
Abstract
Key message
Key genes potentially involved in cacao disease resistance were identified by transcriptomic analysis of important cacao cultivars. Defense gene polymorphisms were identified which could contribute to pathogen recognition capacity.
Abstract
Cacao suffers significant annual losses to the water mold Phytophthora spp. (Oomycetes). In West Africa, P. megakarya poses a major threat to farmer livelihood and the stability of cocoa production. As part of a long-term goal to define key disease resistance genes in cacao, here we use a transcriptomic analysis of the disease-resistant cacao clone SCA6 and the susceptible clone NA32 to characterize basal differences in gene expression, early responses to infection, and polymorphisms in defense genes. Gene expression measurements by RNA-seq along a time course revealed the strongest transcriptomic response 24 h after inoculation in the resistant genotype. We observed strong regulation of several pathogenesis-related genes, pattern recognition receptors, and resistance genes, which could be critical for the ability of SCA6 to combat infection. These classes of genes also showed differences in basal expression between the two genotypes prior to infection, suggesting that prophylactic expression of defense-associated genes could contribute to SCA6’s broad-spectrum disease resistance. Finally, we analyzed polymorphism in a set of defense-associated receptors, identifying coding variants between SCA6 and NA32 which could contribute to unique capacities for pathogen recognition. This work is an important step toward characterizing genetic differences underlying a successful defense response in cacao.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Theobroma cacao L., commonly referred to as cacao, is a tropical tree belonging to the Malvaceae family originating from the Amazon rainforest (Wood et al. 2008; Alverson et al. 1999; Motamayor et al. 2002). As the unique source of the main ingredients of chocolate, cacao is now cultivated throughout the tropics. About 70% of the world’s raw cocoa is supplied by West African countries where cacao is grown by smallholder farmers (Wessel and Quist-Wessel 2015). Export of dried cocoa beans makes up the largest agricultural commodity contribution to foreign exchange earnings and gross domestic product for several producing countries (Wessel and Quist-Wessel 2015). Sustainability of smallholder cocoa production are threatened by the effect of pests and diseases, which are estimated to ruin one-third of the world’s cocoa production annually (Wessel and Quist-Wessel 2015; Ploetz 2016). In the largest exporting countries, such as Côte d’Ivoire and Ghana, prevalence of cacao disease can significantly impact the national economies. Globally, the most widespread disease is Phytophthora pod rot, also called black pod rot, caused by four species of Phytophthora (Surujdeo-Maharaj et al. 2016). The most aggressive of these species, P. megakarya, occurs only in West Africa. In areas invaded by P. megakarya, pod losses can reach 80% of farm production if no disease control is applied (Nyassé et al. 1999; Bailey et al. 2016). Control of black pod is mainly achieved through the use of fungicides; however, application of large quantities of fungicides increase labor and input costs and can also produce toxic effects on humans, wildlife, and the environment. Therefore, breeding disease-resistant varieties is the most effective and economical method of controlling P. megakarya.
The majority of cacao cultivars grown worldwide are susceptible to Phytophthora infection (Eskes and Lanaud 2001). Although significant variation for genetic resistance has been observed in germplasm collections and breeding trials (Blaha and Lotodé 1976; Despreaux et al. 1989; Iwaro et al. 2003; Thevenin et al. 2012), breeding and distribution of resistant varieties is a slow process requiring about 20 years. Conventional breeding methods are hampered by the long growth period of cacao, land requirement for large trials, and gene-environment interactions complicating measurements of plant susceptibility. Over the past two decades, cacao breeders have turned to molecular methods to accelerate crop improvement. A large number of QTLs related to traits of interest including resistance to Phytophthora spp. have been developed from controlled crosses (Crouzillat et al. 1996, 2000a, b; Lanaud et al. 1999, 2004; Motilal and Sounigo 2000; Clement et al. 2003; Flament et al. 2001; Risterucci et al. 2000, 2003; Queiroz et al. 2003, 2006; Brown et al. 2005, 2007; Faleiro et al. 2006; Fernandes et al. 2018) and a meta-QTL analysis examined overlap of regions predicted from many of these studies, identifying consensus QTL believed to be important in defense (Lanaud et al. 2009). Researchers have also applied association mapping strategies (Pugh 2005; Schnell et al. 2005; Marcano et al. 2007, 2009; Royaert et al. 2016). However, because of the relatively small size of mapping populations, QTL and associations identified by these studies typically comprise chromosomal regions containing thousands of genes and thus have limited resolution and utility for marker assisted selection breeding strategies. Molecular characterization of the defense response in other model and crop species offer insight into strategies for identifying genes of particular importance in the interactions of cacao with its pathogens.
The canonical plant defense response involves three stages of activity: recognition of the pathogen, signal transduction, and induction of antimicrobial proteins and chemicals. The recognition stage relies on extracellular receptor-like kinases (RLKs) and receptor-like proteins (RLPs) which detect pathogen/damage-associated molecular patterns (PAMPs and DAMPs, collectively pattern recognition receptors, PRRs) in the apoplast (Tang et al. 2017), or nucleotide binding leucine-rich repeat proteins (NLRs) which detect pathogen effectors secreted into the plant cell (Jones et al. 2016). Within minutes of pathogen perception by a PRR or NLR, signal transduction cascades are triggered which involve Ca2+ burst, ROS burst, activation of MAPK cascades, and transcriptional reprogramming (Cui et al. 2015; Bigeard et al. 2015; Kimura et al. 2017). Phytohormone signaling is also induced, and the disease-associated hormones salicylic acid, jasmonic acid, and ethylene interact with each other and with growth hormones to regulate the plant’s response (Kumar et al. 2015; Vlot et al. 2009; McManus 2012; Yang et al. 2015; Song et al. 2014; De Bruyne et al. 2014; Karasov et al. 2017). These signaling mechanisms ultimately lead to the dynamic and pathogen specific activation of defense mechanisms. These include induction of the pathogenesis-related (PR) genes, which encode proteins with a variety of antimicrobial properties including cell wall and cell membrane disruption and protein and nucleic acid degradation (van Loon and van Strien 1999; van Loon et al. 2006). Another component of the defense response is modification and reinforcement of the cell wall to prevent further invasion (Bellincampi et al. 2014). Programmed cell death can also be induced to prevent further colonization of exposed tissues (Dickman and Fluhr 2013; Coll et al. 2011).
Canonically, disease resistance in plants has been described as a qualitative phenomenon, whereby a PRR or NLR interacts with a pathogen effector directly or indirectly (Mackey et al. 2002; Hoorn and Kamoun 2008), triggering induction of defenses termed the hypersensitive response, marked by localized cell death, which prevents further colonization of plant tissue (Mur et al. 2008). However, plants often exhibit quantitative resistance, sometimes termed disease tolerance, to pathogens, which features delayed disease progression compared to more susceptible varieties of the same species (Kushalappa et al. 2016; Corwin and Kliebenstein 2017; Poland et al. 2009). Quantitative resistance, also known as polygenic resistance, is associated with many genes of small effect, and QTL analysis and subsequent molecular characterization have shown that expression level and polymorphism in PRRs, NLRs, transcription factors, and downstream responders such as PR proteins contribute to quantitative resistance (Corwin and Kliebenstein 2017).
To date, no gene-for-gene interaction has been demonstrated between cacao and any of its pathogens, and therefore described resistance phenotypes in cacao refer to quantitative resistance. Use of -omics strategies in the past decade have greatly advanced the characterization of the arsenal of genes potentially contributing to resistance in cacao. Two annotated cacao genomes have been published (Motamayor et al. 2013; Argout et al. 2011), and another 200 genotypes were recently sequenced and are being analyzed to identify loci associated with resistance to another two cacao diseases, witches’ broom and frosty pod rot, caused by related basidiomycete fungi (McElroy et al. 2018). The version one publication of the Criollo cacao genome included an analysis of cacao’s PRRs identifying 253 leucine-rich repeat receptor-like kinases (LRR-RLKs) and 297 NLRs (Argout et al. 2011). Recent transcriptomic analyses of cacao’s defense response have examined gene regulation during Witches’ Broom development using RNA-Seq (Teixeira et al. 2014), Phytophthora megakarya infection of leaf tissue using a macroarray (Legavre et al. 2015), infection of leaves by Phytophthora palmivora and Colletotrichum theobromicola using a microarray (Fister et al. 2016), and response of leaves to salicylic acid treatment (Fister et al. 2015). Genomic analyses have also been applied to the two most damaging Phytophthora species, P. megakarya and P. palmivora. Both genomes were recently sequenced (Ali et al. 2017a, b) and a transcriptomic approach was used to differentiate the responses induced by inoculation of pods of a susceptible cacao genotype (Ali et al. 2017b). These studies have identified hundreds to thousands of gene involved in the cacao biotic stress response, but further work targeting resistant and susceptible genotypes is required to clarify key molecular players that mediate resistance or tolerance, such that clones with useful variation can more effectively be used in breeding programs.
The goal of this study was to identify genes significantly regulated in response to Phytophthora megakarya early disease progression in a resistant (SCA6) and a susceptible (NA32) cacao genotype and to better understand basal differences in gene expression between these genotypes. Both SCA6 and NA32 are agronomically important genotypes. SCA6 is a well-known source of resistance to Phytophthora (Ali et al. 2016) and cacao’s fungal diseases (Teixeira et al. 2015) and NA32 and its progeny are widely cultivated for good pod quality (Dinarti et al. 2015). Further we sought to characterize allelic variation in defense genes to survey polymorphism at immune-related loci to begin to dissect genetics of defense in cacao.
