesis: analysis using C-13-labelling. Ann Bot 92:557–563
Acknowledgments
The authors are grateful to P. Mathey, C. Jeudy, V. Durey and A.L. Santoni for their excellent technical assistance, C. Lepinay and J. Manifacier for their help during the three harvests, M. P. Bataillé (Université de Caen, France) for C and N analysis, A. Viollet (INRA, Dijon, France) for mycorrhizal infection determination, O. Mathieu (Université Bourgogne, Dijon, France) for his help on N diffusion, M. Delalande (INRA, Mauguio, France) for seeds of M. Truncatula We also thank S. Dequiedt, D. Moreau, A.S. Voisin, M. Prudent and A. Larmure for their useful advices and R. Thompson for the improvement of the English. This work was supported by a PhD grant to A. Zancarini (project n°089201PPO24S00901) from INRA and the Regional Council of Burgundy (France). We thank the Regional Council of Burgundy for support to intregrated Agrale 6 projects (AIB 29000427 & 29000594).
Author information
Authors and Affiliations
Corresponding author
Additional information
Responsible Editor: Jeff R. Powell.
Rights and permissions
About this article
Cite this article
Zancarini, A., Mougel, C., Terrat, S. et al. Combining ecophysiological and microbial ecological approaches to study the relationship between Medicago truncatula genotypes and their associated rhizosphere bacterial communities. Plant Soil 365, 183–199 (2013). https://doi.org/10.1007/s11104-012-1364-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11104-012-1364-7