
Abstract
Aims
To study the relationship between vegetation development and changes in the soil microbial community during primary succession in a volcanic desert, we examined successional changes in microbial respiration, biomass, and community structure in a volcanic desert on Mount Fuji, Japan.
Methods
Soil samples were collected from six successional stages, including isolated island-like plant communities. We measured microbial respiration and performed phospholipid fatty acid (PLFA) analysis, denaturing gradient gel electrophoresis (DGGE) analysis, and community-level physiological profile (CLPP) analysis using Biolog microplates.
Results
Microbial biomass (total PLFA content) increased during plant succession and was positively correlated with soil properties including soil water and soil organic matter (SOM) contents. The microbial respiration rate per unit biomass decreased during succession. Nonmetric multidimensional scaling based on the PLFA, DGGE, and CLPP analyses showed a substantial shift in microbial community structure as a result of initial colonization by the pioneer herb Polygonum cuspidatum and subsequent colonization by Larix kaempferi into central areas of island-like communities. These shifts in microbial community structure probably reflect differences in SOM quality.
Conclusions
Microbial succession in the volcanic desert of Mt. Fuji was initially strongly affected by colonization of the pioneer herbaceous plant (P. cuspidatum) associated with substantial changes in the soil environment. Subsequent changes in vegetation, including the invasion of shrubs such as L. kaempferi, also affected the microbial community structure.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Lava flows and ash deposits from volcanic eruptions create virgin land surfaces on which ecosystem development processes such as plant colonization and soil formation begin. Such areas affected by volcanic activity are referred to as “volcanic deserts”. Volcanic deserts are nutrient-poor ecosystems, and therefore the soil microorganisms function as drivers of carbon and nutrient cycles and soil formation through their role in the decomposition of soil organic matter (e.g. Merilä et al. 2002), and their roles as sources of, and sinks for, labile carbon and nutrients (e.g. Jenkinson and Ladd 1981) are of great importance. This suggests that successional changes in the microbial community will greatly affect soil C and N pools and cycling in volcanic deserts.
Many previous studies have investigated the various processes involved in primary succession, including microbial community changes using a chronosequence approach (e.g. Crews et al. 2001; Schipper et al. 2001; Hopkins et al. 2007; Shillam et al. 2008) or monitoring (e.g. del Moral and Jones 2002; Halvorson and Smith 2009; Jumpponen et al. 2012). However, there are some difficulties in studying successional changes in microbial communities in volcanic deserts based on these approaches. For example, although the chronosequence approach is based on the key assumptions that each of the sites representing different developmental stages had the same initial conditions and has followed the same sequence of changes (Jhonson and Miyanishi 2007), it is difficult or almost impossible in many cases to test this assumption. On the other hand, monitoring of ecosystem development after a volcanic eruption, ensures a common initial condition and allows observation of “actual succession”, but it is limited to relatively short term studies and is unsuitable for understanding primary succession over longer time scales.
One way to mitigate such difficulties is to find a series of different successional stages in the same area (with the same initial conditions including substrate type, slope aspect, and altitudinal location). An example of such an ideal study site is island-like vegetation communities found in alpine and subalpine volcanic deserts. In such areas, seed germination and survival of seedlings are strongly suppressed by harsh environmental conditions (Maruta 1976, 1983) and only a limited number of plant species can colonize the newly created sites (Miyawaki 1971). Therefore, timing of the initial colonization by pioneer plants may differ among positions even within a small area within which some important conditions such as substrate type, slope aspect, and altitudinal location are the same. In addition, plant succession in alpine and subalpine volcanic deserts typically proceeds very slowly (e.g. Miyawaki 1971) because of severe environmental constraints such as a short growing season and low nutrient availability (Maruta 1983; Chiba and Hirose 1993). Consequently, isolated island-like plant communities at different developmental stages, which reflect only the time since first colonization of pioneer plants, are found in subalpine volcanic deserts such as those in Japan (e.g., Maruta 1976; Hirose and Tateno 1984; Masuzawa and Suzuki 1991; Masuzawa 1995; Nishitani and Masuzawa 1996). The presence of these island-like communities at different successional stages in a small area with the same altitudinal location and initial conditions therefore provides a good opportunity to investigate successional changes in the plant community, soil characteristics, and soil microbial community after a volcanic eruption. However, there is little information about the relationships between the shifts in microbial properties such as community structure and the development of island-like communities in subalpine volcanic deserts, except for some studies of mycorrhizal fungal communities (e.g., Nara et al. 2003a, b; Fujiyoshi et al. 2005).
Our objective here was to examine how soil microbial properties such as biomass, community structure, and respiration change through the successional development of island-like communities in a volcanic desert. To achieve this, we performed community-level physiological profile (CLPP) analysis, phospholipid fatty acid (PLFA) analysis, and denaturing gradient gel electrophoresis (DGGE) analysis of 16S rDNA amplified by polymerase chain reaction (PCR) to determine the microbial community composition from functional, taxonomic, and genetic perspectives, respectively. Some recent studies have used two or more of these methods based on different principles and have succeeded in characterizing and identifying the microbial community structure (e.g., Girvan et al. 2003; Bossio et al. 2005; Ibekwe et al. 2007; Xue et al. 2008). This suggests that combining these methods, as is done in this study, should provide a better understanding of soil microbial communities from multiple (functional, taxonomic, and genetic) perspectives.
Materials and methods
Study site
The study was conducted on the southeastern slope of Mount Fuji, the highest mountain in Japan, between 1500 and 1550 m above sea level (35°20′N, 138°47′E). The annual mean air temperature in 2009 was 7.8 °C and the monthly mean air temperature ranged from −2.8 °C in January to 17.9 °C in August. Mean annual precipitation at Gotenba, about 15 km southeast of the study site, averaged 2811 mm between 2000 and 2010.
