
Abstract
In nature, plants are often exposed to combinations of different stresses at the same time, while in many laboratory studies of molecular stress induction phenomena, single stress responses are analyzed. This study aims to identify the common (i.e. more general stress-responsive) and the stress-specific adjustments of the leaf proteome of wild barley to two often co-occurring stress phenomena, i.e. in response to (long-term) drought acclimation (DA) or to (transient) heat stress (HS). In addition, we analyzed those alterations which are specific for the combination of both stresses. Leaf proteome analysis was performed using 2D difference gel electrophoresis followed by protein identification via mass spectrometry with a 1.5 threshold value of changes in relative protein contents. DA resulted in specific upregulation of proteins with cell detoxification functions, water homeostasis maintenance, amino acids synthesis and lipid metabolism and distinct forms of heat shock proteins (HSPs) and proteins with chaperon functions while proteins related to nitrogen metabolism were downregulated. This response was distinguished from the response to transient HS, which included upregulation of a broad range of HSP products. The common response to both stressors revealed upregulation of additional forms of HSPs and the downregulation of enzymes of the photosynthetic apparatus and chlorophyll binding proteins. The simultaneous exposure to both stress conditions resulted mostly in a combination of both stress responses and to unique abundance changes of proteins with yet unclear functions.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
In their natural habitats, plants are exposed to several stresses. These include biological factors like pathogen infections, insect or browsing animals attack, competition by weeds or abiotic stress conditions like water and temperature constraints, salinity, wind, radiation, heavy metal contamination, oxidative stress and others. These stress factors affect plants yield production by more than 50 % (Wang et al. 2003). Drought and high temperature are considered as key stress factors with high potential impact on crop yield. Current climate prediction models indicate a gradual increase in the atmosphere temperature and an increase in the frequency and extent of (transient or long-term) heat stress (HS) periods in the near future for Europe and the Mediterranean (Suzuki et al. 2014). Additionally, high temperatures will be accompanied by extended periods of drought, that could have a negative impact on crop production worldwide and have severe consequences on food security (Rosegrant and Cline 2003; IPCC et al. 2008). Many studies have been conducted to analyze plant response towards one of these stressors (e.g. Fatehi et al. 2012; Ashoub et al. 2013; Budak et al. 2013). However, the combination of two or more stresses like drought and HS is common under field conditions (Shah and Paulsen 2003). It was reported that the responses of tobacco and Arabidobsis to a combination of drought and HS were different from the sum of single responses, meaning that one cannot simply extrapolate from the response of plants to drought or HS applied individually to predict the effect of their combination (Rizhsky et al. 2002, 2004). The dynamic molecular changes of plants to survive stress conditions can be studied on the levels of the transcriptome and the proteome. Transcriptome analyses provide information about gene transcripts alteration levels (Tommasini et al. 2008; Guo et al. 2009). Meanwhile, proteome analyses provide information about changes in protein abundance by modified turnover velocities (Baerenfaller et al. 2012), post translational modification, or involvement of alternative splicing possibilities of transcripts for protein translation. Although being more expensive and difficult than mRNA sequencing techniques, they have the advantage to describe molecular changes directly related to functional alterations of metabolism. Since its discovery (O’Farrell 1975), the two dimensional polyacrylamide gel electrophoresis, 2D PAGE, became a routine approach to analyze proteome changes in response to stresses (Caruso et al. 2009). However, the method is experiencing difficulties in results reproducibility (Bech-Serra et al. 2009). An alternative to the conventional 2D PAGE is the difference gel electrophoresis, DIGE, allowing the sensitive analysis of proteins in a quantitative manner and reducing reproducibility problems. The conventional 2D PAGE or 2D-DIGE followed by mass spectrometry, MS, was successfully used to identify proteome alterations in response several individual stresses including drought in soybean (Alam et al. 2010), heat in wheat (Skylas et al. 2002), salinity in Arabidopsis thaliana and rice (Ndimba et al. 2005; Yan et al. 2005), cold in Arabidopsis thaliana and rice (Amme et al. 2006; Imin et al. 2004), heavy metals in Arabidopsis thaliana (Roth et al. 2006) and others.
Cultivated barley (Hordeum vulgare L.) is among the earliest domesticated crops. It ranks fourth in regard to harvested area and tonnage of the world cereal production (http://faostat.fao.org). It is cultivated in regions where other cereals cannot be grown due to low rainfall rates, e.g. as a winter crop in the Mediterranean and Minor Asia. Its wild ancestor, Hordeum spontaneum (Wild Barley), is mainly distributed in the Mediterranean and Irano-Turanian regions (Harlan and Zohary 1966) as a winter annual species, ranging eastward to Tibet (Choo 2002). Its broad ecological and geographical distribution range supports the accumulation of a high potential for adaptive genetic diversity against various abiotic and biotic stresses beyond what is seen in cultivated barley varieties. It is therefore regarded as a beneficial source for many tolerance genes for breeding programs against stresses (Ellis et al. 2000). This study aims to identify the common and specific changes of wild barley leaf proteome in response to drought, high temperature, and their combination.