Materials and methods
Plant material
The SCA6 and NA32 genotypes were chosen from the collection of parental lines among those used to initiate the breeding program at the Centre National de Recherche Agronomique (CNRA) in Adiopodoume, Côte d’Ivoire. Both clones were originally collected in Peru, but belong to different genetic groups [SCA6 belongs to the Contamana genetic group while NA32 belongs to the Nanay genetic group (Motamayor et al. 2008)]. The performance of these genotypes has been quantified at CNRA using in-field pod rot measurements with mature trees over 6 years and the detached pods and detached leaves in the lab since 2000 (Tahi et al. 2000; Pokou et al. 2008). Of all the ancestral genotypes used in CNRA breeding programs, SCA6 is the most fully characterized resistant genotype, while NA32 is among the most susceptible to P. megakarya and P. palmivora. On a scale of 1 to 5 used to score the leaf test, SCA6 always is rated as 1 (most resistant) to P. palmivora, while NA32 scores on average 4.5 (Pokou et al. 2001, 2008). Leaf and pod based resistance tests of SCA6 tissues inoculated with P. megakarya also classify it as a resistant variety (Nyadanu et al. 2012). Despite its susceptibility, NA32 is widely grown in West Africa because of its high yield (Pokou et al. 2009).
Experimental design
The experiment was carried out in a shade net house at CNRA. Four-month old grafted plants were arranged in two blocks: one sprayed with zoospore suspension and the other with water as a control. Each block was composed of 160 plants arranged in a grid with ~ 20 cm of spacing between plants, with genotypes in alternating rows, and a border of plants on all sides that were not used for sampling. The blocks were subdivided by a 1 m wide walking lane (shade net house organization schematic in Supplemental Fig. S1).
Pathogen maintenance
The P. megakarya isolate SOU 130.B, a virulent strain commonly used in resistance tests in Côte d’Ivoire, was chosen for inoculations. The pathogenicity of the isolate was maintained in the laboratory by regular inoculation of green mature cocoa pods and by re-isolation afterwards on pea agar medium in tubes placed at 26 °C in the dark. Zoospore suspensions were obtained after incubation of culture flasks for 6 days in darkness followed by 10 days with alternating 12 h darkness and fluorescent light. To release zoospores, cultures were flooded with distilled water at 4 °C and incubated with incandescent light for at least 40 min (Tahi et al. 2000).
Application of treatments and sampling
RNA preservation solution (final concentration 3.5 M ammonium acetate, 16.6 mM sodium citrate, 26.6 mM EDTA, pH 5.2), used to prevent RNA degradation, was prepared the morning of plant inoculation. Sampling was performed using scissors to cut petioles to remove one stage C leaf (Fister et al. 2016a, b; Mejia et al. 2012) from each of two randomly selected plants that were then placed into a 50 mL tube of RNA preservation solution and were later pooled for RNA extractions. At each time point, samples from different plants (biological replicates) for each genotype were collected from each block. Time zero samples were collected immediately prior to application of treatment. After this collection, treatments were applied by spraying the blocks with Phytophthora megakarya zoospores (300,000 per mL) or distilled water alone. Each plant was sprayed four times, twice from above and twice from below. Leaf samples were collected at 6, 24, and 72 hai (hours after inoculation). To avoid wound effect on gene expression a different set of plants was used for each time point. After leaves were collected, each experimental plant was removed from the experiment area and was replaced by a healthy, extra plant of the same genotype to prevent transmission of wound signaling among plants and microenvironmental effects from missing plants. Samples were shipped on dry ice in coolers to Pennsylvania State University for subsequent processing.
RNA isolation, RNA-Seq library preparation, and transcriptome sequencing
For each sample, one 3 × 3 cm2 was cut from each of the two pooled leaves and they were washed in sterile water to rinse residual salts from the RNA preservation solution. Together the two squares were ground with a mortar and pestle using liquid nitrogen. RNA was then extracted from the ground tissue according to the PureLink Plant RNA Reagent protocol (Thermo Fisher Scientific, Waltham, MA), with minor modifications. The modifications included using 1 mL of RNA reagent to better resuspend the ground tissue, and all centrifugations were performed at 14,000 rpm to more effectively pellet ground tissue and separate phases after chloroform extractions. Samples were precipitated adding 1/10th volume of 3M sodium acetate and 2.5 volumes of 100% ethanol.
After extraction, RNA samples were treated with RNase-free DNase (ThermoFisher Scientific) according to manufacturer’s instructions, purified with an additional ethanol precipitation as above and resuspended in RNase-free water. Initial measurement of concentration and quality were performed using Qubit and Nanodrop spectrophotometers, respectively.
RNA quality assessment, library preparation, and sequencing were performed at the Pennsylvania State University Genomics Core Facility (University Park, PA). RNA quality was assessed using an Agilent RNA 6000 kit (Agilent Technologies, Santa Clara, CA). Samples with a RIN score of 7.3–8.1 were used in this study. The four replicates with highest RIN for each genotype*time point*treatment combination were selected for library preparation. 200 ng of each sample was used to prepare a barcoded library using the TruSeq Stranded mRNA Library Prep Kit according to the manufacturer’s protocol (Illumina, San Diego, CA). The concentration of each library was determined by qPCR using a Kapa Library Quantification Kit Illumina Platforms according to the manufacturer’s instructions (Kapa Biosystems, Wilmington, MA).
An equimolar pool of all barcoded libraries was made and sequenced on the Illumina HiSeq 2500 in Rapid Mode using 100 nt single-read sequencing. A total of five Rapid Runs were performed producing a total of ~ 1,273,600,000 reads and an average of ~ 19,900,000 reads per sample.
Read processing and gene expression analysis
After sequencing, the 64 samples were processed, first by removing adaptors and trimming low quality sequence using trimmomatic (Bolger et al. 2014). Reads were next mapped to the cacao reference genome sequence (Criollo genome v2.0) using hisat2 (Kim et al. 2015). BEDtools (Quinlan and Hall 2010) was used to produce coverage graphs and data were visualized using IGV (Thorvaldsdóttir et al. 2013) to check for errors. Finally, read counts for each gene were calculated including multimapped reads using featureCounts (Liao et al. 2014). By comparing sequences of reads, one sample, an NA32 6 hai water-treated replicate, was found to have been a mislabeled RNA sample from SCA6 tissue and was excluded from analysis, so the NA32 6 hai water treatment analyses are based on three biological replicates. Comparisons of differential gene expression were performed using DESeq2 with default settings (Love et al. 2014). Correlation analysis of PR and defense-associated receptor gene expression in time 0 samples was performed in JMP 13.0 on DESeq2 normalized counts using the multivariate analysis method with default estimation method. RNA-seq data is available at GEO (GSE116041).
GO annotation and enrichment analysis
GO annotation for Criollo genome v2.0 was performed using BLAST2GO (Conesa et al. 2005; Götz et al. 2008). Cacao polypeptide sequences were compared to the TAIR10 (Berardini et al. 2015) Arabidopsis protein database using BLASTX with an E value cutoff of 1.0E−3. Mapping and annotation were performed using default settings (E-value hit filter of 1.0E−6).
GO enrichment analyses were performed using AgriGO (Du et al. 2010). GSEA (Subramanian et al. 2005) was applied to compare statistically significantly regulated genes (FDR < 0.05) from each comparison to the background set of GO annotation for all cacao genes. These comparisons included all significant genes (FDR < 0.05) from a basal genotype comparison (based on eight samples from each genotype at time zero) and water vs. pathogen treatments for each genotype at 6, 24, and 72 h after inoculation for both genotypes.
Defense gene classification, protein functional domain prediction, and variant analysis
To identify PR genes, we first followed the method presented in (Fister et al. 2016) and found 313 PR genes in version 2 of the Criollo genome (Table S1). Defense associated receptors (Table S2), including NLRs and leucine-rich repeat receptor-like kinases (LRR-RLKs), were identified according to the hidden Markov matrix-based procedure previously outlined (Marden et al. 2017). Other defense associated receptors of interest, including lectin- and malectin-domain containing genes and Feronia genes (Li et al. 2016a, b), which lack LRR domains and were therefore not detected by the HMM, were manually identified using their annotation. All defense associated receptors of interest were submitted to NCBI Batch CDSearch (Marchler-Bauer et al. 2015) to identify conserved functional domains. To identify synonymous and non-synonymous single nucleotide polymorphisms (SNP), we first mapped reads to the predicted coding sequences (CDS) from version 2 of the Criollo genome using CLC Genomics Workbench version 6.5.1. We then identified variable positions using VarScan2 (Koboldt et al. 2012). Synonymous and non-synonymous sites were determined using SNAP (Korber 2000). CLC and VarScan2 were run according to the procedure outlined in Marden et al. (2017) and SNAP was run using default settings. We performed GO enrichment analyses using AgriGO (Du et al. 2010) with a significance threshold of FDR < 0.05. We further investigated polymorphism in several genes of interest by aligning the amino acids sequences of de novo assembled contigs (Haas et al. 2013) using Geneious v10.1.3 (Kearse et al. 2012).
Statistical model for analysis of PR gene expression
To test the hypothesis that PR genes in general are differentially expressed in the two genotypes, we used a standard least squares model (JMP 13.0, SAS) with ‘Genotype,’ ‘Treatment,’ ‘Time Point,’ ‘Genotype*Treatment,’ ‘Genotype*Time Point,’ ‘Treatment*Time Point,’ and ‘Genotype*Treatment*Time Point’ as effects, and with basal expression, calculated from the eight 0 hai samples for each genotype, as a covariate. This analysis used the 313 PR genes in V2 of the Criollo genome, of which 218 PR genes remained after filtering low expressed genes which contribute primarily to noise (genes with average read count < 10 averaged across all samples). To identify pairwise differences among model LS means, a least squares means student’s T test was applied. Next, to test whether signal from the most differentially expressed genes (FDR < 0.05 for either genotype at any time point in the DESeq2 analysis, above) primarily drove the results of the model, we repeated the analysis but excluded that subset of PR genes.