An accumulation of basaltic ejecta composed mainly of scoria (typically 2 to 30 mm wide) from Mount Hoei, which is a parasitic volcano that formed on the mid-slope of Mount Fuji during the 1707 eruption, covers much of the southeastern slope and forms a 3- to 4-km-wide section of the volcanic desert from the summit of Mount Hoei (2702 m) down to an altitude of about 1400 m (Tsuya 1971; Fujiyoshi et al. 2005).
In this area, previous studies have investigated the pattern and mechanisms of plant succession, which begins with isolated island-like communities of the perennial herb Polygonum cuspidatum Sieb. et Zucc. (= Reynoutria japonica Houttuyn) (e.g., Maruta 1976; Hirose and Tateno 1984; Masuzawa and Suzuki 1991; Masuzawa 1995; Nishitani and Masuzawa 1996). As P. cuspidatum spreads outward, its shoot density decreases in the center, thus providing a colonization site for later-successional species (the "central die-back" phenomenon) (Masuzawa 1995). As a result of differences in the timing of the initial colonization by the pioneer P. cuspidatum and its slow growth rate, a number of island-like communities with different successional stages are scattered within the study site. Fujiyoshi et al. (2005) categorized these communities into three successional stages. The first stage (stage I) is pioneer communities dominated by P. cuspidatum. In stage II, P. cuspidatum occupies the peripheral areas of the island-like communities, and herbs and grasses such as Calamagrostis hakonensis Franch. et Savat. and Miscanthus oligostachyus Stapf develop in the central areas. In stage III, shrubs such as Larix kaempferi (Lamb.) Carr. colonize the central areas, but various species of herbaceous plants, including P. cuspidatum, are still observed in the community. Coverage of P. cuspidatum tends to decrease as the island community progresses from stages I to III. In addition to these three successional stages, we defined the bare ground between the islands of vegetation dominated by P. cuspidatum as stage B, the first pioneer stage, and the L. kaempferi forest adjacent to areas that contained the island-like communities as stage F, a late-successional stage. We further subdivided stage III into two sub-stages: peripheral areas dominated by herbaceous plants (stage IIIa) and central areas dominated by shrubs (L. kaempferi, stage IIIb). Overall then, we identified a total of six successional stages (stages B, I, II, IIIa, IIIb, and F).
Soil sampling
Three island-like communities (4 to 6 m in diameter) belonging to each of stages I, II, IIIa, and IIIb were selected, and three study plots (each 1 × 1 m2) were selected for stages B and F (i.e., n = 3 plots for each successional stage). All of these study plots were located within a relatively small area (about 50 m × 100 m). At these study plots, mineral soil samples (< 5 cm below the mineral soil surface) were collected in July 2008 for measurement of microbial respiration and PLFA analysis using a stainless-steel soil corer (Ø = 5 cm, H = 5 cm, volume = 100 cm3). Three core samples per plot were collected and composited. Soil samples for the DGGE and CLPP analyses were collected in a sterilized 15-mL plastic tube at each study plot in July 2009. All collected samples were brought back to the laboratory in a cooler.
Soil pH (H2O) (air-dried soil to water ratio = 1:5 w/w) was measured with a pH meter fitted with a glass electrode (HM-30R; TOA Corp., Kobe, Japan). Soil water content was determined from the sample weights before and after vacuum-drying. The samples were then sieved (<2 mm) to remove plant roots and gravel. Soil organic matter (SOM) contents were determined by combustion at 550 °C for 4 h, and the total carbon and nitrogen contents were measured with a CN analyzer (Sumigraph NC-22; Sumika Chemical Analysis Service, Tokyo, Japan).
Microbial respiration
Microbial respiration (as the CO2 emission rate) was measured at 20 ±0.3 °C in an open-flow system with an infrared gas analyzer (LI-6262; Li-Cor Inc., Lincoln, USA). A detailed description of this system is provided by Bekku et al. (1997). A sieved (<4 mm) fresh soil sample equivalent to between 5 and 20 g dry weight was placed in a cylindrical plastic case (Ø = 9 cm, H = 2.5 cm). After 24 h of equilibration at 20 °C, the plastic case containing the sample was placed in a cylindrical chamber (Ø = 9.2 cm, H = 3 cm) connected to the system. Ambient air containing 400 to 410 ppm CO2 was pumped into the system at a suitable flow rate (100 or 300 mL min−1), depending on the amount of CO2 released from the sample. During measurements, the chamber was placed in a water bath to maintain a constant temperature of 20 ±0.3 °C.
Phospholipid fatty acid analysis
Phospholipid fatty acids (PLFAs) are the main components of the membranes of living cells and the amount and composition of PLFAs have been used as an index of the total microbial biomass and as a fingerprint of the microbial community structure, respectively (e.g., White et al. 1979; Frostegård et al. 1993). This method is culture-independent, and therefore provides unbiased views of the taxonomic structure of complex microbial communities (Ibekwe and Kennedy 1998) although it cannot identify community members at the species or strain levels (Ramsey et al. 2006; Ibekwe et al. 2007).
