
Abstract
The clustered, regularly interspaced, short palindromic repeats (CRISPR)/CRISPR associated (Cas)9 protein system has emerged as a simple and efficient tool for genome editing in eukaryotic cells. It has been shown to be functional in several crop species, yet there are no reports on the application of this or any other genome editing technologies in the cotton plant. Cotton is an important crop that is grown mainly for its fiber, but its seed also serves as a useful source of edible oil and feed protein. Most of the commercially-grown cotton is tetraploid, thus making it much more difficult to target both sets of homeologous alleles. Therefore, in order to understand the efficacy of the CRISPR/Cas9 system to target a gene within the genome of cotton, we made use of a transgenic cotton line previously generated in our laboratory that had a single copy of the green fluorescent protein (GFP) gene integrated into its genome. We demonstrate, for the first time, the use of this powerful new tool in targeted knockout of a gene residing in the cotton genome. By following the loss of GFP fluorescence, we were able to observe the cells that had undergone targeted mutations as a result of CRISPR/Cas9 activity. In addition, we provide examples of the different types of indels obtained by Cas9-mediated cleavage of the GFP gene, guided by three independent sgRNAs. The results provide useful information that will help us target important native genes in the cotton plant in future.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
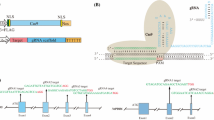
Among the various genome-editing technologies, the CRISPR/Cas9 system from Streptococcus pyogenes has emerged as a simple, yet relatively efficient method to modify a target gene within the genomes of several eukaryotes, including plants and animals. This system, based on the adaptive immune system of prokaryotes against viruses and foreign DNA (Barrangou et al. 2007; Gasiunas et al. 2012; Horvath and Barrangou 2010; Jinek et al. 2012; Koonin and Makarova 2009), has been adapted for targeted genome modification in eukaryotic cells (Cong et al. 2013; Mali et al. 2013a, b). Compared to the older genome editing methods, that include Zinc Finger Nuclease, TALEN and Meganuclease, the CRISPR/Cas9 system is relatively simple (Baltes and Voytas 2015; Voytas and Gao 2014). While the three older technologies require the design and generation of a specific protein sequence that can recognize as well as cleave a target DNA sequence, the CRISPR/Cas9 system, relies on two components—a guide RNA that by RNA-DNA base pairing recognizes the target sequence in the genome and a nuclease that cleaves the target DNA sequence at a specific location within the target. In practice, the guide RNA is supplied as a single transcript that is approximately 100 nucleotides long and is referred to as a single guide RNA (sgRNA). It consists of the first ~20 nt that are complementary to the desired target and the rest comprises of tracrRNA:crRNA that forms a hairpin like structure which interacts with the Cas9 nuclease. This sgRNA/Cas9 complex allows for the targeted cleavage within a desired DNA sequence that is usually termed a protospacer (Cong et al. 2013; Jinek et al. 2012; Mali et al. 2013a). The most important requirement for the recognition/cleavage is that the protospacer sequence should be followed by an NGG sequence (Protospacer Adjacent Motif, PAM) (Jinek et al. 2012; Sternberg et al. 2014). Thus, sgRNA, in theory, can be designed against any DNA sequence as long as it is followed by a PAM sequence. The delivery of the sgRNA can be directly in the form of RNA molecules or through the transcription of a customized DNA cassette. The Cas9 component can be supplied either directly in the form of protein or as an expression cassette (Nelson and Gersbach 2016; Tang and Tang 2016). Its ease of use has resulted in the widespread adoption of the CRISPR/Cas9 method by various laboratories including ones with limited resources. Soon after the efficacy of the CRISPR/Cas9 system was established in eukaryotic cells, this technology was successfully tested in Arabidopsis, tobacco, rice, sorghum and tomato (Brooks et al. 2014; Jiang et al. 2013; Li et al. 2013; Nekrasov et al. 2013; Shan et al. 2013). A number of important crop plants have also been subjected to targeted modifications to introduce useful traits through the use of CRISPR/Cas9 system. Examples include, wheat resistant to powdery mildew, created using both TALENs and CRISPR/Cas9 system (Wang et al. 2014); tomato fruit ripening regulation using CRISPR/Cas9-based editing (Ito et al. 2015); drought tolerant maize through CRISPR/Cas9 (Shi et al. 2016), and herbicide tolerant soybean using CRISPR/Cas9 based editing (Li et al. 2015c).
Cotton is the most important natural fiber crop that also produces a large amount of seeds as a byproduct that is used mainly to obtain edible oil and cattle feed. With the seed-specific gossypol elimination, cottonseed meal protein has the potential to become a feed for the more efficient monogastric animals and could even serve as a direct source of nutrition for humans (Sunilkumar et al. 2006). Thus, cotton represents a unique crop that provides not only fiber, but also feed and food. The cotton plant is also susceptible to a number of pests and diseases, thus requiring a much higher use of chemicals and thus input costs. Hence, it offers a large number of traits that could be improved through the use of precise, targeted mutation/genome modification. Unlike model plant species or some other important food and horticultural crops, progress in molecular breeding of cotton has not occurred at the same pace.