Results
Disease progression
To study the responses of a resistant and a susceptible cacao genotypes to inoculation with Phytophthora megakarya zoospores, young plants were spray inoculated with zoospores or with distilled water as a control. Our strategy was to capture gene expression profiles in the very early stages of infection, to identify disease resistance genes involved in resistance but avoiding the latter stages of infection. Thus, samples were collected for transcriptomic analysis immediately before application of treatment, as well as 6, 24, and 72 hai. However, symptoms of disease progression did not appear until after the sample collection window. By 120 hai, small, isolated lesions appeared on SCA6 leaves, with portions of the midveins and secondary venation showing areas of necrosis (Fig. 1). At 120 hai NA32 leaves were more severely damaged by the pathogen, showing extensive warping and development of large (> 1 cm2) necrotic areas.

Disease progression after inoculation of plants with zoospores. Representative photographs of Stage C leaves of each genotype at time 0 (immediately before inoculation), and at 24 and 120 hai. Scale bars represent 1 cm
Gene induction by Phytophthora megakarya treatment
For the RNA-Seq analyses, on average ~ 19,900,000 reads were generated per sample, and 88.4% mapped unambiguously to the Criollo V2 reference genome sequence (Argout et al. 2017). After removing low expressed genes which contribute to noise, 19,442 genes remained in the analysis. We used normalized counts for these genes to create a correlation matrix and found that global gene expression was largely consistent regardless of genotype, treatment, and time points (mean R = 0.83). Averaging expression values across biological replicates (Supplemental Figs. S2, S3), we found that 6 hai samples had a weaker correlation with all other time points, indicating some diurnal effect resulting from the 6 h shift in sampling time. Within each genotype*time point comparison, pathogen and control samples correlated very highly (R = 0.985) suggesting the pathogen treatment had a weak effect on global transcription.
To identify the number of genes that were responsive to pathogen treatment, we used DESeq2 (Love et al. 2014) to calculate fold change, comparing the pathogen and water treatments at each time point for each of the genotypes. Overall, 50 or fewer genes were detected as statistically significantly regulated (FDR < 0.05), with the exception of the 24 hai time point for SCA6, where 921 genes were below the FDR cutoff (< 0.05) (Fig. 2a) and therefore classified as differentially expressed genes (DEGs). To determine which categories of genes were regulated at 24 hai in SCA6, we used the AgriGO platform (Du et al. 2010; Tian et al. 2017) to perform a Gene Set Enrichment Analysis (Subramanian et al. 2005) based on these 921 DEGs. 71 GO terms were detected as enriched (FDR < 0.05, Table S3). A subset of these are displayed in Fig. 2b, and reflect enrichment of terms typically associated with plant defense responses and terms previously shown to be involved in SCA6’s response to salicylic acid as a defense elicitor (Fister et al. 2015). Many of the 71 are nested terms associated with processes including stress response and redox reactions, and cell compartments, particularly components of the chloroplast. The genes induced and repressed in both genotypes at other time points include a mix of defense associated genes, genes with functions in redox and signaling, and other unrelated molecular pathways (Table S4).

Summary of gene induction detected by RNA-seq analysis. a Table displaying number of differentially expressed genes (FDR < 0.05) at each time point from inoculation vs. control treatment. b Subset of enriched GO terms (with FDR < 0.05) based on input set of 921 DEGs from the 24 hai SCA6 treatment. Y-axis indicates number of DEGs with corresponding GO assignment. Numbers above bars indicate percentage of DEGs in each GO terms calculated from number of genes in background set for the Criollo genome v2.0. Full list of enriched GO terms presented in Table S3. c Volcano plot summarizing gene regulation in the SCA6 24 hai pathogen vs. control comparison. Putative PRR/NLR genes are shown in green, putative PR genes are shown in red. Select PRRs/NLRs and PRs are labelled with abbreviated versions of their annotations or PR gene family, respectively. d Bar graph summarizing statistical model of PR gene expression. Bars represent log10 least squares means calculated from across all PR genes and biological replicates. Error bars represent standard errors. Bars labelled with different letters are significantly different by T test (p < 0.05). e Bar graph summarizing statistical model of PR gene expression as in d, but excluding genes detected as differentially expressed (DE) at any time point in either genotype
Next, we focused our analysis on DEGs in the SCA6 24 hai comparison that putatively belong to gene classes associated with defense. We focused on two broad gene classes: defense associated receptors [including (PRRs) and NLRs] and pathogenesis-related (PR) genes. The analysis revealed that 17 defense-associated receptors were differentially regulated (10 induced, 7 repressed) and 18 PR genes were differentially regulated (15 induced, 3 repressed) (Fig. 2c, Table S5).
While we did not detect dramatic induction of many PR genes at any time point, we hypothesized that pathogen treatment could result in a subtle induction of many PR genes. To test this hypothesis, we constructed a standard least squares model to analyze the expression of PR genes. First we used the model to analyze expression of all 218 PR genes above the background noise level (see methods). This model detected a highly significant genotype*time point*treatment effect (p < 0.0001, Table S6-1), and a posteriori T test revealed that average PR gene expression was significantly higher in 24 and 72 hai SCA6 Phytophthora treated samples than in other sample groups (p < 0.05) (Fig. 2d). We considered that the differentially expressed PR genes could be driving this result. To test more specifically for a subtle difference in PR genes in general, we repeated the analysis, but excluded any PR gene detected as differentially expressed (FDR < 0.05) at any time point for either genotype. Results of these two models do not differ in any meaningful way (Fig. 2e, Table S6-2). Therefore, expression of PR genes in general appear to show a trend toward slight induction in SCA6, which was not observed in NA32. PR gene expression appeared to be upregulated at 6 hai in NA32, but their expression level was lower than SCA6’s PR gene expression at any time point.
Comparison of genotypes in basal state
In addition to induction of defense pathways after infection, basal expression of defense genes plays an important prophylactic role in preventing colonization by microbes. To identify differences in basal gene expression, we analyzed differences in basal expression between the two genotypes by comparing the transcriptomes at the 0 hai time point. We identified 3118 DEGs (FDR < 0.05), with several showing greater than 50-fold higher expression in one or the other genotype (Fig. 3a). The 15 genes with most dramatically higher expression in SCA6 included a guard-cell associated F-Box protein, an anthocyanidin 3-O-glucosyltransferase, two expansins, and two NB-LRRs, while the 15 genes with higher expression in NA32 include two cytochrome P450s and five defense-associated receptors (Table S7).

Summary of basal comparison of genotypes. a Expression profile distribution for all DEGs in 0 hai genotype comparison. b–e Expression profile distributions for genes associated with enriched GO terms. In all distributions, positive values indicate higher expression in SCA6, negative values indicate higher expression in NA32
To categorize the genes with genotype-specific differences in expression, we performed Gene Set Enrichment Analysis of the 3118 DEGs using the AgriGO platform (Du et al. 2010; Tian et al. 2017). Eight GO terms were detected as enriched (FDR < 0.05). These terms included oxidoreductase activity, six terms associated with the chloroplast or its components, and one term associated with the cytoplasm (Table S8). We plotted the distribution of fold differences of genes within several of these GO terms and found that distributions were roughly normal (Fig. 3b–e). Therefore, we did not detect evidence of skewed transcriptional activity, which would indicate generally higher basal expression of these groups of functionally related genes.
We next analyzed basal expression differences in defense-associated receptors and PR genes. Expression levels of the two gene classes were highly correlated between the two genotypes: defense-associated receptors had an R2 of 0.75, and PR genes had an R2 of 0.85, and genes in these families had a wide range in expression level in leaf tissues (Supplemental Fig. 4A, B). Despite the general similarity between the genotypes, we identified 20 defense-associated receptors and three PR genes with higher expression in SCA6. In NA32 we identified 27 defense-associated receptors and three PR genes with higher expression (log2 fold difference > 1.5) (Fig. 4a, Table S9). Several of the defense-associated receptors showed dramatically different expression in the genotypes (Fig. 4b). The defense-associated receptors identified as DEGs tended to have ~ 100–1000 normalized counts in samples from the more highly expressing genotype, while the lower expressing genotype had very few counts (tens or fewer).

Defense response-associated differences in basal transcriptomic profiles between Sca6 and NA32. a Volcano plot displaying DEGs in basal comparison. Putative PRR/NLR genes are labeled in green, PR genes are labeled in red. Positive values on the x-axis indicate log2 fold higher expression in SCA6, negative values indicate higher expression in NA32. Select PRRs/NLRs and PRs are labelled with abbreviated versions of their annotations or PR gene family, respectively. b Table summarizing comparison of RNA-seq counts between the genotypes for select defense-associated receptors. Standard deviations are calculated from eight biological replicates
Global polymorphism analysis
Expression differences between SCA6 and NA32 could help explain their divergent resistance phenotypes. Other dimensions that could help explain resistance and/or susceptibility are the quantity and character of coding variants present in each genotype. To quantify this variation, we mapped reads to the predicted CDS from version 2 of the Criollo genome and identified SNPs using VarScan2. SCA6 had 10,167 total SNPs, 3388 synonymous and 6779 non-synonymous (Fig. 5a). NA32 had 11,458 total SNPs, 4771 synonymous and 6687 non-synonymous. SCA6 and Criollo B97 belong to closely related genetic groups (Contamana and Criollo) (Motamayor et al. 2008), thus the larger number of SNPs observed in NA32 (Iquitos) relative to SCA6 is likely due to greater divergence between Criollo and NA32. However, estimates for both genotypes are consistent with published intra-species comparisons in other crops (Vidal et al. 2012; Yang et al. 2011). Of these SNPs, < 5% (470/10,167) were within defense-associated receptors in SCA6, and < 7% (801/11,458) in NA32. The identity of genes possessing polymorphic sites was not completely congruent between genotypes, though some overlap existed (Fig. 5b). Gene ontology enrichment of functional categories over-represented in our SNP data revealed 125 categories in SCA6 and 167 categories in NA32 (Supplemental Table S10), of which 113 categories were over-represented in both genotypes.