Soil samples used for the PLFA analysis were freeze-dried, sieved (<2 mm) to remove roots and gravel, and stored at –80 °C until analysis. Lipids were extracted using the extraction method of Bligh and Dyer (1959), as modified by White et al. (1979) and Frostegård et al. (1991). Briefly, 3 to 5 g (dry weight) of soil was extracted in a chloroform–methanol–citrate buffer (0.15 M, pH 4.0) (1:2:0.8 v/v/v). The lipids were separated into neutral lipids, glycolipids, and phospholipids on a silicic acid column (Sep-Pak Plus Silica; Waters Corp., Milford, USA). The phospholipids were esterified with 5 % HCl–methanol (Tokyo Kasei Kogyo Co. Ltd., Tokyo, Japan) (Stoffel et al. 1959). The resultant fatty-acid methyl esters were separated using a gas chromatograph/mass spectrometer (GC-17A/GCMS-QP5000; Shimadzu Corp., Kyoto, Japan) equipped with a capillary column (30-m DB-5 ms, phenyl-methyl/silicone; J&W Scientific Inc., Folsom, USA), with helium as the carrier gas. Peak areas were quantified against methyl nonadecanoate fatty acid (19:0) as an internal standard. We used the fatty acid nomenclature of Frostegård et al. (1993) and the total content of PLFA (TotPLFA; nmol g−1) was used to indicate the total microbial biomass (Frostegård et al. 1993). The sums of nine fatty acids (i15:0, a15:0, 15:0, i16:0, 17:0, i17:0, cy17:0, 18:1ω7c, cy19:0) were used as bacterial PLFA (BactPLFA; nmol g−1) (e.g. Federle 1986; Frostegård et al. 1993). Similarly, 18:2ω6 was used as fungal PLFA (FungPLFA; nmol g−1) (e.g. Federle 1986; Frostegård et al. 1993). The proportion of each PLFA (expressed as mol% of total PLFA) was averaged by successional stage.
Denaturing gradient gel electrophoresis
Microbial community analysis using 16S rDNA is also culture-independent and describes the microbial community structure in genetic term. Denaturing gradient gel electrophoresis (DGGE) analysis of 16 S rDNA amplified by PCR has been widely used to detect and identify community members to the species level with high specificity (e.g., Muyzer et al. 1993; Amann et al. 1995; Ibekwe et al. 2007; Yergeau et al. 2007; Li et al. 2011). We analyzed the bacterial community structure using this method.
Total DNA was extracted directly from about 0.5 to 1 g of fresh soil and purified with commercially available kits (Ultra Clean Soil DNA Isolation Kit and Power Clean DNA Clean-UP kit; MO BIO Laboratories Inc., Carlsbad, USA). 16S rDNA genes were PCR-amplified by using the universal primers 341F GC and 534R, with a GC clamp on the forward primer (Muyzer et al. 1993; Watanabe et al. 2001). Each 50-μL reaction vessel contained 1.0 μL of the DNA template, 1.0 μL of each primer (25 pmol μL−1; Invitrogen Life Technologies Corp., Carlsbad, CA, USA), 4.0 μL of dNTP Mixture (2.5 mM each), 0.25 μL of Takara Ex Taq HS (5 units μL−1), and 5.0 μL of 10× Ex Taq Buffer (Takara Bio Inc., Otsu, Japan). The thermal cycle involved an initial pre-denaturing (94 °C for 3 min), followed by 20 cycles comprising a denaturing step (94 °C for 1 min), an annealing step (65 °C decreasing to 55 °C, with 45 s at each temperature), and an extension step (72 °C for 1 min). The annealing temperature was decreased by 1 °C every two cycles until it reached 55 °C, at which point 10 additional cycles were run. We used a 7 min final extension at 72 °C.
For DGGE, we used a DCode System (Bio-Rad Laboratories, Hercules, USA). PCR amplicons (7.5 μL) were loaded on 8 % (v/v) polyacrylamide gels with a 35 % to 65 % denaturant gradient. Gels were run for 16 to 17 h at 60 V and 60 °C in 1 × Tris-acetatic acid-EDTA buffer and stained with SYBR Gold (Invitrogen) in Milli-Q water. The band pattern from each sample was then converted into a binary matrix (with 0 for an absent band and 1 for a visible band), and band patterns from the three replicates per successional stage were integrated into a single binary matrix.
Community-level physiological profiles
Community-level physiological profile (CLPP) analysis is one way of analyzing community structure on the basis of functional groupings (e.g., Garland and Mills 1991; Campbell et al. 1997; Classen et al. 2003). Four types of commercially available Biolog microplates (ECO, GP2, GN2, and FF; Biolog Inc., Hayward, USA) were used to test the community-level carbon-source utilization profiles of the soil microorganisms. The Biolog GP2, GN2, and FF plates each contained 95 separate carbon sources and a blank well with no substrate. In contrast, the Biolog ECO plate consists of three replicates of 31 carbon sources and a blank well. Some carbon sources were contained in two or more types of microplate. Consequently, we examined a total of 140 carbon sources to characterize the utilization potential of these sources for the soil microorganisms. These carbon sources consisted of 50 carbohydrates, 33 carboxylic acids, 7 polymers, 9 amines/amides, 21 amino acids, and 20 miscellaneous compounds (Preston-Mafham et al. 2002).
The Biolog microplates were prepared under sterile conditions within 48 h of sample collection in the field. We generated a 10-mL soil suspension from 1.0 g of fresh soil, thus achieving a 10 % w/v dilution. This solution was vigorously shaken for 10 min and centrifuged at 2500 rpm for 3 min. For stage B soil, we diluted 5 mL of the supernatant with 45 mL of sterilized water (a 1 % w/v dilution). For the other successional stages, we diluted 0.5 mL of the supernatant with 49.5 mL of sterilized water (a 0.1 % dilution). We then added 150 μL of the soil dilution into all wells on each plate and incubated the plates at 25 °C for 7 days. The color development in each well was measured at 24-h intervals as the absorbance at wavelengths of 490 and 750 nm for the FF plate and at 590 nm for the ECO, GP2, and GN2 plates, using a microplate reader (Power Scan HT; DS Pharma Biomedical Co. Ltd., Osaka, Japan). The absorbance data after 5 days were used for further analysis.