With the recent availability of cotton genome sequence (Li et al. 2015a; Zhang et al. 2015), it now becomes possible to utilize molecular tools such as the CRISPR/Cas9 system to investigate the function of genes and to deploy the basic biological information to improve its agronomic performance and quality traits. However, to our knowledge there are no published reports demonstrating the use of any of the aforementioned technologies in the modification of the cotton genome. Currently, most of the genome editing methods rely on cell and tissue culture components during and following the modification to obtain a plant with the desired mutation. The regeneration aspect remains highly genotype dependent and difficult for cotton. Most of the commercial cotton grown around the world is dominated by two species, Gossypium hirsutum and G. barbadense, that are allotetraploids, and therefore, present twice the number of targets that will need to be modified compared to other diploid crops, at least for the traits that require knockout of a gene. Given these limitations, we sought to understand the process of utilizing the CRISPR/Cas9 system to modify the cotton genome. We made use of the Green Fluorescent Protein (GFP) gene as the target that is already integrated into the cotton genome. In this report, we provide the results from experiments wherein we tested three different sgRNA constructs and examples of the mutations achieved at the target sites.
Materials and methods
Plant material and selection of target sequences
A single copy, GFP-expressing line (homozygous, T3 generation) was used to test the genome editing efficiency using the CRISPR/Cas9 system in cotton. This line was generated previously in our laboratory using the pBINmGFP5-ER construct with Agrobacterium mediated transformation protocol (Sunilkumar et al. 2002; Sunilkumar and Rathore 2001). To identify the target sites with different activities we used sgRNA Scorer 1.0 web tool (https://crispr.med.harvard.edu/sgRNAScorer/) as described by Chari et al. (2015).
Vector construction
Plasmid vectors (pTC212, pTC241 and pCGS751) used to assemble the three binary vectors used in this study were kindly provided by Daniel Voytas, University of Minnesota. Sequences corresponding to the selected targets were used to synthesize oligos (Invitrogen, USA) with appropriate overhang sequences (Online Resource 1). Each pair of oligonucleotides were annealed, phosphorylated and introduced into pTC241 using golden gate cloning method with Esp31 restriction enzyme (Thermo Fisher Scientific, USA) as described by Cermak et al. (2015); Cong and Zhang (2015) with slight modifications. pTC212 contains the Cas9 gene codon optimized for Arabidopsis (Baltes et al. 2014). Cas9 and each sgRNA were cloned into the binary vector pCGS751 by Golden Gate cloning method using AarI restriction enzyme (Thermo Fisher Scientific, USA) (Cermak et al. 2015; Cong and Zhang 2015) (Online Resource 2).
Plant transformation
Cotton transformation protocol was followed as described by Rathore et al. (2015) with minor modifications, wherever necessary. GFP expressing cotton seeds were germinated in jars on MSO medium at 28 °C for 10 days under 16 h photoperiod in a growth chamber. Hypocotyl, cotyledonary petiole and cotyledon explants excised from a seedling were infected with Agrobacterium tumefaciens (LBA4404) containing the binary vector. Explants were co-cultivated on P1-AS medium for 3 days under light at 25 °C and then transferred to selection medium containing hygromycin (P1-c4h15) under light at 28 °C in a growth chamber. After 42 days of culture, callus tissue (3 mm or larger) representing an individual transgenic event, growing at cut surface of the explant was carefully excised under a stereomicroscope. Excised calli were placed on P7-c4h15 medium and incubated in the growth chamber under diffused light with monthly subcultures on MSEm medium. Somatic embryos that regenerated were transferred to EG3 medium for germination. T0 plantlets that grew from the somatic embryos on the EG3 medium were placed on MS3 medium for further growth and development.
Fluorescence observations and scoring
Stable transgenic events, in the form of hygromycin-resistant microcalli, growing out from the cut surface of the explant were visualized with a Zeiss M2 BIO Fluorescence Combination Zoom Stereo/Compound microscope. The microscope is equipped with a GFP filter set comprising an exciter filter (BP 470/40 nm), a dichromatic beam splitter (495 nm) and a barrier filter (LP 500 nm). The light source was an HBO 100 W mercury lamp. Photographs were taken with a Zeiss AxioCam color digital camera coupled to the microscope. Images were captured using the Zeiss AxioVision 3.0.6 software. Loss of GFP expression, concomitant with the appearance of plastid-based red fluorescence reflected the activity of CRISPR/Cas9 system. Progression of the loss of GFP fluorescence was observed periodically. At 26 days post infection (dpi), the total number of individual callus lines originating from the explant and the number of events showing the loss of GFP expression were counted. Following the excision of individual events from the explant at 42 dpi, these were cultured further on selection and callus proliferation medium. At 70 dpi, the excised calli were again monitored for fluorescence. Individual lines were placed into three categories based on their fluorescence: (1) completely silenced >90% callus mass showing red fluorescence; (2) chimera 10–90% of the callus mass showing silencing; (3) no silencing (90–100% callus mass showing GFP fluorescence).
Characterization of the mutations
At 120 dpi, genomic DNA was isolated from the calli that were growing on hygromycin medium using a protocol described by Chaudhry et al. (1999), with some modifications. Callus tissue was lysed in the extraction buffer using Tissuelyser II (QIAGEN). Following chloroform:isoamyl alcohol (24:1) extraction, the DNA was precipitated with isopropanol and washed with 70% ethanol. Air-dried DNA pellet was resuspended in 100 µl TE buffer and stored at −20 °C until further use. T0 plants were sacrificed to isolate genomic DNA to characterize the nature of mutations. A common set of primers (CAS9.GFP.M-F and CAS9.GFP.M-R) was used to amplify the GFP fragment that contains all three targets as shown in Fig. 1. Primer sequences are provided in Online Resource 1. PCR was performed using Phusion High-Fidelity DNA polymerase (New England BioLabs Inc.). PCR products were electrophoresed on 1% agarose gel and the amplicon was purified and eluted using QIAquick Gel Extraction kit (QIAGEN). The amplicon was sequenced with both forward and reverse primers by Sanger sequencing. To identify different indels that were generated by CRISPR/Cas9-mediated double stand breaks (DSB) followed by non homologous end joining (NHEJ), we used a web-based tool, CRISP-ID as described by Dehairs et al. (2016). This program is able to resolve two to three different mutations.