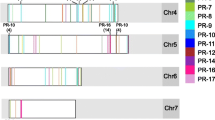
Summary of polymorphism in SCA6 and NA32. a Global analysis of synonymous and non-synonymous SNPs in SCA6 and NA32. Parentheses represent number of polymorphic sites or mean substitutions (± SEM) among defense-associated receptors. SNPs determined by mapping reads to the predicted CDS from version 2 of the Criollo genome and identified variable sites using VarScan2. b Venn diagram representing polymorphic genes in NA32, SCA6, or both. Parentheses represent number of defense-associated receptors belonging to each category. c Amino acid polymorphism in defense-associated receptors of interest. Stars and corresponding annotations represent amino acid differences between SCA6 and NA32, along the with position in the alignment. For example, G242E indicates that at position 242, SCA6 has glycine (G) while NA32 has glutamic acid (E). Tc04v2_g010330: Coiled-coil (CC) nucleotide-binding (NB-ARC) disease resistance protein RGA4; Tc06v2_g002240: NBS-LRR disease resistance protein At4g10780; Tc02v2_g010410: l-type lectin-domain containing receptor kinase IX.1; Tc04v2_g008380: Malectin/receptor-like protein kinase. Gene IDs correspond to Criollo genome v2.0
Analysis of polymorphism in putative defense genes
Lack of defense gene induction in NA32 upon Phytophthora challenge could reflect an inability to recognize the pathogen and allelic variation in defense genes could determine this capability. In order to identify the defense-associated receptors potentially controlling the capacity for recognition, we investigated polymorphism in receptors that were differentially expressed between the genotypes in the basal comparison or were induced by pathogen treatment, were highly polymorphic (≥ 4 non-synonymous mutations), and/or were assigned to one of the disease-resistance consensus QTLs described by Lanaud et al. (2009). We identified four defense associated receptors that fit the majority of these criteria: two canonical NLRs (Tc04v2_g010330 and Tc06v2_g002240), an l-type lectin-domain containing receptor-like kinase IX (Tc02v2_g010410), and a Malectin/receptor-like protein kinase (Tc04v2_g008380). One of the genes, Tc02v2_g010410, also has blast homology with an Arabidopsis gene with the GO term associated with oomycete resistance. Each receptor contained between four and eight polymorphic residues in domains known to participate in ligand binding and subsequent pathogen recognition (Segretin et al. 2014; Wang et al. 2015) (Fig. 5c). Many of the substitutions observed between the two genotypes are non-conservative amino acid changes and are thus more likely to alter the function of the protein.
Discussion
Diseases of cacao significantly reduce the annual cocoa crop, threatening farmer livelihood and the stability of chocolate production. While natural variation in cacao’s disease resistance exists, developing varieties with superior disease resistance with desirable pod and bean characteristics is a slow process. Several QTL mapping studies have identified regions of the cacao genome associated with black pod resistance (Crouzillat et al. 1996, 2000a, b; Lanaud et al. 1999, 2004; Motilal and Sounigo 2000; Clement et al. 2003; Flament et al. 2001; Risterucci et al. 2000, 2003; Queiroz et al. 2003, 2006; Brown et al. 2005, 2007; Faleiro et al. 2006). However, due to small population sizes, the QTLs include hundreds to thousands of genes, many of them with unknown function. Identifying the molecular mechanisms of disease resistance in cacao and narrowing the analysis to specific genes is vital for speeding-up breeding efforts for improving tolerance to a suite of pathogens harmful to cacao. In this study we analyzed the transcriptomes of two important cacao genotypes at basal levels and in response to inoculation with Phytophthora megakarya, a major cacao pathogen (Bailey et al. 2016) and were able to identify key candidate genes with potential roles in cacao’s defense system.
For this experiment, we selected two well-studied cacao genotypes, the broadly resistant SCA6 and the highly Phytophthora susceptible NA32. Defense responses of cacao varieties are characterized through use of attached and detached pod (Iwaro et al. 2005) and leaf assays (Nyassé et al. 1995; Efombagn et al. 2011), as well as through assessment of clones in the field (Pokou et al. 2008), and these metrics are generally well-correlated (Nyadanu et al. 2012; Efombagn et al. 2011). In previous leaf-disc inoculation assays, they consistently scored among the most resistant and most susceptible to Phytophthora spp. in leaf- and pod-based screens (Pokou et al. 2001, 2008; Nyadanu et al. 2012). In our case, we sampled leaf tissues in order to increase biological replication, while recognizing that specific genes involved in foliar and pod reaction to Phytophthora may differ, thus additional evidence would be required to confirm molecular mechanisms in pods. In our experiment, neither genotype developed symptoms of infection during the collection period. However, 5 days after inoculation, NA32 leaves exhibited advanced necrosis. In our experiment SCA6 leaves developed smaller lesions, but not the necrotic flecking typical of hypersensitive response, suggesting quantitative, rather than qualitative, resistance is at play. Interestingly, SCA6 was previously shown to develop a hypersensitive-like response to zoospore inoculation with P. palmivora but not P. megakarya, although the phenotype may be attributed to P. megakarya forming more appressoria, allowing it to more efficiently penetrate unwounded tissue (Ali et al. 2016).
In the canonical plant defense response, recognition of PAMPs or effectors triggers major transcriptional reprogramming which leads to resistance, typically featuring induction of PR genes and other antimicrobial compounds (Bigeard et al. 2015; Wu et al. 2014). These processes have been described in model monocots (Balmer et al. 2013) and dicots (Denoux et al. 2008; Vogel and Somerville 2000). Similar trends have also previously been described in other perennials: disease-resistant rubber tree featured PR gene induction after infection (Li et al. 2016) and a disease-resistant chestnut variety showed earlier and more dramatic induction of a variety of defense associated transcripts than did a susceptible variety (Santos et al. 2017). A disease-resistant apple cultivar was also shown to have constitutively higher basal expression of antimicrobial transcripts than did a susceptible cultivar (Degenhardt et al. 2005). A similar finding was also described in grapevine, where a powdery resistant variety showed higher basal expression of a set of PR genes but minimal transcriptional regulation after inoculation, while a susceptible variety featured major transcriptional changes, including induction of typical defense-response components like PR genes (Fung et al. 2008). Taken together, these results suggest higher basal defense gene expression, more dramatic induction of genes upon pathogen challenge, or both together can explain differences between resistant and susceptible cultivars in interactions of plants with their pathogens.
Expression level of PRRs and NLRs, the upstream genes in the defense response, has also been shown to correlate with disease resistance. Overexpression of a PRR (Mysore et al. 2003) and an NLR (Oldroyd and Staskawicz 1998) in tomato were shown to enhance resistance compared to that mediated by their endogenous expression levels. Expression of a rice PRR under different promoters has also correlated expression level to degree of resistance and induction of downstream defense genes (Cao et al. 2007). Therefore,we were also interested in analyzing both basal expression level and induction of defense-associated receptors.
The two genotypes exhibited significant defense-related gene expression differences prior to pathogen exposure (the basal state; 0 hai time point). We identified 47 putative pattern recognition receptors or resistance genes which were expressed in a genotype-specific manner. These receptor proteins are maintained in large families in plants in order to bind and trigger a response to diverse pathogen molecules, signatures of cellular damage, and modification of other endogenous plant proteins [reviewed in (Cui et al. 2015; Boutrot and Zipfel 2017)]. Our RNA-seq data suggest that these genotypes may not express the same receptors in leaf tissue, which could be one explanation for NA32’s inability to detect and respond to infection. We will test this hypothesis using a transient expression system (Fister et al. 2016a, b) to introduce these receptors more highly expressed in SCA6 into susceptible cacao backgrounds to attempt to complement the susceptibility phenotype.
In our measurement of response to pathogen treatment, GO analysis yielded results that supported previous findings about transcriptomic response of SCA6 to salicylic acid (Fister et al. 2015). In this previous study, SCA6 was demonstrated to have a strong oxidative burst believed to be linked to generation of superoxide and peroxide in the chloroplast which have been linked in enhanced disease resistance in other plants (Van Aken and Van Breusegem 2015). SCA6’s salicylic acid concentration was also previously quantified and compared to a different disease-susceptible cacao variety, and salicylic acid levels did not appear to differ between the genotypes, suggesting that SCA6’s resistance is mediated by factors downstream of or independent from salicylic acid synthesis (Chaves and Gianfagna 2006). At the individual gene level, we were able to detect > twofold induction of genes associated with oxidative burst, as well as 10 PR genes and four defense-associated receptors in SCA6 at 24 hai (Table S5). The PR genes included PR-2 s (β-1,3-Glucanases) and PR-3 s (chitinases), which encode proteins with well-defined roles in degrading pathogen cell walls (van Loon and van Strien 1999; van Loon et al. 2006), including in cacao (Maximova et al. 2006). Furthermore, we detected slight upregulation of average PR gene expression in SCA6 at 24 and 72 hai. Across the whole experiment, average expression of PR genes in the resistant SCA6 genotype at these time points showed a highly significant increase in expression compared to susceptible NA32 (P = 10−17; Supplemental Table S6). It is possible that this small (roughly 10%) induction of the PR class as a whole could impact the pathogen’s ability to colonize the plant. Another possible cause of SCA6’s resistance could be elevated basal expression of PR genes. We did not observe a general higher basal expression of PRs in SCA6, but specific genes were identified with higher expression in SCA6 (Fig. 4, Supplemental Table S9). One of these is a PR-16, an oxalate oxidase encoding gene, a member of a family of genes shown to contribute to ROS burst and Phytophthora defense (He et al. 2013; Tripathy and Oelmüller 2012). The other PR gene with higher basal expression in SCA6 was a PR-1 family member. The molecular activity of PR-1s was recently elucidated in a study showing that the proteins bind sterols and cause pathogen membrane leakage (Gamir et al. 2017). Constitutive overexpression of a PR-1 was previously shown to enhance defense against oomycetes, but not other pathogens, in tobacco (Alexander et al. 1993).