Materials and methods
Plant material and stress treatments
Seeds of wild barley from the northern coastal region of Egypt (Wadi Habies 31°24′ 0″N/27°03″E) obtained from Max-Planck Institute for Breeding Research, Cologne, Germany, were used in this study. Seed material was produced from a single plant by two successive selfing propagation cycles. Seeds were germinated on wet filter papers for 2 days. Four seedlings were transplanted into a 16 × 16 × 20 (depth) cm3 pot filled with 1 kg peat moss medium (Stender AG, Germany) and further regarded as one biological replicate. Seedlings were left to grow in a climate chamber under 16 h photoperiod with light intensity of 400 µmol m−2 s−1 and temperatures of 26 °C (light)/24 °C (dark) and air humidity of 70 %. Soil volumetric water content, VWC, was kept at 30 % using an HH2 moisture meter (Delta-T Devices Ltd, Cambridge, UK). In experimental setup of stress treatments, drought treatment of pot-grown plants was carried out over 3 weeks by withholding water at the stage of two leaves to allow the successive development of an acclimation response, similar to the natural situation. We purposefully did not opt for a treatment with a constant soil water content, since providing water will result in cycles of drought stress and recovery reactions. The soil water potential was measured to be −2.5 MPa (Psypro psychrometer, Wescor, Utah, USA) at the end of drought treatment, prior to sample collection. This long-term drought treatment will be termed “drought acclimation” (DA) (sensu Lambers et al. 2008) to distinguish it from short-term (transient) stress treatments, like the applied HS. To simulate HS conditions, which at least somehow resembled a transient, naturally occurring HS, yet not killing the plants, several preliminary experiments had been carried out to identify conditions which are stressful to the plants (as indicated by the occurrence of a K-band in chlorophyll fluorescence measurements, cf. Oukarroum et al. 2007), but which were not lethal (as indicated by full resumption of growth and successful seed formation after the heat treatment). Additionally, HS treatment was applied in delayed time, at the stage of four leaves, to allow samples collection at the same developmental stage of DA treatment. Finally, the following protocol was developed: plants at the stage of four leaves were transferred into a transparent CERTOMAT H hood (Sartorius, Gottingen, Germany) under a light intensity of 200 µmol m−2 s−1. Temperature was increased gradually by one °C/20 min from 26 °C until 42 °C, to allow a limited adaptation of the plants on the level of protein synthesis. Plants were incubated for further 2 h at this temperature. Leaf temperature was checked by an infrared thermometer and air humidity was maintained by supplying water containers to the center of the hood. For combined stresses, plants at the end of DA treatment were subjected to the same heat treatment regime, but omitting the presence of air humidity supplies. The third leaves of three plants/pot were combined for protein extraction, while the corresponding leaf of the fourth plant was used to measure leaf relative water content, RWC, as described by Smart and Bingham (1974). Three biological replicates/treatment were used for protein extraction and for measuring RWC. Samples were frozen under liquid N2, ground with mortar and pestle and stored at −80 °C. Samples were collected between 4 and 5 p.m.
Protein extraction, 2D-DIGE and MS analysis
Aliquots of 100 mg ground and frozen tissue were transferred into 2 mL centrifuge tubes and pulverized using mixer mill MM400 (Retsch, Haan, Germany) at a frequency of 25 rpm/s for 30 s. Proteins were extracted using 10 % trichloroacetic acid in acetone containing 10 mM dithiothreitol, DTT, following the protocol of Mèchin et al. (2006). Proteins were resuspended in resuspension buffer (RB: 7 M urea, 2 M thiourea, 30 mM TRIS–HCl and 2 % CHAPS, pH 8.3), sonicated for 3 cycles, 5 s each at 35 % power output using the Sonopuls mini 20 sonicator and MS 1.5 probe (Bandelin, Berlin, Germany). Samples were clarified by two consecutive centrifugation steps