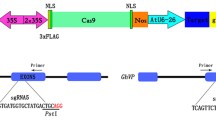
a Schematic representation of the target sites in the GFP coding region showing the location of targets T1, T2 and T3. Black arrows show the primer binding sites. These primers were used to amplify the GFP fragment for sequence analysis. b Three different target sequences were selected based on the scores generated by a web-based sgRNA design tool, sgRNA Scorer [T1 (51.2), T2 (2.2) and T3 (91.2)]. Underlined is the 20-nucleotide target sequence and yellow highlighted nucleotides represent the PAM (protospacer adjacent motif) sequence
In cases where the amplicon contained three or more indels, the PCR product was cloned into a pMiniT vector using NEB PCR cloning kit (New England Biolabs Inc.) and transformed into E. Coli, DH5α. Plasmid DNA was isolated using the QIAprep Spin Miniprep (QIAGEN) plasmid isolation kit from individual colonies that were grown overnight on ampicillin selection medium. The insert within the plasmid was sequenced, using the CAS9.GFP.M-F primer, to identify the nature of mutation.
Results
Selection of target sequences
We used sgRNA Scorer 1.0 (Chari et al. 2015) to identify guide RNA sequences within the GFP gene and obtained 82 possible sequences with varying ranks. Three different target sites were selected from this list for the current study: Target1 (T1: 133–152 bp), Target2 (T2: 241–260 bp), and Target3 (T3: 281–300 bp). These targets were chosen based on their predicted ranking scores of medium, low and high levels (51.2, 2.2 and 91.2, respectively). A schematic representation of the target sites T1, T2 and T3 within the GFP coding sequence is shown in Fig. 1a. Target sequences (underlined) and the respective PAM sequences (yellow highlight) are presented in Fig. 1b. Online Resource 1 shows the custom-built oligos for each of the targets (underlined). For T2 we introduced an additional base, G at the 5′-end of the target sequence as a requirement for the U6 promoter to initiate transcription. The required overhang sequences that facilitate the golden gate method of cloning are not underlined (Online Resource 1). Oligos were annealed and introduced into pTC241. The final assembled vector contains hpt gene as a plant selectable marker, one of the sgRNA cassette, and Cas9 cassette in the same T-DNA (Online Resource 2).
Visual detection of GFP knockout mutations
Hypocotyl, petiole and cotyledon explants from GFP-expressing seedlings were transformed with one of the three constructs and subjected to hygromycin selection. Around 15 dpi, microcalli, representing stable transgenic events, began to appear on the cut surface of the explants. After that, the explants were closely monitored under a fluorescence microscope for GFP silencing. First mutation reflecting the silencing of the GFP gene was observed with sgRNA3 construct at 20 dpi. At 26 dpi, total number of microcalli that grew on each explant were counted and their fluorescence status was also scored. Forty explants in each category were scored, with the exception of petiole explants transformed with sgRNA2 construct. In this case, only 24 explants were scored because one plate was lost due to contamination. A particular event was considered silenced whether it showed partial or complete silencing in the callus mass. Transformation efficiencies varied among the three constructs and different type of explants (Fig. 2). Overall, the highest efficiencies were observed with sgRNA3 at 49.8% of silenced events, followed by 43.4% for sgRNA2 and 25.1% for sgRNA1, that showed the lowest percentage of silenced events. Thus, sgRNA3 was the most efficient, while sgRNA1 showed the least activity. Periodic monitoring of the explants had indicated that as the microcalli grew, the silenced tissue within these grew at a degree higher than what could be accounted for by the tissue growth. To confirm this observation, we followed some explants up to 40 dpi. An example is presented in Fig. 3 that shows a dramatic increase in the proportion of silenced tissue compared to the growth of the callus itself, especially by day 40.

Efficiency of Cas9-mediated mutation of the GFP gene in the callus tissue originating from hypocotyl, petiole and cotyledon explants observed at 26 days post transformation. Three different guide RNAs (sgRNA1, sgRNA2 and sgRNA3) were used, each targeting a different region of the GFP coding sequence. Numbers shown under each explant: number of independent transgenic events scored for the presence or absence of GFP fluorescence/number of explants examined

Progression of CRISPR/Cas9-mediated mutation of GFP gene in the callus tissue originating from a cotton hypocotyl piece transformed with the sgRNA2 construct. The same hypocotyl explant was photographed with the aid of a fluorescence microscope at a 26 dpi, b 30 dpi, and c 40 dpi. Blue arrow shows the growth of callus showing GFP fluorescence. Red arrow shows the callus wherein GFP has undergone mutation and rendered non-functional
Individual transgenic events were excised from all the explants at 42 dpi and subcultured on callus proliferation medium under hygromycin selection. At 70 dpi, these calli were scored for their fluorescence status. Figure 4 shows examples from three categories used for scoring. Figure 4b shows a callus that showed complete silencing of GFP, while the image depicted in Fig. 4d shows chimeric callus. GFP fluorescence remains undiminished in the callus shown in Fig. 4f suggesting that the GFP gene remained unmutated and functional. Highest number of completely silenced calli (43%) was observed with sgRNA3, followed by sgRNA2 (40%), and sgRNA1 (6%) (Fig. 5). sgRNA1 also showed the highest percentage of chimeric calli and more events without any mutations. Based on the very low efficiency of GFP silencing with sgRNA1, the culture lines for Target 1 were not advanced further for regeneration. Only the lines obtained from targeting with sgRNA2 and sgRNA3 were maintained in culture to induce somatic embryogenesis followed by recovery of T0 plantlets.