In contrast to SCA6, the susceptible NA32 genotype did not exhibit a strong transcriptional response to pathogen treatment at any time point we surveyed. As explained above, genotypic differences in expression levels are one potential explanation for this result. Other dimensions include the quantity and character of coding variants that renders NA32 unable to detect and respond to this strain of pathogen. To that end, we mapped reads to the predicted coding sequences of the Criollo V2 genome in order to identify SNPs in each genotype (Fig. 5a). SCA6 possessed 2,683 genes with at least one polymorphic site, while NA32 possessed 3,658. The identity of these genes was not completely congruent between SCA6 and NA32 (Fig. 5b) and GO enrichment revealed functional categories unique to each genotype (Supplemental Table S10). These categories include many of the same GO terms that were enriched in the expression data for SCA6, including response to abiotic stress, photosynthesis, response to osmotic stress, and response to chemical stimulus. Overlap between gene expression and SNP profiles indicates these categories may play an important role in shaping defense response.
Lastly, we identified four defense-associated receptors that may be potential mediators of P. megakarya recognition in SCA6 (Fig. 5c). As is common in NLR genes (Poland et al. 2009; Segretin et al. 2014; Allen et al. 2004; Karasov et al. 2014), polymorphic residues in the ligand binding domains of each receptor could contribute to differential recognition of pathogens between the two genotypes. NBS-LRR genes have specifically been shown to have altered effector recognition specificity upon single amino acid substitutions in the LRR domain (Segretin et al. 2014). While NLR mediated resistance is usually thought of as qualitative, examples of NBS-LRR mediated quantitative resistance, or field resistance, have been described (Rietman et al. 2012). Thus, the PRRs and NLRs we identified may provide quantitative resistance against certain isolates of P. megakarya. Homologs of both the l-type lectin-domain containing receptor-like kinase and the malectin/receptor-like protein kinase in other plant species have been implicated in resistance to various Phytophthora species. In one study, two l-type lectin receptor kinases (LecRK) were demonstrated to provide resistance to both Phytophthora brassicae and Phytophthora capsici when overexpressed in both Arabidopsis thaliana and Nicotiana benthamiana (Wang et al. 2015). Moreover, both LecRKs were also shown to provide resistance when stably expressed in transgenic N. benthamiana, suggesting they provide resistance to Phytophthora species in long-diverged plant lineages (Wang et al. 2016). The malectin/receptor-like protein kinase identified in our study shares homology with ANX1 and ANX2. These two genes have recently been shown to be negative regulators of PRR-mediated immunity via interference with FLS2–BAK1 complex formation (130). It is possible that differential expression and polymorphism in the cacao malectin/receptor-like protein kinase we identified could alter this negative regulation, thereby promoting resistance or susceptibility in SCA6 or NA32, respectively. While it is plausible that SCA6 defense is partially or even completely controlled by the defense associated receptors we have identified, more functional analyses are required to understand the basis of resistance between these two cacao genotypes. The CRISPR/Cas9 gene editing technique has been applied to cacao (Fister et al. 2018), and it offers a potential strategy for studying association of particular receptors with pathogen recognition, allowing for targeted mutagenesis of domains or even residues of interest.
Improved understanding of the genetics underlying the interaction of cacao with its pathogens, including Phytophthora spp., is essential to reduce losses to farmer income and stabilize the world’s cocoa supply. Genomic and transcriptomic analyses of useful clones is essential to understanding cacao’s defense response and breeding of superior clones. In this study, we identified candidate defense genes which will be the subject of functional analyses to verify their role in protecting the plant from infection. Previous analyses of defense in cacao have not characterized polymorphism in putative defense genes and doing so is critical for guiding breeding efforts.
Data availability
Data Deposition Information: Our transcriptome data were stored on the NCBI GEO platform and given the accession number GSE116041. Data are available here: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE116041 and will be made publicly accessible after publication of the manuscript.
References
Alexander D, Goodman RM, Gut-Rella M, Glascock C, Weymann K, Friedrich L et al (1993) Increased tolerance to two oomycete pathogens in transgenic tobacco expressing pathogenesis-related protein 1a. Proc Natl Acad Sci USA 90(15):7327–7331. https://doi.org/10.1073/pnas.90.15.7327
Ali SS, Amoako-Attah I, Bailey RA, Strem MD, Schmidt M, Akrofi AY et al (2016) PCR-based identification of cacao black pod causal agents and identification of biological factors possibly contributing to Phytophthora megakarya’s field dominance in West Africa. Plant Pathol 65(7):1095–1108. https://doi.org/10.1111/ppa.12496
Ali SS, Shao J, Lary DJ, Kronmiller BA, Shen D, Strem MD et al (2017a) Phytophthora megakarya and Phytophthora palmivora, closely related causal agents of cacao black pod rot, underwent increases in genome sizes and gene numbers by different mechanisms. Genome Biol Evol 9(3):00
Ali SS, Shao J, Lary DJ, Strem MD, Meinhardt LW, Bailey BA (2017b) Phytophthora megakarya and P. palmivora, causal agents of black pod rot, induce similar plant defense responses late during infection of susceptible cacao pods. Front Plant Sci. https://doi.org/10.3389/fpls.2017.00169
Allen RL, Bittner-Eddy PD, Grenville-Briggs LJ, Meitz JC, Rehmany AP, Rose LE et al (2004) Host–parasite coevolutionary conflict between Arabidopsis and downy mildew. Science (New York) 306(5703):1957–1960
Alverson WS, Whitlock BA, Nyffeler R, Bayer C, Baum DA (1999) Phylogeny of the core Malvales: evidence from ndhF sequence data. Am J Bot. https://doi.org/10.2307/2656928
Argout X, Salse J, Aury J-M, Guiltinan MJ, Droc G, Gouzy J et al (2011) The genome of Theobroma cacao. Nat Genet 43(2):101–108
Argout X, Martin G, Droc G, Fouet O, Labadie K, Rivals E et al (2017) The cacao Criollo genome v2.0: an improved version of the genome for genetic and functional genomic studies. BMC Genom 18(1):730. https://doi.org/10.1186/s12864-017-4120-9
Bailey BA, Ali SS, Akrofi AY, Meinhardt LW (2016) Phytophthora megakarya, a causal agent of black pod rot in Africa. Caco diseases. Springer, New York, pp 267–303
Balmer D, Planchamp C, Mauch-Mani B (2013) On the move: induced resistance in monocots. J Exp Bot 64(5):1249–1261. https://doi.org/10.1093/jxb/ers248
Bellincampi D, Cervone F, Lionetti V (2014) Plant cell wall dynamics and wall-related susceptibility in plant–pathogen interactions. Front Plant Sci. https://doi.org/10.3389/fpls.2014.00228
Berardini TZ, Reiser L, Li D, Mezheritsky Y, Muller R, Strait E et al (2015) The Arabidopsis information resource: making and mining the “gold standard” annotated reference plant genome. Genesis 53(8):474–485. https://doi.org/10.1002/dvg.22877
Bigeard J, Colcombet J, Hirt H (2015) Signaling mechanisms in pattern-triggered immunity (PTI). Mol Plant 8(4):521–539. https://doi.org/10.1016/j.molp.2014.12.022
Blaha G, Lotodé R (1976) Un critère primordial de sélection du cacaoyer au Cameroun: la résistance à la pourriture brune des cabosses (Phytophthora palmivora). Variations des réactions à la maladie en liaison avec les données écologiques et l’état physiologique des fruits. Café Cacao thé 20(2):97–116
Bolger AM, Lohse M, Usadel B (2014) Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30(15):2114–2120. https://doi.org/10.1093/bioinformatics/btu170
Boutrot F, Zipfel C (2017) Function, discovery, and exploitation of plant pattern recognition receptors for broad-spectrum disease resistance. Annu Rev Phytopathol 55(1):257–286. https://doi.org/10.1146/annurev-phyto-080614-120106
Brown JS, Schnell R, Motamayor J, Lopes U, Kuhn DN, Borrone JW (2005) Resistance gene mapping for witches’ broom disease in Theobroma cacao L. in an F2 population using SSR markers and candidate genes. J Am Soc Hortic Sci 130(3):366–373
Brown JS, Phillips-Mora W, Power EJ, Krol C, Cervantes-Martinez C, Motamayor JC (2007) Mapping QTLs for resistance to frosty pod and black pod diseases and horticultural traits in Theobroma cacao L. Crop Sci. https://doi.org/10.2135/cropsci2006.11.0753