at 19,000 g for 15 min each at 20 °C and protein concentration was measured (Bradford 1976). An aliquot of 50 µg protein/biological replicate was labeled with fluorescent Cy3, or Cy5 N-hydroxysuccinimide esters following the manufacturer’s recommendations (Lumiprobe, Hannover, Germany) according to the scheme in Table 1. The internal standard consisted of a mixed pool of all biological samples in the experiment at identical protein concentrations, labeled with Cy2. After labeling, volumes were adjusted to 170 µL/reaction mix with RB, an equal volume of 2 × loading buffer (RB containing 2 % DTT, 1 % IPG buffer 4–7) was added, and 18 cm IPG strips 4–7 were hydrated with the sample for the first dimension of the 2 D electrophoresis (overnight at 36 kVh). A parallel strip was rehydrated using 900 µg internal standard to isolate sufficient amounts of proteins for the Matrix-assisted laser desorption/ionization (MALDI) Orbitrap MS analysis. Following electrofocusing and prior to separation on a 18 cm 8–14 % gradient SDS-PAGE (Laemmli 1970), casted into low-fluorescence glass plates, strips were equilibrated for 15 min in 10 mL of SDS equilibration solution [ES: 6 M urea, 75 mM Tris–HCl pH 8.8, 29.3 % (v/v) glycerol, 2 % (w/v) SDS and 0.002 % (w/v) bromophenol blue] containing 1 % DTT, followed by additional 15 min in ES containing 2.5 % (w/v) iodoacetamide. After electrophoresis, gels were scanned using a Typhoon 9410 scanner (GE-Healthcare, Freiburg, Germany) prior to spot visualization by silver staining (Chevallet et al. 2006). One-way ANOVA test and T test analysis were carried out on images to distinguish the significance of up- and downregulation of protein spots using DeCyder 2-D Differential Analysis Software (GE Healthcare). The preparative gels with 900 µg internal standard proteins, run in parallel, were stained overnight using colloidal Coomassie blue stain (Neuhoff et al. 1988). Tryptic digestion was carried out on significantly differentially expressed protein spots with at least 1.5-fold threshold changes in relative fluorescence signal intensity and excised from polyacrylamide gels as described by Ashoub et al. (2013) and the included references. Extracted peptides were spotted on a MALDI target using α-cyano-4-hydroxy-cinnamic acid (CHCA, Bruker Daltonics, Bremen, Germany) as MALDI matrix (3 mg/ml in 70 % acetonitrile, 0.1 % trifluoroacetic acid). MS measurements were conducted using two different mass spectrometers to allow identification of the proteins when searching against huge, unreviewed NCBI databases since the barley proteome has neither been fully sequenced nor reviewed. Peptide mass fingerprint (PMF) spectra were acquired with a hybrid LTQ-Orbitrap XL mass spectrometer equipped with a MALDI ion source (Thermo Scientific, Bremen, Germany) due to its high mass accuracy, which is needed for identification via PMF. Twenty-five scans were recorded from each spot with fixed laser energy in positive ion mode between 820 and 4,000 m/z with a resolution of 30,000 at 400 m/z. For subsequent calibration, a tryptic BSA digest was additionally spotted onto the MALDI target to achieve a mass accuracy of ± 3 ppm. Spectra files obtained from the MALDI-Orbitrap measurements were deisotoped using the vendor’s software tool Xtract and saved as MGF files. Peptide mass fingerprint analysis was performed using the Mascot database search engine v2.4 (Matrix Science Ltd.) using the following settings: ±3 ppm peptide mass tolerance, tryptic enzyme specificity with a maximum of one missed cleavage, carbamidomethylation of cysteine residues as fixed modification and methionine oxidation as variable modification. PMF, spectra were searched against the NCBInr database restricted to Viridiplantae (green plants). Additionally, MS/MS measurements were performed to confirm the identifications via PMF. For those experiments, a 4800 MALDI TOF/TOF Analyzer (AB Sciex, Darmstadt, Germany) was chosen due to its superior fragmentation properties (Rietschel et al. 2009). Settings for MALDI-TOF/TOF–MS/MS measurements, spectra processing and data analysis were used as described by Ashoub et al. (2013) (Fig. 1).