Silencing of the GFP gene in the callus tissue, 70 days following transformation. The callus was excised from the transformed explants 42 days following transformation and grown under hygromycin selection. Three different types of callus tissues are depicted. a, b Completely silenced, c, d chimeric callus showing silencing in a part of the tissue, e, f no silencing observed. For each callus, a bright field micrograph and its corresponding fluorescence image are shown

Fluorescence status of individual callus scored at 70 days following transformation in response to three different sgRNA constructs. Calli were observed visually under a fluorescence microscope and classified under three different categories. Green 90–100% of callus mass showing GFP florescence; Yellow chimeric callus (10–90% callus showing silencing); and Red 90–100% of callus mass showing silencing of GFP
Molecular characterization of the mutations
To analyze the nature of mutations in the GFP gene caused by CRISPR/Cas9, leading to its silencing, ten individual completely silenced calli from sgRNA1 and sgRNA3 transformations and 20 such calli from sgRNA2 transformation were selected for sequencing. Also, ten calli obtained following transformation with the sgRNA2 construct that were still showing GFP expression were selected to examine if these carried any mutations at all. PCR was performed on the genomic DNA of selected calli using the primers, CAS9.GFP.M-F and CAS9.GFP.M-R. Representative gel profile that shows the amplification of the GFP fragment for both sets of calli is shown in Online Resource 3.
DNA sequencing was performed on PCR products using the same forward and reverse primers that were also used to amplify the fragment. No mutations were observed in any of the ten calli that continued to show GFP expression at 120 dpi. All the GFP-silenced calli showed various types of indels. One silenced event, resulting from transformation with the sgRNA2 vector, showed only one type of mutation. This mutation observed in the event T2_1 is shown in Fig. 6 and Online Resource 4B. This was surprising given the fact that the GFP-expressing material that was subjected to CRISPR/Cas9 treatment was homozygous and thus had two allelic copies of the GFP transgene. Event T2_1 represented the only example of a homozygous mutation that we found out of 40 silenced callus lines that were examined. The DNA from the remaining silenced calli showed bi-allelic (two types of mutations, one on each allelic copy) or more than two mutations. Direct Sanger sequencing of the region surrounding the target showed multiple, overlapping peaks resulting from more than one indels, thus making it difficult to resolve the sequences. Examples of chromatograms showing no mutation, homozygous, bi-allelic or chimeric indels are shown in Online Resource 4. To distinguish these mutation types, we used a web-based tool CRISP-ID, which can predict up to three allelic mutations from a single Sanger sequencing trace (Dehairs et al. 2016). The mutations that we were able to resolve using this program, are shown in Fig. 6. Examples include indels generated by targeting each of the three selected sequences. In some cases, this program was unable to resolve sequences even when only two overlapping peaks were visible on the chromatogram. In cases where we were unable to resolve the mutations, the PCR product was cloned into pMiniT vector and transformed into E. coli. Plasmid isolated from individual colony cultures were sequenced. Some examples of the results obtained for each of the three sgRNAs are shown in Online Resource 5. For callus T1_14 which resulted from sgRNA1-mediated mutation, two types of indels are seen. Also, in the case of callus T2_9, resulting from sgRNA2-mediated mutation, two types of indels were detected. Callus T3_27 was obtained following transformation with a vector containing sgRNA3. In this instance, we were able to detect four different types of indels.

Examples of mutations ascertained by sequencing PCR products that were amplified from the genomic DNA of calli at 120 dpi. Loci T1, T2, T3 were targeted using sgRNA1, sgRNA2 & sgRNA3, respectively. Underlined target sequence in the wild-type GFP gene; yellow highlighted PAM sequence; WT wild-type sequence; Callus number: 1, 2, 3, 5, 6, 7, 12, 13, 15, 18, 19, 21, 25, 26, 28, 30; A, B, C: various mutations detected in the same callus. Homozygous mutation is indicated with an asterisk symbol. Types of indels are shown on the right side of each sequence. Nucleotide substitution or insertion is shown in lower case
Molecular characterization of mutations in T0 plantlets
Similar analysis for mutations was conducted on DNA from T0 plantlets obtained from cultures resulting from sgRNA2- and sgRNA3-mediated mutations. Nine T0 plantlets obtained from individual culture lines following sgRNA2-mediated knockout of GFP were examined for the nature of mutations. Two plants showed homozygous mutations, while the remaining seven showed bi-allelic indels. From the cultures obtained following targeting with sgRNA3, we obtained six T0 plantlets. In this case we observed a homozygous mutation in one plant and bi-allelic indels in the remaining five plants. Mutations obtained from each of these plants were easily resolved by CRISP-ID and are shown in Fig. 7. Examples of chromatograms showing homozygous and bi-allelic indels are shown in Online Resources 6 and 7.