Cao Y, Ding X, Cai M, Zhao J, Lin Y, Li X et al (2007) The expression pattern of a rice disease resistance gene Xa3/Xa26 is differentially regulated by the genetic backgrounds and developmental stages that influence its function. Genetics 177(1):523–533. https://doi.org/10.1534/genetics.107.075176
Chaves FC, Gianfagna TJ (2006) Necrotrophic phase of Moniliophthora perniciosa causes salicylic acid accumulation in infected stems of cacao. Physiol Mol Plant Pathol 69(1):104–108. https://doi.org/10.1016/j.pmpp.2007.02.003
Clement D, Risterucci AM, Motamayor JC, N’Goran J, Lanaud C (2003) Mapping quantitative trait loci for bean traits and ovule number in Theobroma cacao L. Genome. https://doi.org/10.1139/g02-118
Coll NS, Epple P, Dangl JL (2011) Programmed cell death in the plant immune system. Cell Death Differ 18(8):1247–1256
Conesa A, Gotz S, Garcia-Gomez J, Terol J, Talon M, Robles M (2005) Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 21(18):3674–3676
Corwin JA, Kliebenstein DJ (2017) Quantitative resistance: more than just perception of a pathogen. Plant Cell 29(4):655–665. https://doi.org/10.1105/tpc.16.00915
Crouzillat D, Lerceteau E, Petiard V, Morera J, Rodriguez H, Walker D (1996) Theobroma cacao L.: a genetic linkage map and quantitative trait loci analysis. Theor Appl Genet. https://doi.org/10.1007/bf00225747
Crouzillat D, Ménard B, Mora A, Phillips W, Pétiard V (2000a) Quantitative trait analysis in Theobroma cacao using molecular markers. Euphytica 114(1):13–23
Crouzillat D, Phillips W, Fritz PJ, Pétiard V (2000b) Quantitative trait loci analysis in Theobroma cacao using molecular markers. Inheritance of polygenic resistance to Phytophthora palmivora in two related cacao populations. Euphytica 114(1):25–36
Cui H, Tsuda K, Parker JE (2015) Effector-triggered immunity: from pathogen perception to robust defense. Annu Rev Plant Biol 66(1):487–511. https://doi.org/10.1146/annurev-arplant-050213-040012
De Bruyne L, Höfte M, De Vleesschauwer D (2014) Connecting growth and defense: the emerging roles of brassinosteroids and gibberellins in plant innate immunity. Molecular plant 7(6):943–959. https://doi.org/10.1093/mp/ssu050
Degenhardt J, Al-Masri AN, Kürkcüoglu S, Szankowski I, Gau AE (2005) Characterization by suppression subtractive hybridization of transcripts that are differentially expressed in leaves of apple scab-resistant and susceptible cultivars of Malus domestica. Mol Genet Genomics 273(4):326–335
Denoux C, Galletti R, Mammarella N, Gopalan S, Werck D, De Lorenzo G et al (2008) Activation of defense response pathways by OGs and Flg22 elicitors in Arabidopsis seedlings. Mol Plant 1(3):423–445. https://doi.org/10.1093/mp/ssn019
Despreaux D, Clement D, Partiot M (1989) Black pod disease of cocoa in Cameroon: analysis of a resistant character observed in field. Agronomie (France)
Dickman MB, Fluhr R (2013) Centrality of host cell death in plant–microbe interactions. Annu Rev Phytopathol 51:543–570
Dinarti D, Susilo AW, Meinhardt LW, Ji K, Motilal LA, Mischke S et al (2015) Genetic diversity and parentage in farmer selections of cacao from Southern Sulawesi, Indonesia revealed by microsatellite markers. Breed Sci 65(5):438–446. https://doi.org/10.1270/jsbbs.65.438
Du Z, Zhou X, Ling Y, Zhang Z, Su Z (2010) agriGO: a GO analysis toolkit for the agricultural community. Nucleic Acids Res 38:W64–W70. https://doi.org/10.1093/nar/gkq310
Efombagn MIB, Bieysse D, Nyassé S, Eskes A (2011) Selection for resistance to Phytophthora pod rot of cocoa (Theobroma cacao L.) in Cameroon: repeatability and reliability of screening tests and field observations. Crop Prot 30(2):105–110
Eskes A, Lanaud C (2001) Cocoa. In: André C, Michel J, Serge H, Dominique N (eds) Tropical plant breeding. CIRAD, Montpellier pp 78–105
Faleiro FG, Queiroz VT, Lopes UV, Guimarães CT, Pires JL, Yamada MM et al (2006) Mapping QTLs for witches’ broom (Crinipellis perniciosa) resistance in cacao (Theobroma cacao L.). Euphytica 149(1–2):227–235
Fernandes LdS, Royaert S, Corrêa FM, Mustiga GM, Marelli J-P, Corrêa RX et al (2018) Mapping of a major QTL for Ceratocystis wilt disease in an F1 population of Theobroma cacao. Front Plant Sci. https://doi.org/10.3389/fpls.2018.00155
Figueira A, Albuquerque P, Leal-Jr G (2006) Genetic mapping and differential gene expression of Brazilian alternative resistance sources to Witches’ Broom (causal agent Crinipellis perniciosa). In 15th international Cocoa research conference
Fister AS, O’Neil ST, Shi Z, Zhang Y, Tyler BM, Guiltinan MJ et al (2015) Two Theobroma cacao genotypes with contrasting pathogen tolerance show aberrant transcriptional and ROS responses after salicylic acid treatment. J Exp Bot 66(20):6245–6258. https://doi.org/10.1093/jxb/erv334
Fister AS, Shi Z, Zhang Y, Helliwell EE, Maximova SN, Guiltinan MJ (2016a) Protocol: transient expression system for functional genomics in the tropical tree Theobroma cacao L. Plant Methods 12(1):1–13. https://doi.org/10.1186/s13007-016-0119-5
Fister AS, Mejia LC, Zhang Y, Herre EA, Maximova SN, Guiltinan MJ (2016b) Theobroma cacao L. pathogenesis-related gene tandem array members show diverse expression dynamics in response to pathogen colonization. BMC Genome 17:363. https://doi.org/10.1186/s12864-016-2693-3
Fister AS, Landherr L, Maximova SN, Guiltinan MJ (2018) Transient expression of CRISPR/Cas9 machinery targeting TcNPR3 enhances defense response in Theobroma cacao. Front Plant Sci. https://doi.org/10.3389/fpls.2018.00268
Flament MH, Kebe I, Clément D, Pieretti I, Risterucci AM, N’Goran JAK (2001) Genetic mapping of resistance factors to Phytophthora palmivora in cocoa. Genome. https://doi.org/10.1139/gen-44-1-79
Fung RWM, Gonzalo M, Fekete C, Kovacs LG, He Y, Marsh E et al (2008) Powdery mildew induces defense-oriented reprogramming of the transcriptome in a susceptible but not in a resistant grapevine. Plant Physiol 146(1):236–249. https://doi.org/10.1104/pp.107.108712
Gamir J, Darwiche R, Hof P, Choudhary V, Stumpe M, Schneiter R et al (2017) The sterol-binding activity of PATHOGENESIS-RELATED PROTEIN 1 reveals the mode of action of an antimicrobial protein. Plant J 89(3):502–509. https://doi.org/10.1111/tpj.13398
Götz S, García-Gómez JM, Terol J, Williams TD, Nagaraj SH, Nueda MJ et al (2008) High-throughput functional annotation and data mining with the Blast2GO suite. Nucleic Acids Res 36(10):3420–3435
Haas BJ, Papanicolaou A, Yassour M, Grabherr M, Blood PD, Bowden J et al (2013) De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat Protoc 8(8):1494
He X, Miyasaka SC, Fitch MM, Khuri S, Zhu YJ (2013) Taro (Colocasia esculenta) transformed with a wheat oxalate oxidase gene for improved resistance to taro pathogen Phytophthora colocasiae. HortScience 48(1):22–27
Iwaro A, Bekele F, Butler D (2003) Evaluation and utilisation of cacao (Theobroma cacao L.) germplasm at the International Cocoa Genebank, Trinidad. Euphytica 130(2):207–221
Iwaro A, Thévenin J-M, Butler DR, Eskes A (2005) Usefulness of the detached pod test for assessment of cacao resistance to Phytophthora pod rot. Eur J Plant Pathol 113(2):173–182
Jones JDG, Vance RE, Dangl JL (2016) Intracellular innate immune surveillance devices in plants and animals. Science. https://doi.org/10.1126/science.aaf6395
Karasov TL, Kniskern JM, Gao L, DeYoung BJ, Ding J, Dubiella U et al (2014) The long-term maintenance of a resistance polymorphism through diffuse interactions. Nature 512(7515):436
Karasov TL, Chae E, Herman JJ, Bergelson J (2017) Mechanisms to mitigate the trade-off between growth and defense. Plant Cell 29(4):666–680. https://doi.org/10.1105/tpc.16.00931
Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S et al (2012) Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28(12):1647–1649. https://doi.org/10.1093/bioinformatics/bts199
Kim D, Langmead B, Salzberg SL (2015) HISAT: a fast spliced aligner with low memory requirements. Nat Methods 12(4):357–360. https://doi.org/10.1038/nmeth.3317
Kimura S, Waszczak C, Hunter K, Wrzaczek M (2017) Bound by fate: the role of reactive oxygen species in receptor-like kinase signaling. Plant Cell 29(4):638–654. https://doi.org/10.1105/tpc.16.00947
Koboldt DC, Zhang Q, Larson DE, Shen D, McLellan MD, Lin L et al (2012) VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res 22(3):568–576. https://doi.org/10.1101/gr.129684.111
Korber B (2000) HIV signature and sequence variation analysis. Comput Anal HIV Mol Seq 4:55–72
Kumar D, Chapagai D, Dean P, Davenport M (2015) Biotic and abiotic stress signaling mediated by salicylic acid. In: Pandey KG (ed) Elucidation of abiotic stress signaling in plants: Functional genomics perspectives, vol 1. Springer, New York, pp 329–346
Kushalappa AC, Yogendra KN, Karre S (2016) Plant innate immune response: qualitative and quantitative resistance. Crit Rev Plant Sci 35(1):38–55
Lachenaud P, Eskes A, N’Goran JAK, Clément D, Kébé BI.(2001) Premier cycle de sélection récurrente en Côte d’Ivoire et choix des géniteurs du second cycle. Proceedings of the 13th international cocoa research conference