Silver stained image of H. spontaneum leaf proteins and a magnification of spot areas (in boxes). First dimension: 18-cm IEFstrips pH 4–7, second dimension: 8–14 % gradient SDS-PAGE. Lines indicate up- or downregulated protein spots subjected to MALDI–MS analysis
Results and discussion
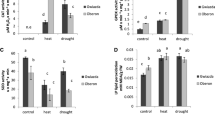
In this study, we used wild barley to identify leaf proteins that are specifically and commonly changed in response to DA and HS, applied alone, and to the simultaneous combination of both stresses. In cereals, most studies were conducted on cultivated plant species due to their commercial importance. However, less is known about the response of their wild relatives to stresses. Studies on (crossable) ancestors may yield invaluable information on available genetic material for improving cultivated cereals. The leaf RWC was used as a parameter to indicate the homogeneity of the water status of the biological replicates prior to protein analysis. RWC of drought and combined stressed plants were reduced to 76 ± 3.9 and 73 ± 1.2 % respectively, in comparison to 96 ± 1.1 % of control plants. In the HS plants, RWC (95 ± 1.6 %) was not significantly changed when compared to control plants. As an alternative to the conventional 2D PAGE, the 2D-DIGE was applied as a quantitative approach to detect protein changes of two samples within a single gel. By application of a specific combination scheme of biological replicates (Table 1), we were able to identify statistically significant differentially expressed spots among treatments. We further introduced the 1.5-fold change threshold of the fluorescence signal of the identified spots between treatments and control to consider protein alterations strong enough prior to be subjected to mass spectrometry (MS) analysis. Protein analysis indicated that out of 1,292 predicted spots by DeCyder 2-D Differential Analysis Software, 103 were statistically differentially expressed in regard to DA, 54 spots in regard to HS and 104 spots in regard to the combination of both stresses. Out of these, 63 proteins underwent an alteration by at least 1.5-fold and were subjected to MS and analysis database search. To confirm protein identification, MS was performed using two different mass spectrometers (cf. Methods section). Peptides mass fingerprint (PMF) and MS/MS fragment spectra were obtained from the same samples. Out of the 63 analysed proteins, only those 45 protein spots that passed the PMF score threshold value and/or were verified by MS/MS are considered in this work. The PMF data for all the identified 63 spots are presented in supplementary Table 1. Proteins identified by MS including their accession numbers, names (where applicable), their up and downregulation levels and their putative functions were categorized to groups in regard to the applied stress and are summarized in Table 2. Out of the confirmed spots, DA resulted in a specific upregulation of fourteen protein spots while six protein spots were downregulated. HS resulted in a specific upregulation of seven protein spots and downregulation of one protein spot. As a common response to both stresses, four protein spots were upregulated and eight protein spots were downregulated. The unique response of combined stresses (i.e. not found under one stress alone) consisted of the upregulation of one protein spot and downregulation of four protein spots.
Proteome alteration in response to stress conditions
Specific responses to drought acclimation
Under drought stress, stomata are closed to reduce water loss. This results in the decrease of carbon dioxide uptake, reduction of photosynthesis efficiency, an increase in excess excitation energy, and, ultimately, in the accumulation of several ROS and methylglyoxal, which negatively affect proteins, lipids and DNA functions. Under these conditions, repair and detoxification mechanisms are activated to prevent damage (Smirnoff, 1993). DA treatment resulted in the upregulation of proteins involved in cell detoxification. For instance, the putative lactoylglutathione lyase (#41) glutathione S-transferase (#47) were upregulated. They act as components of the glyoxalase pathway and in antioxidant formation. On the level of lipids protection, tocopherol cyclase (#28) was upregulated. Tocopherols are lipophilic antioxidants and protect against lipid-soluble byproducts of oxidative stress (Smirnoff, 1993). In mitochondria the mitochondrial NADH-ubiquinone oxidoreductase (#10) and mitochondrial formate dehydrogenase (#33) were upregulated. The mitochondrial formate dehydrogenase plays a detoxifying role (Li et al., 2002) and 75 kDa mitochondrial NADH-ubiquinone oxidoreductase belongs to the ubiquinone cycle, which plays a role in the plant mitochondrial electron transport chain. It is reduced to ubiquinol which acts as a scavenger of ROS and to the superoxide radical which is produced during mitochondrial electron transport in plants (Rich and Bonner 1978). On the other hand, during drought stress, plant water content is reduced which requires osmotic adjustment to maintain the metabolic activities of the cells. Betaine aldehyde dehydrogenase (#24) was upregulated during DA. It converts betaine aldehyde to glycine betaine, which mediates osmotic adjustment and is suggested to have positive effects on enzyme and thylakoid membrane integrity and in maintaining photosynthesis efficiency (Ashraf and Foolad 2007). Although its actual role in plant osmotolerance remains to be elucidated, it is thought to play a role in detoxifying free radicals by forming stable complexes with them (Bose et al. 2014).
DA resulted in the upregulation of enzymes which play a role in the biosynthesis of sulfur compounds. Methionine synthase (#8) as well as S-adenosylmethionine synthase 2 and 3 (#29, #31) were upregulated. Methionine synthase is involved in the synthesis of the amino acid methionine, a key amino acid in protein synthesis and in gene regulation through DNA methylation. It is also involved in the S-adenosylmethionine biosynthesis and regeneration cycle. S-adenosylmethionine synthase is responsible for the formation of S-adenosylmethionine, the methyl donor for transmethylation reactions of proteins, nucleic acids, polysaccharides and fatty acids and the propylamino donor in polyamine biosynthesis. The metabolite is also involved in the control of gene expression, cell proliferation and the production of vitamins important for the development of stress tolerance reactions (Amir 2008). Among proteins involved in lipid metabolism, lipoxygenase 6 (#3, #4) was upregulated. Lipoxygenases are common enzymes in plants, involved in a number of diverse features including growth and development, pest resistance, and senescence or responses to wounding. It was suggested that ethylene, a regulatory signal under drought stress is released by lipid peroxidation through lipoxygenase activity (Smirnoff 1993). Grebner et al. (2013) reported that lipoxygenase 6-derived oxylipins are important for the responses to stresses in Arabidopsis.