Examples of mutations ascertained by sequencing PCR products that were amplified from the genomic DNA obtained from T0 plants. Loci T2, and T3 were targeted using sgRNA2 and sgRNA3, respectively. Underlined target sequence in the wild-type GFP gene; yellow highlighted PAM sequence; WT wild-type sequence; Plant numbers: P1-P15; A, B: bi-allelic mutations detected in a single plant. Homozygous mutations are indicated with an asterisk symbol. Types of indels are shown on the right side of each sequence. Nucleotide insertion is shown in lower case
Discussion
The CRISPR/Cas9 system represents a revolutionary new genome-editing tool that has found widespread adoption by the scientific community to conduct both basic and applied research. Compared to the older genome editing technologies, it is simpler, more flexible, permits multiplexing, and above all more affordable (Char et al. 2017; Li et al. 2013, 2015b; Voytas and Gao 2014; Xing et al. 2014). The original defense system from the prokaryotes has been simplified and streamlined such that only two components, Cas9 enzyme and a customized sgRNA, are required to generate an indel (DSB followed by NHEJ) at the target site in the genome. This ability to introduce DSB at a precise target site has been further extended to create a precise nucleotide substitution or insertion of a desired DNA sequence through homology-dependent repair (HDR). The CRISPR/Cas9 system has been used successfully in many plant species (Bortesi and Fischer 2015; Seth and Harish 2016). As stated earlier, none of the genome editing technologies has been used to modify the cotton genome. This is largely due to the recalcitrant nature of cotton transformation, the unavailability of cotton genome sequence (until recently), and its large genome size. With the recent availability of the draft genome sequence of G. hirsutum and some information on its transcriptome, it should be possible to initiate studies designed to improve this unique crop plant (Yan et al. 2016). As a first step towards addressing this deficiency, we conducted the study described herein to understand the process and to learn the limitations of the technology for cotton improvement. We made use of a transgenic cotton line that has a single copy T-DNA insert containing a GFP expression cassette.
Efficiency of Cas9-mediated editing depends on the target site, nucleotide composition, GC content, and secondary structures of sgRNA (Liang et al. 2016; Ma et al. 2015). To optimize the frequency of mutations, multiple target sites with different cleavage efficiencies need to be tested in the desired gene (Baysal et al. 2016). We selected three different target sequences with predicted low, medium and high efficiencies using a web-based tool sgRNA Scorer 1.0 (Chari et al. 2015). Based on the efficiency predictions, we assembled sgRNA1 that has a medium score of 51.2, sgRNA2 with a low score of 2.2, and sgRNA3 with a high score of 91.2. The use of GFP gene as the target allowed us to follow the putative mutations easily. Although earliest loss of GFP fluorescence was observed at 20 dpi, it is possible that the mutation responsible for the phenotype had occurred much earlier. At 26 dpi, GFP silenced calli were counted under a fluorescence microscope to ascertain the differences in the activity levels among the three sgRNAs. Overall, sgRNA3 resulted in the highest levels of silencing phenotype, closely followed by sgRNA2, whereas sgRNA1 proved to be the least efficient. Thus, for sgRNA3, the observed efficiency was consistent with that predicted by the sgRNA Scorer 1.0 tool. Interestingly, sgRNA2 performed almost as efficiently as sgRNA3 even though its predicted efficiency was only 2.2. sgRNA1 with a predicted score of 51.2, in the medium range, proved to be the least efficient among the three sgRNAs tested in this study. The results depicted in Fig. 3 show that there is a progressive increase in the number of transgenic events that show GFP silencing. The results depicted in this figure also show that from 30 dpi to 40 dpi, the area of the callus that shows silencing of GFP has grown at much faster rate than the actual growth of the callus itself. One possible explanation for this is that the GFP protein is highly stable in the cotton cells and it takes 1–2 weeks for the fluorescence signal to completely disappear. As discussed earlier, another possibility is that in a single transgenic event, following several cell divisions, more than one cell undergoes CRISPR/Cas9-mediated mutations independently, at different time points. It will take an intensive investigation to determine the causes underlying these observations, however, it is beyond the scope of current study.
In order to avoid the mixing of different transgenic events, individual transgenic calli growing on the cut surfaces of explants were excised carefully and grown further on selection/proliferation medium. Excision of independent events from the original explant and further subculture of each is a normal practice that we follow for any cotton transformation experiment. The cultures, each growing separately, were scored again at 70 dpi to ascertain if the differences in the efficiency among the three sgRNAs persisted. Overall, we observed 53–73% of editing efficiency, taking into account both chimera and completely silenced calli, using the three different sgRNAs. Again, highest silenced events were observed with sgRNA3 showing 43% completely silenced calli, closely followed by sgRNA2 at 40%, while sgRNA1 was the least efficient at 6%. Such discrepancies between the predicted and observed efficiencies have also been observed in some other studies. Baysal et al. (2016) tested two sgRNAs against the rice OsBEIIb gene. The predicted score for one of the sgRNAs was 96%, however, it did not result in any mutations. Another sgRNA, with a predicted score of 39%, provided a mutation efficiency of only 5%. A study conducted by Wang et al. (2016) also reported discrepancies between the bioinformatically predicted sgRNA scores for several targets in the wheat genome and the observed editing efficiencies. Currently, there are several tools available to design sgRNAs other than sgRNA Scorer 1.0 that was used to select the target sequences in the current study (Doench et al. 2014; Heigwer et al. 2014; Prykhozhij et al. 2015; Wong et al. 2015), and also some especially for designing plant sgRNAs and to avoid off-target effects in certain plant species (Brazelton et al. 2015; Lei et al. 2014; Xie et al. 2014). More recently, Wong et al. (2015) developed a guide RNA designing tool (WU-CRISPR), based on many novel features that underlie the potency of highly effective sgRNAs. This was done by reanalyzing a pool of 1841 sgRNAs in a public dataset (Doench et al. 2014). They considered different characteristics of sgRNA such as GC content, secondary structure, contiguous stretch of the same nucleotides, and free accessibility of the seed region for target recognition (i.e. avoiding the use of U at position 19 and use of C or U at position 20 in the guide sequence). When the GFP sequence was analyzed using this particular tool, we obtained only 13 possible target sequences. Only one of the sequences targeted with sgRNA3 in the current study was found in this list, with a potency score of 77. The same sequence received a ranking of 91.2% in the sgRNA Scorer 1.0. Targets T1 and T2 were excluded by WU-CRISPR because T1 has four contiguous thymines and the 19th base in T2 is a thymine. Despite the fact that T2 was not selected as a target by WU-CRISPR, sgRNA2 was almost as effective as sgRNA3 in knocking out GFP. All 13 target sequences identified by WU-CRISPR were present in the set of 81 target sequences predicted by sgRNA Scorer 1.0. However, the potency score assigned to each of these sequences by the two bioinformatics tools was different. As the capabilities of the sgRNA designing tools are improved and more plant species are included in the search criteria to avoid off-target effects, it may become possible to select the most efficient target sequences for a gene of interest.