Absorbance in the blank well was subtracted from the absorbance readings in all other wells. For the FF plate data, absorbance was corrected by subtracting the 750-nm reading from the 490-nm reading, because the 750-nm reading measures the turbidity that results from mycelia production by soil fungi (Kubicek et al. 2003; Papaspyridi et al. 2011). After these corrections, the absorbance data were converted into binary data; wells for which the corrected absorbance was greater than or equal to twice that in the blank well were given a score of 1, and the others were given a score of 0. The resultant binary matrixes from the three replicates per successional stage were integrated into a single binary matrix by scoring wells with at least one of the three values equal to 1 as having a value of 1, and scoring all other wells as having a value of 0.
Statistical analysis
We used one-way ANOVA to test for significant differences in soil and microbial properties among the successional stages, and used Tukey's test to determine the rank order. Significance was defined at P < 0.05.
Differences in microbial community structure among the successional stages were examined based on PLFA, DGGE, and CLPP analyses with the nonmetric multidimensional scaling (NMDS) ordination technique. NMDS is an ordination method that iteratively searches for the best way to represent the data in a reduced number of dimensions so that the distances in the ordination diagram reflect the dissimilarities in community structure of the original samples. This method is considered better than many other ordination techniques because it does not assume a linear relationship among variables (Clarke 1993; Hannam et al. 2006). For the PLFA composition data (averaged mol% data matrix), the arcsine square-root transformation was used because the mol% data were non-normal (Hannam et al. 2006) and a dissimilarity matrix of PLFA composition using the Bray-Curtis dissimilarities coefficient was calculated. In contrast, dissimilarity matrices for DGGE and CLPP data (integrated binary data matrix) were calculated using the Jaccard dissimilarity coefficient designed for this type of data. Generated dissimilarity matrices were subjected to NMDS using R software (version 2.15.0; R Development Core Team 2012) and we obtained two-dimensional ordination graphs. The dimensions (axes) in the graph have no special significance and can be rotated or mirrored without influencing the relative distances between the points. The closer the points on this graph, the more similar they are in composition. As a measure of the goodness-of-fit of the reproduced distances to the observed distances, the “stress value” of NMDS is used. When stress values are <0.1, the NMDS plot is considered to be an acceptable representation of the original data (Clarke 1993). In our analyses, the values were <0.1 in all cases.
Results
Soil properties
Table 1 summarizes some key soil properties for each successional stage. Average soil pH (H2O) levels fell within a narrow range of 5.6 to 6.3. Soil water content, total C and N contents, and the SOM content were all very low in the bare ground (stage B) and tended to increase with increasing successional stage. Total C, N, and SOM contents in stage F were nearly twice those in the earlier stages (stages I to IIIb). The soil C/N ratio in stage B was significantly lower than values in the other successional stages (I − F).
Microbial respiration rate
Microbial respiration per gram of soil differed significantly among the successional stages (one-way ANOVA, P < 0.05; Fig. 1). The microbial respiration rate in Stage B (0.4 μg CO2–C g−1 h−1) was significantly lower than those in the other stages (Tukey's test, P < 0.05) and tended to increase with increasing successional stage until a relatively constant respiration rate was reached in Stages II, IIIa and F.

Microbial respiration rate per gram of soil from the six successional stages (at 20 °C). Values represent means ± SD (n = 3). Bars labeled with different letters differ significantly (Tukey's test, P < 0.05)
PLFA content and composition
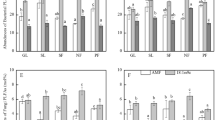
Twenty-seven PLFAs in total were detected, with ten of them detected in Stage B, but more (16–23 PLFAs) were detected in the other stages (Stages I − F). The total PLFA (TotPLFA) content in the soil differed significantly among the successional stages (one-way ANOVA, P < 0.01; Fig. 2a). An especially marked increase in TotPLFA was observed between stages B and I (from 6 to 94 nmol g−1; Tukey’s test, P = 0.16). In addition, TotPLFA increased significantly between stages IIIb and F (from 125 to 255 nmol g−1; Tukey’s test, P < 0.05). In contrast, the content did not differ significantly among stages I to IIIb (94 to 125 nmol g−1; Tukey’s test, P > 0.95). Contents of BactPLFA and FungPLFA showed similar trends to that of TotPLFA content, with both of them being lowest in stage B and highest in stage F (Fig. 2b,c). Especially in Stage B, the FungPLFA content was very small (below the detectable limit) and the ratio of BactPLFA to FungPLFA (F/B ratio) in stage B was significantly lower than ratios in the later successional stages (I − F) (Fig. 2d, Tukey’s test, P < 0.05).

a Total PLFA (TotPLFA), b bacterial PLFA (FungPLFA), and c fungal PLFA (BactPLFA) contents and d the ratio of BactPLFA to FungPLFA (F/B ratio) in soil from the six successional stages. Values represent means ± SD (n = 3). Bars labeled with different letters differ significantly (Tukey's test, P < 0.05)
The TotPLFA, BactPLFA, and FungPLFA contents all showed high positive correlations (r > 0.91) with some soil properties such as soil water content, and soil organic matter, total C, and total N contents (P < 0.001). Although the soil C/N ratio was also significantly correlated with PLFA contents (P < 0.05), the correlation coefficient was relatively small (r < 0.62). In contrast, the soil C/N ratio showed a relatively high positive correlation with the F/B ratio (r = 0.8, P < 0.001).
In the NMDS using Bray-Curtis dissimilarities based on the averaged PLFA composition data, soil from the pioneer stage (B) was apparently segregated from soils from the other successional stage (I − F) along the first dimension (Fig. 3a; Smirnov-Grubbs’ test, P < 0.01). The remaining five stages gradually changed from the upper to lower part of the graph along the second dimension (y-axis) of the NMDS (Fig. 3a).