Drought affects the cellular homeostasis and causes protein dysfunction. Therefore, cells utilize efficient mechanisms to allow proteins to maintain their functional conformation and to prevent the aggregation of denatured proteins under stress. During DA, protein disulfide isomerase (#21) was upregulated. It is involved in catalyzing protein folding by formation and breakage of disulfide bonds between cystein residues within proteins. Heat shock proteins (HSP) 70-4L (#2), with an increased Mr of 94 kDa, was distinctively upregulated. It is distinguished from the cognate HSP 70 species that are constitutively expressed in plants under normal conditions and alter their abundance in response to heat (#16) or combined stress (#12, #14) or from the HSP 70 form which is upregulated as a common response to both stresses (#13, #15). The members of the HSP 70 family play important roles in preventing aggregation and supporting the refolding of non-native proteins under normal and stress conditions (Frydman 2001). They are also involved in organelle protein import and translocation processes and in the proteolytic degradation of unstable proteins by targeting the proteins to lysosomes or proteasomes (Hartl 1996). The overexpression of HSP 70 genes correlates positively with the acquisition of thermotolerance and results in enhanced tolerance to salt, water deficiency and high-temperature stress in plants (Wang et al. 2004). On the other hand, chloroplast proteins related to nitrogen metabolism [ferredoxin-nitrite reductase (#22), chloroplastic ferredoxin-NADP reductase (#40), glutamine synthetase isoform GS1-2 (#37) were downregulated during DA. Ferredoxin-nitrite reductase and glutamine synthetase are involved in nitrogen metabolism. Since plants under abiotic stress show a deficiency in nitrogen metabolism (Bernard and Habash 2009), we assume that the observed effect of DA was due to a decrease of nitrogen uptake from the soil under drought. In addition, proteins involved in photosynthesis [ATP synthesis: ATP synthase CF1 α (#23) and carbon metabolism: ribulose-bisphosphate carboxylase (Rubisco) activase (RA), large isoform (#30)] were downregulated. Their function is further discussed in the part of combined stress response.
Specific responses to heat stress
Heat negatively affects protein stability and enzyme functions of plants. As a response, plants produce HSPs and proteins with chaperon functions to restore the correct configuration of proteins, to prevent aggregation and to degrade damaged proteins (Wang et al. 2004). In this study HSP 90 (#6), a cognate HSP 70 species (#16), the chloroplast 26 kDa HSP (#45), sHSPs with Mr of 17.6 kDa (#54) and a protein involved in photosynthesis maintenance, chloroplastic ATP-dependent zinc metalloprotease FTSH (#11) were upregulated in response to HS. HSP 90 species have a general role in the refolding of misfolded proteins that can accumulate during plant development or in response to various biotic and abiotic stress conditions, sensing the environment and mediating appropriate phenotypic plasticity (Sangster and Queitsch 2005). sHSPs confer thermotolerance by selectively binding and stabilizing proteins, preventing their aggregation at elevated temperatures, protecting enzymes against heat induced inactivation and facilitating protein refolding by other chaperones (Ganea 2001). Chloroplastic ATP-dependent zinc metalloprotease FTSH 1 is involved in the removal of unassembled or oxidatively damaged D1 proteins from photosystem II, PSII (Adam et al. 2001) and has been suggested to contribute to heat tolerance in grapevine (Rocheta et al. 2014).
In addition, chloroplast 30S ribosomal protein 1 (#42) was upregulated. It is involved in protein synthesis which might reflect the need to produce more proteins with various functions under heat stress conditions.
Common responses to DA and heat stress
Members of the HSP 70 family were upregulated under both treatments. Cognate HSP 70 (#12 and #14), and HSP70 (#13 and #15) were upregulated. On the other hand, proteins involved in photosynthesis were affected as a common response to both DA and HS treatment: rubisco activase (RA) small isoform (# 32, # 35 and #36), ATP synthase β (#25, #26), and type I chlorophyll a/b-binding protein (#48, #49) as well as chloroplast 30S ribosomal protein 1 (#43) were downregulated. In sunflower, photosynthetic assimilation of CO2 is reduced in response to the loss of ATP synthase activity under moderate drought and additionally by decreasing Rubisco activity under severe drought stress conditions (Tezara et al. 1999). RA, which regulates Rubisco activity, is sensitive to high temperature. Therefore, inactivation (and degradation) of RA provides a probable biochemical explanation for the inactivation of Rubisco and thus photosynthesis at high temperatures (Feller et al. 1998) as well as under drought stress. Seki et al. (2002) reported the downregulation of 13 Arabidopsis thaliana chlorophyll a/b-binding proteins transcripts in response to drought, cold and high salinity conditions. High temperature leads to the dissociation of the inner antenna pigment protein complexes from the central core of the PSII (Georgieva 1999), thus making the chl a/b binding proteins accessible to degradation.