In order to confirm whether the silencing of the GFP was due to mutations caused by the CRISPR/Cas9 system, we examined the sequence of the PCR amplicon corresponding to the GFP gene in the calli that showed complete silencing and also those that did not show any silencing. Some examples of the indels generated in the GFP-silenced calli in response to each of the three sgRNAs are shown in Fig. 6 and Online Resource 5. Online Resource 4 shows representative sequencing chromatograms for three types of mutations (homozygous, bi-allelic and chimeric) detected in individual callus lines. As mentioned earlier, only one type of mutation was observed in one of the transgenic events (T2_1). A 39 bp deletion of exactly the same sequence from both allelic copies of the resident GFP gene was detected in this line, suggesting that this line represented a homozygous mutation. The slightly smaller size of PCR amplicon obtained from this event also provides some indication of the relatively large deletion from both the GFP alleles (lane #11, Online Resource 3). In cotton, regeneration of plants occurs via somatic embryogenesis following a long period of culture (6–10 months). Since a somatic embryo originates from a single cell, the GFP knockout T0 plants should show no more than two types of indels, unlike some of the callus lines described above. Indeed, the results presented in Fig. 7 show that 12 plants have two different indels each (seven plants for target 2 and five for target 3). Two plants out of nine examined for target 2 and one out of six for target 3 showed homozygous mutations. Zhang et al. (2014) found that 3.8% of all the T0 rice plants (7.7% of all the plants with targeted mutations) carried homozygous mutations. In another study on rice, Ma et al. (2015) observed a much higher rate of homozygous mutations. Of all the mutant plants, 27.8% carried homozygous mutations. In an investigation on tomato, Pan et al. (2016) reported that an average of 11.36% T0 plants contained homozygous mutations for the SlPIF4 gene. Thus, the homozygous mutation observed in our study appears to be a common occurrence. One explanation for our result is that CRISPR/Cas9-mediated DSB followed by NHEJ resulted in exactly the same indel in each allelic copy of the GFP in the chromosome pair. However, culture line T2_1 showed a homozygous mutation of 39 bp deletion. Given the relatively large size of the deletion, it is unlikely that DSB followed by NHEJ resulted in such a homozygous mutation. Another possible explanation for homozygous mutation was put forward by Ma et al. (2015). They suggested that following the DSB-NHEJ-mediated indel in one allele, homology directed repair mechanism uses this mutated copy as a template to generate the same mutation in the second allele in the sister chromosome that has undergone a Cas9-sgRNA-directed DSB at the target site. Those callus lines that show chimeric indels (more than two types of mutations) may be a result of mixing of two separate transformation events. It is also possible that in a given transformation event, that has undergone multiple cell divisions, the CRISPR/Cas9 system leads to mutations at different time points in two or more cells. Based on their results on rice, Zhang et al. (2014) and Xu et al. (2015) found that the WT copy of the target gene in heterozygous and chimeric plants could continue to mutate either in T0 or T1 generation. Similarly, Pan et al. (2016) observed that in chimeric tomato plants that comprised of cells/tissues with and without mutations, the CRISPR/Cas9 complex continued to generate mutations in the WT alleles. Thus, in the current study, it is not surprising to find a group of cells that carry multiple mutations in the callus tissue. None of the callus lines that continued to exhibit GFP expression, showed a mutation in the target sequence suggesting that the cells in these lines carried ‘WT’ copies of the GFP gene. This could be due to multiple reasons. It may be that these events lacked the intact T-DNA, with full or partial loss of sgRNA/Cas9 cassettes. As described above, it is also possible that the sgRNA/Cas9 complex had not yet generated mutations in the GFP genes present in the cells of these events. It was beyond the scope of the current investigation to follow these events on a long-term basis.
In conclusion, we have demonstrated the ability of the CRISPR/Cas9 system to generate DSB-based mutations at a desired target site within the genome of cotton cells. The results also suggest the efficiencies of several sgRNAs should be examined beforehand for each gene of interest to find the best possible protospacer sequence(s). Even though we observed mutations with all three of the sgRNAs, in silico predictions of the sgRNA efficiencies did not match those observed in this study. Commercial kits have become available to examine the cleavage efficiency of sgRNAs in vitro. The most optimum sgRNA thus identified can then be used to target the gene of interest in vivo. Obviously, the lack of off-target effect for the chosen sgRNA is another important consideration. Being a tetraploid, the commercial cotton will present twice the number of targets (even in case of single copy genes) that need to be mutated compared to the GFP transformants examined in the current study. Thus, it becomes all the more important to select the best possible protospacer sequence within the gene of interest. Certainly the long culture period required for regeneration of a cotton plant will help ensure that all four target sites of a given native gene of interest are mutated in some of the plants that are recovered. The results obtained from wheat (Wang et al. 2016, 2014; Zhang et al. 2016), a hexaploid, suggest that it should be possible to apply this powerful new technology for the improvement of cotton. Specifically, Zhang et al. (2016) used a gene gun-based transient expression of Cas9 and sgRNA to target six different genes in hexaploid wheat and obtained efficient editing of each of these genes with no detectable transgene integration. When TaLOX2 was the target, they obtained as many as 76 mutation events from 800 embryos that were bombarded, and of these 34 plants carried mutations on all six alleles. Although we have used the Agrobacterium-mediated stable transformation to conduct gene editing in cotton in the current study, it may also be worth exploring particle bombardment-based transient expression to target native genes in this species.