Differences in microbial or bacterial community structure across the six successional stages (B–F) according to nonmetric multidimensional scaling (NMDS) based on a PLFA, b DGGE, and c CLPP data. The NMDS was conducted on the Bray-Curtis dissimilarity for PLFA data and Jaccard dissimilarity for DGGE and CLPP data
16S rDNA PCR-DGGE analysis
The genetic structures of the soil bacterial communities were compared by means of 16S rDNA PCR-DGGE analysis and distinct banding patterns were obtained that reflected the composition of the bacterial community in each successional stage. The number of distinct bands ranged from 16 to 19 in stages B to IIIa, whereas more bands were detected in stages IIIb (23) and F (34). Figure 3b presents results of the NMDS analysis using Jaccard dissimilarities based on the 16S rDNA PCR-DGGE banding patterns (binary matrix data). The first dimension of NMDS (x-axis) separated stage B from other stages (I − F) (Smirnov-Grubbs’ test, P = 0.07), whereas the second dimension (y-axes) separated stages IIIb and F from other stages (B − IIIa) (Smirnov-Grubbs’ test, P = 0.08 − 0.09). Thus the six successional stages were divided into three groups; stage B, stages I − IIIa, and stages IIIb − F.
Carbon utilization pattern as an index of CLPP
The number of carbon sources used by the soil microbial community was very small in stage B (21 of 140 carbon sources) but increased in stage I (91 carbon sources). In contrast, the soil microbial community in stages II to F used most (125 to 133) of the 140 carbon sources that were tested. The NMDS using Jaccard dissimilarities based on the community-level carbon-source use pattern (binary matrix data; Fig. 3c) showed that stage B was clearly separated from the other stages (I − F) along the first dimension (Smirnov-Grubbs’ test, P < 0.01). However, the classification of the remaining stages differed from those based on the PLFA and DGGE analyses (Fig. 3a, b) and divided the stages into two groups: stage I and stages II to F along the second dimension (Smirnov-Grubbs’ test, P < 0.01).
Discussion
Ecosystem development due to primary succession is accompanied by the accumulation of soil organic matter, with studies reporting that soil C, N, and SOM contents increase as succession proceeds in various successional series, including in glacier forelands (Bekku et al. 1999; Tscherko et al. 2003; Allison et al. 2007), post-glacial uplift islands (Aikio et al. 2000), and volcanic deserts (e.g., Halvorson et al. 1991; Crews et al. 2001; Halvorson and Smith 2009). Our results also showed increases in these soil properties with succession (Table 1). In addition, the quality of the SOM appears to shift during succession, because the soil C/N ratio, which is an index of the microbial availability of SOM (Kaye and Hart 1997), changed during succession, especially between stages B and I (Table 1).
Microbial biomass tended to increase as the plant communities developed (Fig. 2a), and it was strongly correlated with the soil water content, and total C, N, and SOM contents. These results suggest that soil development including the accumulation of SOM and an increase in water holding capacity along the primary succession was important for the increase in soil microbial biomass. In particular, water content, and total C, N and SOM contents jumped between stages B and I (Table 1), which agreed well with the large increase in microbial biomass during this period (Fig. 2a). The same could hold true for the changes between stage IIIb and F. Therefore, the “establishment period” of each island community (i.e., stages B to I) and the “maturation period” (i.e., stages III to F), when communities containing shrubs such as Larix kaempferi connect with each other, were especially important for determining the increase in size of the microbial community (Fig. 2).
The increase in microbial biomass during succession led to an increase in the soil microbial respiration rate per gram of soil (Fig. 1). However, the microbial respiration rate per gram of biomass, expressed here as the microbial respiration rate per nmol PLFA, showed the opposite trend, decreasing as succession progressed (Fig. 4). Some previous studies have also reported that the microbial respiration rate per unit biomass (the "metabolic quotient," qCO2) decreased during succession (e.g., Wardle and Ghani 1995; Ohtonen et al. 1999). Insam and Haselwandter (1989) suggested that the decrease in qCO2 during succession resulted from an increase in energy efficiency of the microbial community, because the more favorable conditions for microbial survival during later successional stages reduce the maintenance energy requirements.

Microbial respiration rate per nmol PLFA in soils from the six successional stages (at 20 °C). Values represent means ± SD (n = 3). Bars labeled with different letters differ significantly (Tukey's test, P < 0.05)
In addition to the microbial biomass and respiration rate (or activity), successional changes in the microbial community structure were observed on the basis of the PLFA, DGGE, and CLPP analyses (Fig. 3). These three methods all showed that the microbial community structure in stage B was distinct from those in subsequent stages (Fig. 3). In addition, the number of PLFAs detected, and the number of carbon sources used in the CLPP analysis, were lower in stage B than in the other stages, indicating the low level of richness of the microbial community at this stage. Ibekwe et al. (2007) studied the microbial community structure in a volcanic desert on Mount St. Helens and reported that soils from bare ground had smaller and less diverse microbial communities than the soils associated with plants. These results suggest that colonization by the first pioneer plants (i.e., establishment of an island community) has a strong impact on the microbial community structure as well as on the microbial biomass in a volcanic desert. Our stage B had a very small FungPLFA content and therefore had a low F/B ratio (Fig. 2c,d), suggesting that the microbial community in stage B was relatively dominated by bacteria. Thereafter, the FungPLFA content and F/B ratio increased in the later successional stages (I − F), indicating that the microbial community structure shifted from bacterial- to fungal-dominated. Stoichiometric differences between bacteria and fungi would partly explain this type of shift in microbial community structure. Fungal biomass is known to have a greater mean C/N ratio than bacterial biomass, and therefore fungi are expected to have lower nitrogen requirements (Strickland and Rousk 2010). Therefore, an environment with a higher soil C/N ratio (stage B) would be more expedient for bacteria compared with fungi. Because fungi are more efficient in their energy use than bacteria and form a large inactive biomass (Ohtonen et al. 1999), such a shift in the microbial community structure would increase the energy-use efficiency of the overall microbial community in later successional stages and decrease the microbial respiration rate per biomass as described above.