Specific responses to combined stresses
While, in general, heat or drought responsive proteins showed the same behavior under the combined stresses, we also identified alteration of certain proteins, whose response was unique for the combined stresses and included some barley predicted proteins of yet unknown function (Table 2). One protein spot (#1) was upregulated and several proteins with mostly yet unidentified functions (#20, #38, #55, and #56) were downregulated, whose sequences are homologous to predicted proteins in H. vulgare or other plant species.
Comparative barley proteome analyses
Proteins detected by barley proteome analyses in other studies in response to stresses differ according to the genotype under study and degree and period of stress. Nevertheless, a comparison with results for H. vulgare under drought stress in soil or poly ethylene glycol treatments (Ashoub et al. 2013; Kauser et al. 2013) or under combined drought and heat stresses (Rollins et al. 2013) reveals a common response between both species. For example, methionine synthetase, HSP70 species, glutathione transferase, S-adenosylmethionine synthase, lipoxygenase, disulfide isomerse, betaine aldehyde dehydrogenase were upregulated in response to drought stress in all cited studies, while RA was downregulated. On the other hand, ATP synthase α subunit was downregulated in response to drought stress or combined drought treatment and HS in H. spontaneum, but showed upregulation in one genotype of H. vulgare. Several proteins involved in detoxification mechanisms, glyoxylase I, tocopherol cyclase, were not detected in the three cited studies. HSP 70, HSP 90 and ATP-dependent zinc metalloprotease were detected in response to HS by Rollins et al. (2013), but they reported no detected changes of sHSPs. Apart from genetic differences between H. spontaneum and H. vulgare, this difference may be due to the different HS temperatures applied (42 °C in the present study vs. 36 °C in Rollins et al. 2013).
Conclusion
Several studies have been conducted on cultivated barley in response to individual abiotic stress conditions. However, only a limited number of studies have been carried out on combined stresses or on its wild relative. To our knowledge, this is the first study that provides information about the proteome changes of Mediterranean wild barley in response to combined drought and heat stresses. Exposure to a prolonged non-lethal drought stress selectively stimulated the over expression of detoxification enzymes which ameliorate survival under stomatal closure and the subsequent production of reactive oxygen species. Production of HSPs or other proteins with chaperon function was stimulated under both types of stresses. In addition to chaperon species commonly expressed under both stress conditions, stress specific chaperons could be identified. A short time of heat exposure to 42 °C was sufficient to activate the production of a wide range of HSPs as a protection mechanism for other proteins. Both stress conditions showed a negative impact on ATP generation and photosynthesis, as suggested by the downregulation of ATP synthase and Rubisco activase. The combined stresses resulted in unique protein expression alterations which could not be detected by studying each individual stress.
Abbreviations
- BADH:
-
Betaine aldehyde dehydrogenase
- DIGE:
-
Difference gel electrophoresis
- DA:
-
Drought acclimation
- ES:
-
Equilibration solution
- HSPs:
-
Heat shock proteins
- HS:
-
Heat stress
- RWC:
-
Leaf relative water content
- MS:
-
Mass spectrometry
- MALDI:
-
Matrix-assisted laser desorption/ionization
- PMF:
-
Peptide mass fingerprint
- PSII:
-
Photosystem II
- ROS:
-
Reactive oxygen species
- RB:
-
Resuspension buffer
- RA:
-
Rubisco activase
- SHSPs:
-
Small heat shock proteins
- 2D PAGE:
-
Two dimensional polyacrylamide gel electrophoresis
- VWC:
-
Volumetric water content
References
Adam Z, Adamska I, Nakabayashi K et al (2001) Chloroplast and mitochondrial proteases in Arabidopsis. A proposed nomenclature. Plant Physiol 125:1912–1918
Alam I, Sharmin SA, Kim KH, Yang JK, Choi MS, Lee BH (2010) Proteome analysis of soybean roots subjected to short-term drought stress. Plant Soil 333:491–505
Amir R (2008) Towards improving methionine content in plants for enhanced nutritional quality. Funct Plant Sci Biotechnol 2:36–46
Amme S, Matros A, Schlesier B, Mock HP (2006) Proteome analysis of cold stress response in Arabidopsis thaliana using DIGE-technology. J Exp Bot 57:1537–1546