References
Baltes NJ, Voytas DF (2015) Enabling plant synthetic biology through genome engineering. Trends Biotechnol 33:120–131
Baltes NJ, Gil-Humanes J, Cermak T, Atkins PA, Voytas DF (2014) DNA replicons for plant genome engineering. The Plant Cell 26:151–163
Barrangou R et al (2007) CRISPR provides acquired resistance against viruses in prokaryotes. Science 315:1709–1712
Baysal C, Bortesi L, Zhu C, Farré G, Schillberg S, Christou P (2016) CRISPR/Cas9 activity in the rice OsBEIIb. Mol Breed 36:1–11. doi:10.1007/s11032-016-0533-4
Bortesi L, Fischer R (2015) The CRISPR/Cas9 system for plant genome editing and beyond. Biotechnol Adv 33:41–52
Brazelton VA et al (2015) A quick guide to CRISPR sgRNA design tools. GM Crops and Food 6:266–276. doi:10.1080/21645698.2015.1137690
Brooks C, Nekrasov V, Lippman ZB, Van Eck J (2014) Efficient gene editing in tomato in the first generation using the clustered regularly interspaced short palindromic repeats/CRISPR-associated9 system. Plant Physiol 166:1292–1297
Cermak T, Starker CG, Voytas DF (2015) Efficient design and assembly of custom TALENs using the golden gate platform. In: Pruett-Miller SM (ed) Chromosomal mutagenesis. Springer New York, New York, NY, pp 133–159
Char SN et al (2017) An Agrobacterium-delivered CRISPR/Cas9 system for high-frequency targeted mutagenesis in maize. Plant Biotechnol J 15(2):257–268
Chari R, Mali P, Moosburner M, Church GM (2015) Unraveling CRISPR-Cas9 genome engineering parameters via a library-on-library approach. Nat Meth 12:823–826
Chaudhry B, Yasmin A, Husnain T, Riazuddin S (1999) Mini-scale genomic DNA extraction from cotton. Plant Mol Biol Rep 17:1–7
Cong L, Zhang F (2015) Genome engineering using CRISPR-Cas9 system. In: Pruett-Miller SM (ed) Chromosomal mutagenesis. Springer New York, New York, NY, pp 197–217
Cong L et al (2013) Multiplex genome engineering using CRISPR/Cas systems. Science 339:819–823
Dehairs J, Talebi A, Cherifi Y, Swinnen JV (2016) CRISP-ID: decoding CRISPR mediated indels by Sanger sequencing. Sci Rep 6:28973. doi:10.1038/srep28973
Doench JG et al (2014) Rational design of highly active sgRNAs for CRISPR-Cas9-mediated gene inactivation. Nat Biotech 32:1262–1267
Gasiunas G, Barrangou R, Horvath P, Siksnys V (2012) Cas9–crRNA ribonucleoprotein complex mediates specific DNA cleavage for adaptive immunity in bacteria. Proc Natl Acad Sci 109:E2579–E2586
Heigwer F, Kerr G, Boutros M (2014) E-CRISP: fast CRISPR target site identification. Nat Methods 11:122–123
Horvath P, Barrangou R (2010) CRISPR/Cas, the immune system of bacteria and archaea. Science 327:167–170
Ito Y, Nishizawa-Yokoi A, Endo M, Mikami M, Toki S (2015) CRISPR/Cas9-mediated mutagenesis of the RIN locus that regulates tomato fruit ripening. Biochem Biophys Res Commun 467:76–82
Jiang W, Zhou H, Bi H, Fromm M, Yang B, Weeks DP (2013) Demonstration of CRISPR/Cas9/sgRNA-mediated targeted gene modification in Arabidopsis, tobacco, sorghum and rice. Nucleic Acids Res:1–12. doi:10.1093/nar/gkt780
Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E (2012) A programmable dual-RNA–Guided DNA endonuclease in adaptive bacterial immunity. Science 337:816–821
Koonin EV, Makarova KS (2009) CRISPR-Cas: an adaptive immunity system in prokaryotes. F1000 Biol Rep 1:95. doi:10.3410/b1-95
Lei Y, Lu L, Liu H-Y, Li S, Xing F, Chen L-L (2014) CRISPR-P: a web tool for synthetic single-guide RNA design of CRISPR-system in plants. Mol Plant 7:1494–1496
Li J-F et al (2013) Multiplex and homologous recombination-mediated genome editing in Arabidopsis and Nicotiana benthamiana using guide RNA and Cas9. Nat Biotech 31:688–691
Li F et al (2015a) Genome sequence of cultivated Upland cotton (Gossypium hirsutum TM-1) provides insights into genome evolution. Nat Biotech 33:524–530
Li J-F, Zhang D, Sheen J (2015b) Targeted plant genome editing via the CRISPR/Cas9 technology. Plant Funct Genomics Methods Protoc 1284:239–255
Li Z et al (2015c) Cas9-guide RNA directed genome editing in soybean. Plant Physiol 169:960–970
Liang G, Zhang H, Lou D, Yu D (2016) Selection of highly efficient sgRNAs for CRISPR/Cas9-based plant genome editing. Sci Rep 6:21451. doi:10.1038/srep21451
Ma X et al (2015) A robust CRISPR/Cas9 system for convenient, high-efficiency multiplex genome editing in monocot and dicot plants. Mol Plant 8:1274–1284
Mali P, Esvelt KM, Church GM (2013a) Cas9 as a versatile tool for engineering biology. Nat Meth 10:957–963
Mali P et al (2013b) RNA-guided human genome engineering via Cas9. Science 339:823–826
Nekrasov V, Staskawicz B, Weigel D, Jones JDG, Kamoun S (2013) Targeted mutagenesis in the model plant Nicotiana benthamiana using Cas9 RNA-guided endonuclease. Nat Biotech 31:691–693