After stage I, successional changes in microbial community structure, as shown in the NMDS graphs, differed between the PLFA and DGGE analyses (Fig. 3a,b), which may be partly explained by two different methodological reasons; that is, differences in targeted taxon (whole communities in PLFA vs. bacterial communities in DGGE) and resolution (PLFA cannot detect structural change at a species level, Ramsey et al. 2006; Ibekwe et al. 2007) between these two methods. The PLFA analysis showed that the microbial community structure changed gradually after stage I (Fig. 3a). Changes in species and/or composition of plant communities will cause changes in the quality of the SOM, although we did not detect this through a change in the C/N ratio, because different plant species produce litter and rhizosphere deposition with different qualities (e.g., Xuluc-Tolosa et al. 2003; Osono et al. 2006) and the quality of SOM is strongly affected by litter quality (e.g., Pennanen et al. 2001). Such a shift in SOM quality would lead to differences in microbial community structure. Some previous studies also suggested that differences in individual species, composition, and diversity of plant communities affected the microbial community structure via changes in soil carbon quality (Carney and Matson 2006; Eisenhauer et al. 2010). In addition, differences in symbiotic microorganisms among the plant species could also affect the results. For example, P. cuspidatum and L. kaempferi are known to form different types of mycorrhizae; the former associates with arbuscular mycorrhizal fungi, whereas the latter forms ectomycorrhizae as well as arbuscular mycorrhizae (Fujiyoshi et al. 2005). This would affect the microbial community structure detected by the PLFA analysis. The DGGE analysis showed a substantial shift in the bacterial community structure between stages IIIa and IIIb, which could also reflect differences in SOM quality because the dominant vegetation type largely shifted from herbaceous plants, including P. cuspidatum, to shrub species such as L. kaempferi during this period and L. kaempferi is known to produce high-lignin, recalcitrant leaf litter (e.g. Kunito et al. 2009).
In contrast to the PLFA and DGGE analyses, the CLPP analysis did not show clear differences in community structure among stages II to F (Fig. 3c). These results indicate that the taxonomic groups or members of each microbial community changed, but that the functional changes in the overall microbial community are negligible during the later stages of succession. In diverse microbial communities (well developed communities), the ability of specific members (or taxonomic groups) to utilize carbon from a specific carbon source would be expected to overlap (or perhaps be compensated) by other member(s) or group(s), which would lead to stabilization of the carbon utilization profile of the whole microbial community. The lack of changes in CLPP may also be due to methodological reasons. The CLPP analysis is a culture-dependent method, and it is known that only a very small proportion of soil microorganisms are able to be cultured (Atlas and Bartha 1998). Therefore, CLPP analysis based on Biolog microplates is likely to be relatively insensitive to slight changes in microbial community structure. In addition, most of the tested carbon sources were utilized by the microbial communities after stage II, which also make it difficult to detect differences in carbon source utilization patterns.
Our results show that succession in the soil microbial community in a volcanic desert was closely related to the development of isolated island-like plant communities. Colonization by the first pioneer herbaceous plant (P. cuspidatum) changed the soil environment dramatically, including both the amount and quality of the SOM, and this caused an increase in microbial biomass, a shift in the microbial community structure, and changes in the physiological activity of this community. In addition, subsequent invasion of the central part of the island-like communities by shrubs such as L. kaempferi was an important event, especially for the bacterial community structure.
References
Aikio S, Väre H, Strömmer R (2000) Soil microbial activity and biomass in the primary succession of a dry heath forest. Soil Biol Biochem 32:1091–1100
Allison VJ, Condron LM, Peltzer DA, Richardson SJ, Turner BL (2007) Changes in enzyme activities and soil microbial community composition along carbon and nutrient gradients at the Franz Josef chronosequences, New Zealand. Soil Biol Biochem 39:1770–1781
Amann RL, Ludwig W, Schleifer KH (1995) Phylogenetic identification and in situ detection of individual microbial cells without cultivation. Microbiol Rev 59:143–169
Atlas RM, Bartha R (1998) Microbial ecology: fundamentals and applications. Benjamin Cummings, Dordrecht
Bekku Y, Koizumi H, Oikawa T, Iwaki H (1997) Examination of four methods for measuring soil respiration. Appl Soil Ecol 5:247–254
Bekku Y, Kume A, Nakatsubo T, Masuzawa T, Kanda H, Koizumi H (1999) Microbial biomass in relation to primary succession on arctic degraciated moraines. Polar Biosci 12:47–53
Bligh EG, Dyer WJ (1959) A rapid method of total lipid extraction and purification. Can J Biochem Physiol 37:911–917
Bossio DA, Girvan MS, Verchot L, Bullimore J, Borelli T, Albrecht A, Scow KM, Ball AS, Pretty JN, Osborn AM (2005) Soil microbial community response to land use change in an agricultural landscape of western Kenya. Microb Ecol 49:50–62