Ashoub A, Beckhaus T, Berberich T, Karas M, Brüggemann W (2013) Comparative analysis of barley leaf proteome as affected by drought stress. Planta 237:771–781
Ashraf M, Foolad MR (2007) Roles of glycine betaine and proline in improving plant abiotic stress resistance. Environ Exper Bot 59:206–216
Baerenfaller K, Massonnet C, Walsh S et al (2012) System-based analysis of Arabidopsis leaf growth reveals adaptation to water deficit. Mol Syst Biol 8:606
Bech-Serra JJ, Borthwick A, Colomé N, Consortium PR, Albar JP, Wells M, del Sánchez Pino M, Canals F (2009) A multi-laboratory study assessing reproducibility of a 2D-DIGE differential proteomic experiment. J Bimol Tech 20:293–296
Bernard SM, Habash DZ (2009) The importance of cytosolic glutamine synthetase in nitrogen assimilation and recycling. New Phytol 182:608–620
Bose J, Rodrigo-Moreno A, Shabala S (2014) ROS homeostasis in halophytes in the context of salinity stress tolerance. J Exp Bot 65:1241–1257
Bradford MM (1976) Rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72:248–254
Budak H, Akpinar BA, Unver T, Turktas M (2013) Proteome changes in wild and modern wheat leaves upon drought stress by two-dimensional electrophoresis and nanolc-esi–ms/ms. Plant Mol Biol 83:89–103
Caruso G, Cavaliere C, Foglia P, Gubbiotti R, Guarino C, Samperi R, Laganà A (2009) Analysis of drought responsive proteins in wheat (Triticum sativumdurum) by 2D-PAGE and MALDITOF mass spectrometry. Plant Sci 177:570–576
Chevallet M, Luche S, Rabilloud T (2006) Silver staining of proteins in polyacrylamide gels. Nat Protoc 1:1852–1858
Choo TM (2002) Genetic resources of Tibetan barley in China. Crop Sci 42:1759–1760
Ellis R, Foster B, Handley L, Gordon D, Russell J, Powell W (2000) Wild barley: a source of genes for crop improvement in the 21st century? J Exp Bot 51:9–17
Fatehi F, Hosseinzadeh A, Alizadeh H, Brimavandi T, Struik PC (2012) The proteome response of salt-resistant and salt-sensitive barley genotypes to long-term salinity stress. Mol Biol Rep 39:6387–6397
Feller U, Crafts-Brandner S, Salvucci ME (1998) Moderately high temperatures inhibit ribulose-1,5-bisphosphate carboxylase/oxygenase (Rubisco). Activase-mediated activation of Rubisco. Plant Physiol 116:539–546
Frydman J (2001) Folding of newly translated proteins in vivo: the role of molecular chaperones. Annu Rev Biochem 70:603–647
Ganea E (2001) Chaperone-like activity of alpha-crystallin and other small heat shock proteins. Curr Protein Pept Sci 2:205–225
Georgieva K (1999) Some mechanisms of damage and acclimation of the photosynthetic apparatus to high tempreture. Bulg J Plant Physiol 25:89–99
Grebner W, Stingl NE, Oenel A, Mueller MJ, Berger S (2013) Lipoxygenase 6-dependent oxylipin synthesis in roots is required for abiotic and biotic stress resistance of Arabidopsis thaliana. Plant Physiol 161:2159–2170
Guo P, Baum M, Grando S, Ceccarelli S, Bai G, Li R, von Korff M, Varshney RK, Graner A, Valkoun J (2009) Differentially expressed genes between drought-tolerant and drought-sensitive barley genotypes in response to drought stress during the reproductive stage. J Exp Bot 60:3531–3544
Harlan JR, Zohary D (1966) Distribution of wild wheats and barley. Science 153:1074–1080
Hartl FU (1996) Molecular chaperones in cellular protein folding. Nature 381:571–580
Imin N, Kerim T, Rolfe BG, Weinman JJ (2004) Effect of early cold stress on the maturation of rice anthers. Proteomics 4:1873–1882
IPCC, Kundzewicz ZW, Palutikof J, Wu S (2008) Climate change and water. Technical paper of the Intergovernmental Panel on Climate Change. Cambridge University Press, Cambridge
Kausar R, Arshad M, Shahzad A, Komatsu S (2013) Proteomics analysis of sensitive and tolerant barley genotypes under drought stress. Amino Acids 44:345–359
Laemmli UK (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680–685
Lambers H, Chapin FS III, Pons TL (2008) Assumptions and approaches. In: Lambers H, Chapin FS III, Pons TL (eds) Plant physiological ecology, 2nd edn. Springer, Heidelberg, pp 1–9
Li R, Moore M, Bonham-Smith PC, King J (2002) Overexpression of formate dehydrogenase in Arabidopsis thaliana resulted in plants tolerant to high concentrations of formate. J Plant Physiol 159:1069–1076
Mèchin V, Damerval C, Zivy M (2006) Total protein extraction with TCA-acetone. Methods Mol Biol 355:1–8
Ndimba BK, Chivasa S, Simon WJ, Slabas AR (2005) Identification of Arabidopsis salt and osmotic stress responsive proteins using two-dimensional difference gel electrophoresis and mass spectrometry. Proteomics 5:4185–4196