Nelson CE, Gersbach CA (2016) Engineering delivery vehicles for genome editing. Annu Rev Chem Biomol Eng 7:637–662. doi:10.1146/annurev-chembioeng-080615-034711
Pan C et al (2016) CRISPR/Cas9-mediated efficient and heritable targeted mutagenesis in tomato plants in the first and later generations. Sci Rep 6:24765. doi:10.1038/srep24765
Prykhozhij SV, Rajan V, Gaston D, Berman JN (2015) CRISPR multitargeter: a web tool to find common and unique CRISPR single guide RNA targets in a set of similar sequences. PloS ONE 10:e0119372. doi:10.1371/journal.pone.0119372
Rathore KS, Campbell LM, Sherwood S, Nunes E (2015) Cotton (Gossypium hirsutum L.). In: Wang K (ed) Agrobacterium protocols: Vol 2. Springer New York, New York, NY, pp 11–23. doi:10.1007/978-1-4939-1658-0_2
Seth K, Harish (2016) Current status of potential applications of repurposed Cas9 for structural and functional genomics of plants. Biochem Biophys Res Commun 480:499–507
Shan Q et al (2013) Targeted genome modification of crop plants using a CRISPR-Cas system. Nat. Biotech 31:686–688
Shi J et al (2016) ARGOS8 variants generated by CRISPR-Cas9 improve maize grain yield under field drought stress conditions. Plant Biotechnol J. doi:10.1111/pbi.12603
Sternberg SH, Redding S, Jinek M, Greene EC, Doudna JA (2014) DNA interrogation by the CRISPR RNA-guided endonuclease Cas9. Nature 507:62–67
Sunilkumar G, Rathore KS (2001) Transgenic cotton: factors influencing Agrobacterium-mediated transformation and regeneration. Mol Breed 8:37–52
Sunilkumar G, Mohr L, Lopata-Finch E, Emani C, Rathore KS (2002) Developmental and tissue-specific expression of CaMV 35S promoter in cotton as revealed by GFP. Plant Mol Biol 50:463–479
Sunilkumar G, Campbell LM, Puckhaber L, Stipanovic RD, Rathore KS (2006) Engineering cottonseed for use in human nutrition by tissue-specific reduction of toxic gossypol. Proc Natl Acad Sci 103:18054–18059
Tang W, Tang AY (2016) Applications and roles of the CRISPR system in genome editing of plants. J For Res. doi:10.1007/s11676-016-0281-7
Voytas DF, Gao C (2014) Precision genome engineering and agriculture: opportunities and regulatory challenges. PLoS Biol 12:e1001877
Wang Y, Cheng X, Shan Q, Zhang Y, Liu J, Gao C, Qiu JL (2014) Simultaneous editing of three homoeoalleles in hexaploid bread wheat confers heritable resistance to powdery mildew. Nat Biotechnol 32:947–951
Wang W, Akhunova A, Chao S, Akhunov E (2016) Optimizing multiplex CRISPR/Cas9-based genome editing for wheat. bioRxiv. doi:10.1101/051342
Wong N, Liu W, Wang X (2015) WU-CRISPR: characteristics of functional guide RNAs for the CRISPR/Cas9 system. Genome Biol 16:1. doi:10.1186/s13059-015-0784-0
Xie K, Zhang J, Yang Y (2014) Genome-wide prediction of highly specific guide RNA spacers for CRISPR–Cas9-mediated genome editing in model plants and major crops. Mol Plant 7:923–926
Xing H-L et al (2014) A CRISPR/Cas9 toolkit for multiplex genome editing in plants. BMC Plant Biol 14:327. doi:10.1186/s12870-014-0327-y
Xu R-F et al (2015) Generation of inheritable and “transgene clean” targeted genome-modified rice in later generations using the CRISPR/Cas9 system. Sci Rep 5:11491. doi:10.1038/srep11491
Yan R et al (2016) Progress in genome sequencing will accelerate molecular breeding in cotton (Gossypium spp.). 3 Biotech 6:217. doi:10.1007/s13205-016-0534-3
Zhang H et al (2014) The CRISPR/Cas9 system produces specific and homozygous targeted gene editing in rice in one generation. Plant Biotechnol J 12:797–807
Zhang T et al (2015) Sequencing of allotetraploid cotton (Gossypium hirsutum L. acc. TM-1) provides a resource for fiber improvement. Nat Biotech 33:531–537
Zhang Y et al (2016) Efficient and transgene-free genome editing in wheat through transient expression of CRISPR/Cas9 DNA or RNA. Nat Commun 7:12617. doi:10.1038/ncomms12617
Acknowledgements
The base vectors for assembling the transformation constructs were kindly provided by Dr. Daniel Voytas, University of Minnesota.
Author contributions
KR: conception and design, acquisition of data, analysis and interpretation of data, drafting and revising the article. MJ: conception and design, performing the experiments, acquisition of data, analysis and interpretation of data, drafting and revising the article. LC: performing the experiments, acquisition of data.
Funding
This research was supported by funding from Texas A&M AgriLife Research.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Janga, M.R., Campbell, L.M. & Rathore, K.S. CRISPR/Cas9-mediated targeted mutagenesis in upland cotton (Gossypium hirsutum L.). Plant Mol Biol 94, 349–360 (2017). https://doi.org/10.1007/s11103-017-0599-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11103-017-0599-3