Campbell CD, Grayston SJ, Hirst DJ (1997) Use of rhizosphere carbon sources in sole carbon source tests to discriminate soil microbial communities. J Microbiol Meth 30:33–41
Carney KM, Matson PA (2006) The influence of tropical plant diversity and composition on soil microbial communities. Microb Ecol 52:226–238
Chiba N, Hirose T (1993) Nitrogen acquisition and use in three perennials in the early stage of primary succession. Funct Ecol 7:287–292
Classen AT, Boyle SI, Haskins KE, Overby ST, Hart SC (2003) Community-level physiological profiles of bacteria and fungi: plate type and incubation temperature influences on contrasting soils. FEMS Microbiol Ecol 44:319–328
Clarke KR (1993) Non-parametric multivariate analyses of changes in community structure. Aust J Ecol 18:117–143
Crews TE, Kurina LM, Vitousek PM (2001) Organic matter and nitrogen accumulation and nitrogen fixation during early ecosystem development in Hawaii. Biogeochemistry 52:259 − 279
del Moral R, Jones C (2002) Vegetation development on pumice at Mount St. Helens, USA. Plant Ecol 162:9–22
Eisenhauer N, Beßler H, Engels C, Gleixner G, Habekost M, Milcu A, Partsch S, Sabais ACW, Scherber C, Steinbeiss S, Weigelt A, Weisser WW, Scheu S (2010) Plant diversity effects on soil microorganisms support the singular hypothesis. Ecology 91:485−496
Federle TW (1986) Microbial distribution in soil – new techniques. In: Gantar M, Megusar F (eds) Perspectives in microbial ecology. Slovene Society for Microbiology, Ljubljana, pp 493–498
Frostegård Å, Tunlid A, Bååth E (1991) Microbial biomass measured as total lipid phosphate in soils of different organic content. J Microbiol Meth 14:151–163
Frostegård Å, Tunlid A, Bååth E (1993) Phospholipid fatty acid composition, biomass, and activity of microbial communities from two soil types experimentally exposed to different heavy metals. Appl Environ Microbiol 59:3605–3617
Fujiyoshi M, Kagawa A, Nakatsubo T, Masuzawa T (2005) Successional changes in mycorrhizal type in the pioneer plant communities of a subalpine volcanic desert on Mt. Fuji, Japan. Polar Biosci 18:60–72
Garland JL, Mills AL (1991) Classification and characterization of heterotrophic microbial communities on the basis of patterns of community-level sole-carbon-source utilization. Appl Environ Microbiol 57:2351–2359
Girvan MS, Bullimore J, Pretty JN, Osborn AM, Ball AS (2003) Soil type is the primary determinant of the composition of the total and active bacterial communities in arable soils. Appl Environ Microbiol 69:1800–1809
Halvorson JJ, Smith JL (2009) Carbon and nitrogen accumulation and microbial activity in Mount St. Helens pyroclastic substrates after 25 years. Plant Soil 315:211–228
Halvorson JJ, Smith JL, Franz EH (1991) Lupine influence on soil carbon, nitrogen and microbial activity in developing ecosystems at Mount St. Helens. Oecologia 87:162–170
Hannam KD, Quideau SA, Kishchuk BE (2006) Forest floor microbial communities in relation to stand composition and timber harvesting in northern Alberta. Soil Biol Biochem 38:2565–2575
Hopkins DW, Badalucco L, English LC, Meli SM, Chudek JA, Ioppolo A (2007) Plant litter decomposition and microbial characteristics in volcanic soils (Mt Etna, Sicily) at different stages of development. Biol Fetil Soils 43:461–469
Hirose T, Tateno M (1984) Soil nitrogen patterns induced by colonization of Polygonum cuspidatum on Mt. Fuji. Oecologia 61:218–223
Ibekwe AM, Kennedy AC (1998) Phospholipid fatty acid profiles and carbon utilization patterns for analysis of microbial community structure under field and greenhouse conditions. FEMS Microbiol Ecol 26:151–163
Ibekwe AM, Kennedy AC, Halvorson JJ, Yang CH (2007) Characterization of developing microbial communities in Mount St. Helens pyroclastic substrate. Soil Biol Biochem 39:2496–2507
Insam H, Haselwandter K (1989) Metabolic quotient of the soil microflora in relation to plant succession. Oecologia 79:174–178
Jenkinson DS, Ladd JN (1981) Microbial biomass in soil: measurement and turnover. In: Paul EA, Ladd JN (eds) Soil biochemistry 5. Marcel Dekker, New York, pp 415–471
Jhonson EA, Miyanishi K (2007) Disturbance and succession. In: Jhonson EA, Miyanishi K (eds) Plant disturbance ecology: the process and response. Academic, New York, pp 1–14
Jumpponen A, Brown SP, Trappe JM, Cázares E, Strömmer R (2012) Twenty years of research on fungal − plant interactions on Lyman Glacier forefront – lessons learned and questions yet unanswered. Fung Ecol 5:430–442
Kaye JP, Hart SC (1997) Competition for nitrogen between plants and soil microorganisms. Trends Ecol Evol 12:139–143
Kubicek CP, Bissett J, Druzhinina I, Kullnig-Gradinger C, Szakacsc G (2003) Genetic and metabolic diversity of Trichoderma: a case study on South-East Asian isolates. Fungal Genet Biol 38:310–319
Kunito T, Akagi Y, Park HD, Toda H (2009) Influences of nitrogen and phosphorus addition on polyphenol oxidase activity in a forested Andisol. Eur J Forest Res 128:361–366
Li Y, Wang L, Zhang W, Wang H, Fu X, Le Y (2011) The variability of soil microbial community composition of different types of tidal wetland in Chongming Dongtan and its effect on soil microbial respiration. Ecol Eng 37:1276–1282
Maruta E (1976) Seedling establishment of Polygonum cuspidatum on Mt. Fuji. Jpn J Ecol 26:101–105
Maruta E (1983) Growth and survival of current-year seedlings of Polygonum cuspidatum at the upper distribution limit on Mt. Fuji. Oecologia 60:316–320
Masuzawa T (1995) Successional process on Mt. Fuji: from alpine herbaceous perennials to dwarf scrub. In: Box EO, Peet RK, Masuzawa T, Yamada I, Fujiwara K, Maycock PF (eds) Vegetation science in forestry. Kluwer Academic Publishers, Netherlands, pp 423–431