Neuhoff V, Arold N, Taube D, Ehrhardt W (1988) Improved staining of proteins in polyacrylamide gels including isoelectric focusing gels with clear background at nanogram sensitivity using Coomassie Brilliant Blue G-250 and R-250. Electrophoresis 9:255–262
O’Farrell PH (1975) High resolution two-dimensional electrophoresis of proteins. J Biol Chem 250:4007–4021
Oukarroum A, El Madidi S, Schansker G, Strasser RJ (2007) Probing the responses of barley cultivars (Hordeum vulgare L.) by chlorophyll a fluorescence OLKJIP under drought stress and re-watering. Environ Exper Bot 60:438–446
Rich PR, Bonner WD Jr (1978) The sites of superoxide anion generation in higher plant mitochondria. Arch Biochem Biophys 188:206–213
Rietschel B, Baeumlisberger D, Arrey TN, Bornemann S, Rohmer M, Schuerken M, Karas M, Meyer B (2009) The benefit of combining nLC-MALDI-Orbitrap MS data with nLC-MALDI-TOF/TOF data for proteomic analyses employing elastase. J Proteome Res 8:5317–5324
Rizhsky L, Hongjian L, Mittler R (2002) The combined effect of drought stress and heat shock on gene expression in tobacco. Plant Physiol 130:1143–1151
Rizhsky L, Liang HJ, Shuman J, Shulaev V, Davletova S, Mittler R (2004) When defense pathways collide. The response of Arabidopsis to a combination of drought and heat stress. Plant Physiol 134:1683–1696
Rocheta M, Becker JD, Coito JL, Carvalho L, Amâncio S (2014) Heat and water stress induce unique transcriptional signatures of heat-shock proteins and transcription factors in grapevine. Funct Integr Genomics 14:135–148
Rollins J, Habte E, Templer S, Colby T, Schmidt J, von Korff M (2013) Leaf proteome alterations in the context of physiological and morphological responses to drought and heat stress in barley (Hordeum vulgare L.). J Exp Bot 64:3201–3212
Rosegrant M, Cline S (2003) Global food security: challenges and policies. Science 302:1917–1919
Roth U, von Roepenack-Lahaye E, Clemens S (2006) Proteome changes in Arabidopsis thaliana roots upon exposure to Cd +2 . J Exp Bot 57:4003–4013
Sangster TA, Queitsch C (2005) The HSP90 chaperone complex, an emerging force in plant development and phenotypic plasticity. Curr Opin Plant Biol 8:86–92
Seki M, Narusaka M, Ishida J et al (2002) Monitoring the expression profiles of 7000 Arabidopsis genes under drought, cold and high-salinity stresses using a full-length cDNA microarray. Plant J 31:279–292
Shah NH, Paulsen GM (2003) Interaction of drought and high temperature on photosynthesis and grain-filling of wheat. Plant Soil 257:219–226
Skylas DJ, Cordwell SJ, Hains PG, Larsen MR, Basseal DJ, Walsh BJ, Blumenthal C, Rathmell W, Copeland L, Wrigley CW (2002) Heat shock of wheat during grain filling: proteins associated with heat-tolerance. J Cereal Sci 35:175–188
Smart RE, Bingham GE (1974) Rapid estimates of relative water content. Plant Physiol 53:258–260
Smirnoff N (1993) The role of active oxygen in the response of plants to water deficit and desiccation. New Phytol 125:27–58
Suzuki N, Rivero RM, Shulaev V, Blumwald E, Mittler R (2014) Abiotic and biotic stress combinations. New Phytol 203:32–43
Tezara W, Mitchell VJ, Driscoll SD, Lawlor DW (1999) Water stress inhibits plant photosynthesis by decreasing coupling factor and ATP. Nature 401:914–917
Tommasini L, Svensson JT, Rodriguez EM, Wahid A, Malatrasi M, Kato K, Wanamaker S, Resnik J, Close TJ (2008) Dehydrin gene expression provides an indicator of low temperature and drought stress: transcriptome-based analysis of Barley (Hordeum vulgare L.). Funct Integr Genomics 8:387–405
Wang W, Vinocur B, Altman A (2003) Plant responses to drought, salinity and extreme temperatures: towards genetic engineering for stress tolerance. Planta 218:1–14
Wang W, Vinocur B, Shoseyov O, Altman A (2004) Role of plant heat-shock proteins and molecular chaperones in the abiotic stress response. Trends Plant Sci 9:244–252
Yan S, Tang Z, Su W, Sun W (2005) Proteomic analysis of salt stress-responsive proteins in rice root. Proteomics 5:235–244
Acknowledgments
This work was supported by a DAAD (German Academic Exchange Service) grant to A. Ashoub under the Alumni Re-Invitation Program.
Conflict of interest
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Ashoub, A., Baeumlisberger, M., Neupaertl, M. et al. Characterization of common and distinctive adjustments of wild barley leaf proteome under drought acclimation, heat stress and their combination. Plant Mol Biol 87, 459–471 (2015). https://doi.org/10.1007/s11103-015-0291-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11103-015-0291-4