Lanaud C, Risterucci AM, Pieretti I, Falque M, Bouet A, Lagoda PJL (1999) Isolation and characterization of microsatellites in Theobroma cacao L. Mol Ecol 8(12):2141–2143
Lanaud C, Risterucci AM, Pieretti I, N’goran JA, Fargeas D (2004) Characterisation and genetic mapping of resistance and defence gene analogs in cocoa (Theobroma cacao L.). Mol Breeding 13(3):211–227
Lanaud C, Fouet O, Clément D, Boccara M, Risterucci AM, Surujdeo-Maharaj S et al (2009) A meta-QTL analysis of disease resistance traits of Theobroma cacao L. Mol Breeding 24(4):361–374. https://doi.org/10.1007/s11032-009-9297-4
Legavre T, Ducamp M, Sabau X, Argout X, Fouet O, Dedieu F et al (2015) Identification of Theobroma cacao genes differentially expressed during Phytophthora megakarya infection. Physiol Mol Plant Pathol 92:1–13
Li C, Wu H-M, Cheung AY (2016a) FERONIA and her pals: functions and mechanisms. Plant Physiol 171(4):2379–2392. https://doi.org/10.1104/pp.16.00667
Li X, Bi Z, Di R, Liang P, He Q, Liu W et al (2016b) Identification of powdery mildew responsive genes in Hevea brasiliensis through mRNA differential display. Int J Mol Sci 17(2):181. https://doi.org/10.3390/ijms17020181
Liao Y, Smyth GK, Shi W (2014) featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. https://doi.org/10.1093/bioinformatics/btt656
Love MI, Huber W, Anders S (2014) Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15(12):550. https://doi.org/10.1186/s13059-014-0550-8
Mackey D, Holt BF, Wiig A, Dangl JL (2002) RIN4 interacts with Pseudomonas syringae type III effector molecules and is required for RPM1-mediated resistance in Arabidopsis. Cell 108(6):743–754. https://doi.org/10.1016/S0092-8674(02)00661-X
Mang H, Feng B, Hu Z, Boisson-Dernier A, Franck C, Meng X et al (2017) Differential regulation of two-tiered plant immunity and sexual reproduction by ANXUR receptor-like kinases. Plant Cell 29:3140–3156
Marcano M, Pugh T, Cros E, Morales S, Páez EAP, Courtois B et al (2007) Adding value to cocoa (Theobroma cacao L.) germplasm information with domestication history and admixture mapping. Theor Appl Genet 114(5):877–884
Marcano M, Morales S, Hoyer MT, Courtois B, Risterucci AM, Fouet O et al (2009) A genomewide admixture mapping study for yield factors and morphological traits in a cultivated cocoa (Theobroma cacao L.) population. Tree Genet Genomes 5(2):329–337
Marchler-Bauer A, Derbyshire MK, Gonzales NR, Lu S, Chitsaz F, Geer LY et al (2015) CDD: NCBI’s conserved domain database. Nucleic Acids Res 43(D1):D222–D226. https://doi.org/10.1093/nar/gku1221
Marden JH, Mangan SA, Peterson MP, Wafula E, Fescemyer HW, Der JP et al (2017) Ecological genomics of tropical trees: how local population size and allelic diversity of resistance genes relate to immune responses, cosusceptibility to pathogens, and negative density dependence. Mol Ecol 26(9):2498–2513. https://doi.org/10.1111/mec.13999
Maximova S, Marelli J-P, Young A, Pishak S, Verica J, Guiltinan M (2006) Over-expression of a cacao class I chitinase gene in Theobroma cacao L. enhances resistance against the pathogen, Colletotrichum gloeosporioides. Planta 224(4):740–749. https://doi.org/10.1007/s00425-005-0188-6
McElroy MS, Navarro AJR, Mustiga G, Stack C, Gezan S, Peña G et al (2018) Prediction of cacao (Theobroma cacao) resistance to Moniliophthora spp. diseases via genome-wide association analysis and genomic selection. Front Plant Sci. https://doi.org/10.3389/fpls.2018.00343
McManus MT (2012) Annual plant reviews. The plant hormone ethylene. Wiley, Hoboken
Mejia L, Guiltinan M, Shi Z, Landherr L, Maximova S (2012) Expression of designed antimicrobial peptides in Theobroma cacao L. trees reduces leaf necrosis caused by Phytophthora spp. Small Wonders Pept Dis Control. https://doi.org/10.1021/bk-2012-1095.ch018. 1905.
Motamayor JC, Risterucci AM, Lopez PA, Ortiz CF, Moreno A, Lanaud C (2002) Cacao domestication I: the origin of the cacao cultivated by the Mayas. Heredity. https://doi.org/10.1038/sj.hdy.6800156
Motamayor JC, Lachenaud P, da Silva e Mota JW, Loor R, Kuhn DN, Brown JS et al (2008) Geographic and genetic population differentiation of the Amazonian chocolate tree (Theobroma cacao L). PLoS ONE 3(10):e3311. https://doi.org/10.1371/journal.pone.0003311
Motamayor J, Mockaitis K, Schmutz J, Haiminen N, Livingstone D, Cornejo O et al (2013) The genome sequence of the most widely cultivated cacao type and its use to identify candidate genes regulating pod color. Genome Biol 14(6):R53. https://doi.org/10.1186/gb-2013-14-6-r53
Motilal LA, Sounigo O (2000) Theobroma cacao L.: genome linkage map and QTLs for Phytophthora palmivora resistance.
Mur LA, Kenton P, Lloyd AJ, Ougham H, Prats E (2008) The hypersensitive response; the centenary is upon us but how much do we know? J Exp Bot 59(3):501–520. https://doi.org/10.1093/jxb/erm239
Mysore KS, D’Ascenzo MD, He X, Martin GB (2003) Overexpression of the disease resistance gene Pto in tomato induces gene expression changes similar to immune responses in human and fruitfly. Plant Physiol 132(4):1901–1912. https://doi.org/10.1104/pp.103.022731
Nyadanu DA, Adomako R, Kwoseh B, Lowor C, Dzahini-Obiatey ST, Akrofi H, Owusu Ansah A, Assuah F (2012) Histological mechanisms of resistance to black pod disease in cacao (Theobroma cacao L.). J Plant Sci 7(2):89–94
Nyassé S, Cilas C, Herail C, Blaha G (1995) Leaf inoculation as an early screening test for cocoa (Theobroma cacao L.) resistance to Phytophthora black pod disease. Crop Prot 14(8):657–663. https://doi.org/10.1016/0261-2194(95)00054-2
Nyassé S, Grivet L, Risterucci A-M, Blaha G, Berry D, Lanaud C et al (1999) Diversity of Phytophthora megakarya in Central and West Africa revealed by isozyme and RAPD markers. Mycol Res 103(10):1225–1234
Oldroyd GED, Staskawicz BJ (1998) Genetically engineered broad-spectrum disease resistance in tomato. Proc Natl Acad Sci 95(17):10300. https://doi.org/10.1073/pnas.95.17.10300
Ploetz R (2016) The impact of diseases on cacao production: a global overview. Springer, Cacao Diseases, pp 33–59
Pokou ND, N’Goran JAK, Kébé IS, Eskes A, Tahi GM, Sangaré A (2008) Levels of resistance to Phytophthora pod rot in cocoa accessions selected on-farm in Côte d’Ivoire. Crop Prot 27(3–5):302–309. https://doi.org/10.1016/j.cropro.2007.07.012
Pokou ND, N’Goran JAK, Lachenaud P, Eskes AB, Montamayor JC, Schnell R et al (2009) Recurrent selection of cocoa populations in Côte d’Ivoire: comparative genetic diversity between the first and second cycles. Plant Breeding 128(5):514–520. https://doi.org/10.1111/j.1439-0523.2008.01582.x
Poland JA, Balint-Kurti PJ, Wisser RJ, Pratt RC, Nelson RJ (2009) Shades of gray: the world of quantitative disease resistance. Trends Plant Sci 14(1):21–29
Pugh T (2005) Etude des déséquilibres de liaison dans une collection de cacaoyers (Theobroma cacao L.) appartenant au groupe Criollo/Trinitario et application au marquage génétique des caractères d’intérêt. Soutenue le 11
Queiroz V, Guimarães C, Anhert D, Schuster I, Daher R, Pereira M et al (2003) Identification of a major QTL in cocoa (Theobroma cacao L.) associated with resistance to witches’ broom disease. Plant Breeding 122(3):268–272
Quinlan AR, Hall IM (2010) BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26(6):841–842. https://doi.org/10.1093/bioinformatics/btq033
Rietman H, Bijsterbosch G, Cano LM, Lee H-R, Vossen JH, Jacobsen E et al (2012) Qualitative and quantitative late blight resistance in the potato cultivar Sarpo Mira is determined by the perception of five distinct RXLR effectors. Mol Plant Microbe Interact 25(7):910–919
Risterucci A-M, Paulin D, N’Goran J, Ducamp M, Lanaud C (2000) Mapping of quantitative trait loci (QTLs) for resistance to Phytophthora in Theobroma cacao L. Ingenic Newslett 5:9–10
Risterucci AM, Paulin D, Ducamp M, N’Goran JAK, Lanaud C (2003) Identification of QTLs related to cocoa resistance to three species of Phytophthora. Theor Appl Genet 108(1):168–174. https://doi.org/10.1007/s00122-003-1408-8
Royaert S, Jansen J, da Silva DV, de Jesus Branco SM, Livingstone DS, Mustiga G et al (2016) Identification of candidate genes involved in Witches’ broom disease resistance in a segregating mapping population of Theobroma cacao L. in Brazil. BMC Genom 17(1):1–16. https://doi.org/10.1186/s12864-016-2415-x
Santos C, Duarte S, Tedesco S, Fevereiro P, Costa RL (2017) Expression profiling of Castanea genes during resistant and susceptible interactions with the oomycete pathogen Phytophthora cinnamomi reveal possible mechanisms of immunity. Front Plant Sci. https://doi.org/10.3389/fpls.2017.00515
Schnell R, Olano C, Brown J, Meerow A, Cervantes-Martinez C, Nagai C et al (2005) Retrospective determination of the parental population of superior cacao (Theobroma cacao L.) seedlings and association of microsatellite alleles with productivity. J Am Soc Hortic Sci 130(2):181–190