Masuzawa T, Suzuki J (1991) Structure and succession of alpine perennial community (Polygonum cuspidatum) on Mt. Fuji. Proc NIPR Symp Polar Biol 4:155–160
Merilä P, Strömmer R, Fritze H (2002) Soil microbial activity and community structure along a primary succession transect on the land-uplift coast in western Finland. Soil Biol Biochem 34:1647–1654
Miyawaki A (1971) Vegetation of Mt. Fuji. Fujisan (Mt. Fuji). Fuji-Kyuko, Tokyo, pp 665 − 721 (in Japanese with English summary)
Muyzer G, de Waal EC, Uitierlinden AG (1993) Profiling of complex microbial populations by denaturing gradient gel electrophoresis analysis of polymerase chain reaction-amplified genes coding for 16S rRNA. Appl Environ Microbiol 59:695–700
Nara K, Nakaya H, Hogetsu T (2003a) Ectomycorrhizal sporocarp succession and production during early primary succession on Mount Fuji. New Phytol 158:193–206
Nara K, Nakaya H, Zhou WZ, Hogetsu T (2003b) Underground primary succession of ectomycorrhizal fungi in a volcanic desert on Mount Fuji. New Phytol 159:743–756
Nishitani S, Masuzawa T (1996) Germination characteristics of two species of Polygonum in relation to their altitudinal distribution on Mt. Fuji, Japan. Arct Antarct Alp Res 28:104–110
Ohtonen R, Fritze H, Pennanen T, Jumpponen A, Trappe J (1999) Ecosystem properties and microbial community changes in primary succession on a glacier forefront. Oecologia 119:239–246
Osono T, Mori A, Uchida M, Kanda H (2006) Chemical property of live and dead leaves of tundra plant species in Oobloyah Valley, Ellesmere Island, high arctic Canada. Mem NIPR Spec Issue 59:144–155
Papaspyridi LM, Katapodis P, Gonou-Zagou Z, Kapsanaki-Gotsi E, Christakopoulos P (2011) Growth and biomass production with enhanced beta-glucan and dietary fibre contents of Ganoderma australe ATHUM 4345 in a batch-stirred tank bioreactor. Eng Life Sci 11:65–75
Pennanen T, Strommer R, Markkola A, Fritze H (2001) Microbial and plant community structure across a primary succession gradient. Scand J Forest Res 16:37–43
Preston-Mafham J, Boddy L, Randerson PF (2002) Analysis of microbial community functional diversity using sole-carbon-source utilisation profiles - a critique. FEMS Microbiol Ecol 42:1–14
R Development Core Team (2012) R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna. ISBN 3-900051-07-0, http://www.R-project.org/
Ramsey PW, Rilling MC, Feris KP, Holben WE, Gannon JE (2006) Choice of methods for soil microbial community analysis: PLFA maximizes power compared to CLPP and PCR-based approaches. Pedobiologia 50:275–280
Schipper LA, Degens BP, Sparling GP, Duncan LC (2001) Soil Biol Biochem 33:2093–2103
Shillam L, Hopkins DW, Badalucco L, Laudicina VA (2008) Structural diversity and enzyme activity of volcanic soils at different stages of development and response to experimental disturbance. Soil Biol Biochem 40:2182–2185
Stoffel W, Chu F, Ahrens EH (1959) Analysis of long-chain fatty acids by gas-liquid chromatography. Micromethod for preparation of methyl ester. Anal Chem 31:307–308
Strickland MS, Rousk J (2010) Considering fungal:bacterial dominance in soils e Methods, controls, and ecosystem implications. Soil Biol Biochem 42:1385–1395
Tscherko D, Hammesfahr U, Zeltner G, Kandeler E, Böcker R (2003) Plant succession and rhizosphere microbial communities in a recently deglaciated alpine terrain. Basic Appl Ecol 6:367–383
Tsuya H (1971) Topography and geology of volcano Mt. Fuji. Fujisan (Mount Fuji). Fuji-Kyuko, Tokyo, pp 1 − 149 (in Japanese with English summary)
Wardle DA, Ghani A (1995) A critique of the microbial metabolic quotient (qCO2) as a bioindicator of disturbance and ecosystem development. Soil Biol Biochem 27:1601–1610
Watanabe K, Kodama Y, Harayama S (2001) Design and evaluation of PCR primers to amplify bacterial 16S ribosomal DNA fragments used for community fingerprinting. J Microbiol Meth 44:253–262
White DC, Davis WM, Nickels JS, King JD, Bobbie RJ (1979) Determination of the sedimentary microbial biomass by extractible lipid phosphate. Oecologia 40:51–62
Xue D, Yao HY, Ge DY, Huang CY (2008) Soil microbial community structure in diverse land use systems: a comparative study using biolog, DGGE, and PLFA analyses. Pedosphere 18:653–663
Xuluc-Tolosa FU, Vester HFM, Ramírez-Marcial N, Castellanos-Albores J, Lawrence D (2003) Leaf litter decomposition of tree species in three successional phases of tropical dry secondary forest in Campeche, Mexico. Forest Ecol Manag 174:401–412
Yergeau E, Bokhorst S, Huiskes AHL, Boschker HTS, Aerts R, Kowalchuk GA (2007) Size and structure of bacterial, fungal and nematode communities along anAntarctic environmental gradient. FEMS Microbiol Ecol 59:436–451
Acknowledgements
We thank Dr N. Sakurai of Hiroshima University for providing the laboratory facilities. The comments from the editor and four anonymous reviewers greatly improved the manuscript. This study was partly supported by a Grant-in-Aid for Scientific Research from the Japan Society for the Promotion of Science.
Author information
Authors and Affiliations
Corresponding author
Additional information
Responsible Editor: Nico Eisenhauer.
Rights and permissions
About this article
Cite this article
Yoshitake, S., Fujiyoshi, M., Watanabe, K. et al. Successional changes in the soil microbial community along a vegetation development sequence in a subalpine volcanic desert on Mount Fuji, Japan. Plant Soil 364, 261–272 (2013). https://doi.org/10.1007/s11104-012-1348-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11104-012-1348-7