Segretin ME, Pais M, Franceschetti M, Chaparro-Garcia A, Bos JI, Banfield MJ et al (2014) Single amino acid mutations in the potato immune receptor R3a expand response to Phytophthora effectors. Mol Plant Microbe Interact 27(7):624–637
Song S, Qi T, Wasternack C, Xie D (2014) Jasmonate signaling and crosstalk with gibberellin and ethylene. Curr Opin Plant Biol 21:112–119
Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA et al (2005) Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA 102(43):15545–15550
Surujdeo-Maharaj S, Sreenivasan T, Motilal L, Umaharan P (2016) Black pod and other Phytophthora induced diseases of cacao: history, biology, and control. Cacao diseases. Springer, New York, pp 213–266
Tahi M, Kebe I, Eskes AB, Ouattara S, Sangare A, Mondeil F (2000) Rapid screening of cacao genotypes for field resistance to Phytophthora palmivora using leaves, twigs, and roots. Eur J Plant Pathol 106(1):87–94. https://doi.org/10.1023/a:1008747800191
Tang D, Wang G, Zhou J-M (2017) Receptor kinases in plant-pathogen interactions: More than pattern recognition. Plant Cell 29(4):618–637. https://doi.org/10.1105/tpc.16.00891
Teixeira PJPL, Thomazella DPdT, Reis O, do Prado PFV, do Rio MCS, Fiorin GL et al (2014) High-resolution transcript profiling of the atypical biotrophic interaction between Theobroma cacao and the fungal pathogen Moniliophthora perniciosa. Plant Cell 26:4245–4269. https://doi.org/10.1105/tpc.114.130807
Teixeira PJPL, Thomazella DPT, Pereira GAG (2015) Time for chocolate: current understanding and new perspectives on cacao Witches’ broom disease research. PLoS Pathog. https://doi.org/10.1371/journal.ppat.1005130
Thevenin JM, Rossi V, Ducamp M, Doare F, Condina V, Lachenaud P (2012) Numerous clones resistant to Phytophthora palmivora in the “Guiana” genetic group of Theobroma cacao L. PLoS ONE 7(7):e40915. https://doi.org/10.1371/journal.pone.0040915
Thorvaldsdóttir H, Robinson JT, Mesirov JP (2013) Integrative genomics viewer (IGV): high-performance genomics data visualization and exploration. Brief Bioinform 14(2):178–192
Tian T, Liu Y, Yan H, You Q, Yi X, Du Z et al (2017) agriGO v2.0: a GO analysis toolkit for the agricultural community, 2017 update. Nucleic Acids Res 45(W1):W122–W129. https://doi.org/10.1093/nar/gkx382
Tripathy BC, Oelmüller R (2012) Reactive oxygen species generation and signaling in plants. Plant Signal Behav 7(12):1621–1633. https://doi.org/10.4161/psb.22455
van Loon LC, van Strien EA (1999) The families of pathogenesis-related proteins, their activities, and comparative analysis of PR-1 type proteins. Physiol Mol Plant Pathol 55(2):85–97. https://doi.org/10.1006/pmpp.1999.0213
Van Aken O, Van Breusegem F (2015) Licensed to kill: mitochondria, chloroplasts, and cell death. Trends Plant Sci 20(11):754–766. https://doi.org/10.1016/j.tplants.2015.08.002
van Loon LC, Rep M, Pieterse CMJ (2006) Significance of inducible defense-related proteins in infected plants. Annu Rev Phytopathol 44(1):135–162. https://doi.org/10.1146/annurev.phyto.44.070505.143425
van der Hoorn RAL, Kamoun S (2008) From guard to decoy: a new model for perception of plant pathogen effectors. Plant Cell 20(8):2009–2017. https://doi.org/10.1105/tpc.108.060194
Vidal RO, Nascimento LCd MC, Mondego J, Amarante Guimarães Pereira G, Falsarella Carazzolle M (2012) Identification of SNPs in RNA-seq data of two cultivars of Glycine max (soybean) differing in drought resistance. Genet Mol Biol 35(1):331–334
Vlot AC, Dempsey DMA, Klessig DF (2009) Salicylic acid, a multifaceted hormone to combat disease. Annu Rev Phytopathol 47(1):177–206. https://doi.org/10.1146/annurev.phyto.050908.135202
Vogel J, Somerville S (2000) Isolation and characterization of powdery mildew-resistant Arabidopsis mutants. Proc Natl Acad Sci USA 97(4):1897–1902
Wang Y, Cordewener JH, America AH, Shan W, Bouwmeester K, Govers F (2015) Arabidopsis lectin receptor kinases LecRK-IX. 1 and LecRK-IX. 2 are functional analogs in regulating Phytophthora resistance and plant cell death. Mol Plant Microbe Interact 28(9):1032–1048
Wang Y, Nsibo DL, Juhar HM, Govers F, Bouwmeester K (2016) Ectopic expression of Arabidopsis L-type lectin receptor kinase genes LecRK-I. 9 and LecRK-IX. 1 in Nicotiana benthamiana confers Phytophthora resistance. Plant Cell Rep 35(4):845–855
Wessel M, Quist-Wessel PMF (2015) Cocoa production in West Africa, a review and analysis of recent developments. NJAS 74–75:1–7. https://doi.org/10.1016/j.njas.2015.09.001
Wood GAR, Lass R. Cocoa (2008) Longman scientific & technical. Wiley, New York
Wu L, Chen H, Curtis C, Fu ZQ (2014) Go in for the kill: how plants deploy effector-triggered immunity to combat pathogens. Virulence 5(7):710–721. https://doi.org/10.4161/viru.29755
Yang SS, Tu ZJ, Cheung F, Xu WW, Lamb JF, Jung H-JG et al (2011) Using RNA-Seq for gene identification, polymorphism detection and transcript profiling in two alfalfa genotypes with divergent cell wall composition in stems. BMC Genomics 12(1):199
Yang Y-X, Ahammed GJ, Wu C, Fan S-y, Zhou Y-H (2015) Crosstalk among jasmonate, salicylate and ethylene signaling pathways in plant disease and immune responses. Curr Protein Pept Sci 16(5):450–461
Acknowledgements
Thank you to Dr. Craig Praul at the Penn State Genomics Core facility for providing library preparation and transcriptome sequencing services. Thank you to Dr. Istvan Albert at the Penn State Bioinformatics Support Center for advice regarding processing of RNA-seq data. Thank you to Lena Sheaffer for assistance with project management. Thank you to Affian Kacou and Amah Yao for arranging the experiment in shade net house. We thank Irie Boua, Herve Kouakou, Lassana Bakayoko, and Koffi Cyrille at CNRA for their support in maintaining plant and inoculating plant material.
Funding
This work was supported by the Fulbright International exchange Program Number G-1-0005, a Grant from the USDA Foreign Agriculture Service, The Pennsylvania State University College of Agricultural Sciences, the Huck Institutes of the Life Sciences, the Penn State Endowed Program in Molecular Biology of Cacao, NSF Plant Genome Research Award 1546863 and by the USDA National Institute of Food and Agriculture, Federal Appropriations under Project PEN04569 and Accession Number 1003147.
Author information
Authors and Affiliations
Contributions
DNP designed the experiments, oversaw generation of plant material, inoculated and collected leaf samples, extracted RNA, and drafted the manuscript. ASF inoculated and collected samples, performed statistical analyses of RNA-seq data, and wrote and edited the manuscript. NW analyzed polymorphism in the cacao genotypes, identified candidate defense-associated receptor genes, and wrote and edited the manuscript. MT propagated the grafted plants from stock trees in the germplasm collection. KC prepared pathogen inoculum and zoospore suspension for inoculation. AS performed bioinformatic analysis of RNA-seq data and calculated differential gene expression. JHM contributed to data analyses and edited the manuscript. SNM and MJG contributed to experimental design, oversaw experimental progress, and contributed to manuscript writing and editing.
Corresponding author
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Supplemental Figure S1
. Shade net house layout. Pink and blue stripes represent rows of SCA6 and NA32 plants. Black borders around striped areas represent border plants (either genotype) which were excluded from sampling. 1 m wide walking paths between blocks are indicated. (PDF 57 KB)
Supplemental Figure S2
. Heatmap of transcriptome correlations. Analysis is based on mean expression value calculated from biological replicates. S/N indicates genotype; 0,6,24,72 indicates time point; C/P indicates control or pathogen treatment. (PDF 16 KB)
Supplemental Figure S3
. Heatmap of transcriptome correlations. S/N indicates genotype; 0,6,24,72 indicates time point; C/P indicates control or pathogen treatment. Final number (1-5) indicates biological replicate. (PDF 46 KB)
Supplemental Figure S4
. Scatterplots showing correlations between expression levels of A) defense associated receptors and B) PR genes between NA32 and SCA6 samples collected at time zero. Measurements are the average log10 transformed DESeq2 normalized counts calculated from eight replicates at time zero. (PDF 98 KB)
Rights and permissions
About this article

Cite this article
Pokou, D.N., Fister, A.S., Winters, N. et al. Resistant and susceptible cacao genotypes exhibit defense gene polymorphism and unique early responses to Phytophthora megakarya inoculation. Plant Mol Biol 99, 499–516 (2019). https://doi.org/10.1007/s11103-019-00832-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11103-019-00832-y