
Abstract
MicroRNAs (miRNAs) play critical regulatory roles mainly through cleaving their target mRNAs or repressing gene translation during plant development. Grapevines are among the most economically important fruit crops with available whole genome sequences. Studies on grapevine miRNAs (Vv-miRNAs) are also widely available. However, studies on the regulation mode of Vv-miRNAs on their target mRNAs during grapevine development have not been studied well, especially at the transcriptome-wide level. Here, six small RNA and mRNA libraries from various grapevine tissues were constructed for Illumina and Degradome sequencing. Subsequently, we systematically analyzed the spatiotemporal variations in the regulation of the target genes of regulation of Vv-miRNAs. In total, 242 known and 132 novel Vv-miRNAs and 193 target mRNAs were identified, including 103 target mRNAs for known and 90 target mRNAs for novel miRNAs, were validated in one or more of the tissues examined. More than 50 % of novel miRNAs were expressed exclusively in the flowers and berries, where they cleaved their target genes in a tissue-specific manner, especially, the breadth of their cleavage sites in flower tissues. Moreover, six novel miRNAs in berries responded to exogenous gibberellin and/or ethylene under a quantitative real time RT-PCR analysis, which confirmed their regulatory functions during berry development. Up to 93.6 % of the known miRNAs were highly conserved in various tissues, where their expression levels exhibited dynamic variations during grapevine development. Significantly, some Vv-miRNA families had one key member that acted as the main regulator of their target genes during grapevine development.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
MicroRNAs (miRNAs) are short, single-stranded, non-coding RNAs that regulate the expression of their target genes mainly through cleavage and/or translation inhibition of the target mRNA during or after transcription. Numerous studies have indicated that miRNAs play important roles in plant development and responses to environmental stresses (Bartel 2004; Mallory and Vaucheret 2004; Carrington and Ambros 2003; Leung and Sharp 2010). Although many miRNAs have been discovered and identified using bioinformatics and/or experimental approaches in various plant species (Jaillon et al. 2007; Sunkar et al. 2008; Fahlgren et al. 2007; Moxon et al. 2008a, b; Carra et al. 2009; Pantaleo et al. 2010; Wang et al. 2011a), the number of identified tissue-, species-, and developmental stage-specific miRNAs is still limited because of their low accumulations levels compared with conserved miRNAs, thereby hindering detection through traditional sequencing. The advent of high-throughput sequencing technologies has allowed the mining of these specific low-abundance miRNAs (Sunkar et al. 2008; Pantaleo et al. 2010; Szittya et al. 2008; Song et al. 2010; Mica et al. 2010; Wang et al. 2011b). Although many grapevine miRNAs (Vv-miRNAs) have been identified through deep sequencing (Pantaleo et al. 2010; Mica et al. 2010; Wang et al. 2011b), they only comprise a small fraction of the potential grapevine miRNA population and more novel Vv-miRNAs in diverse tissues or developmental stages still need to be mined and investigated to gain further insights into the functions of grapevine miRNAs and their modes of action during development on a transcriptome-wide scale.
In plants, the high miRNA:mRNA complementarity and slicing of target mRNAs has made Degradome sequencing a highly efficient and suitable approach for functional miRNA analysis and for validating miRNA targets (Pantaleo et al. 2010; Allen et al. 2010). Although 5′ RNA ligase mediated amplification of cDNA ends (5′-RLM-RACE) can also be used to verify target genes by detecting miRNA cleavage sites on target mRNAs and the cleavage products, it requires prior prediction of target genes, it has a low throughput as an identification method, and it is excessively time consuming and labor intensive (Thomson et al. 2011). By contrast, Degradome sequencing employs new next-generation deep sequencing approaches. Thus, it is capable of detecting target cleavage products having low accumulation levels and generating mass data without the need to predict the target genes, which can provide important information for the large-scale verification of target mRNAs. Degradome sequencing of mRNA libraries from various tissues and developmental stages can reveal the types and numbers of target genes, as well as the frequencies of the miRNA cleaved targets and their cleavage sites on a transcriptome-wide scale. The sequencing results can then be analyzed for dynamic variations in the regulation modes of miRNAs on their target genes during plant development.
Grapevine is a widely grown fruit crop with a high nutritional value and economic importance through direct berry consumption and beverage production; it has long played an important role in the human diet and health. Grapevines are also a preferable experimental system for molecular studies because it is the first fruit crop to have its entire genome sequenced (Jaillon et al. 2007; Velasco et al. 2007) and it has the fruit with the highest number of publicly available expressed sequence tag (EST) sequences. Several studies on Vv-miRNAs and their target genes have been carried out (Jaillon et al. 2007; Wang et al. 2011a, b; Lu et al. 2008; Lazzari et al. 2009). Despite these advances in Vv-miRNA research, all the identified Vv-miRNAs are derived from one or a few tissues or stages of Vitis vinifera or V. vinifera × Vitis labrusca hybrids (hVVs), with only a limited number of their target genes have been validated experimentally. This finding suggests the need for more systematic studies on more Vv-miRNAs, their target genes, and their modes of action in a wider array of tissues and/or growing-stages.
Although V. vinifera PN40024 is used as the reference genome for grapevines, it is highly homologous to the hVV grapevine genome, and thus we could obtain a large number of valuable information from the hVV grapevine using the PN40024 reference genome even though some of the V. labrusca miRNA information is lost. In our previous studies, we identified Vv-miRNAs through deep sequencing of one small RNA library from a mixture of flowers and berries (Wang et al. 2011b). Moreover, based on our previous results, we selected 18 Vv-miRNAs from flowers and berries and then validated and analyzed the expression of their targets using RLM-RACE and poly(A) polymerase-mediated 3′ rapid amplification of cDNA ends (PPM-RACE) technology during the developmental stages of hVV grapes (Wang et al. 2013). We found that the accumulation abundance of the cleavage products of various miRNA from the same family differed during different stages of grapevine development. However, the mechanisms by which Vv-miRNA regulate their targets in different grapevine tissues and developmental stages are still unclear at the transcriptome-wide level. Such studies on the dynamic variations of regulatory modes of Vv-miRNAs are necessary for understanding the functions of Vv-miRNAs and their target genes.
To adequately characterize the variations in regulatory roles of Vv-miRNA, we systematically studied the variations in the modes of action of Vv-miRNAs on their target mRNAs using Illumina and Degradome sequencing based on our earlier two works (Wang et al. 2011b, 2013). We deep sequenced six small RNA and mRNA libraries (sRNA and mRNA libraries) from the young and mature leaves, inflorescences, flowers, and young and mature berries of the grapevine cv. ‘Summer Black’ (hVV; Fig. 1a). Bioinformatics analysis was then performed to analyze the dynamic variations in the regulatory roles of various Vv-miRNAs on their target genes at the transcriptome scale and comprehensively determine the regulatory roles of miRNAs. Our findings provide information on the functional genomics of grapevines, especially their miRNA-based regulatory systems, and it will help us better understand grapevine growth and development.

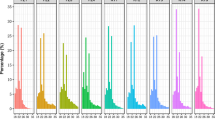
Sequencing flow chart, length distribution of small RNAs, categories and percentages of small RNAs and frequency of redundant and unique miRNAs from grapevine tissue sRNA libraries. a Flow chart of small RNA and mRNA library construction and sequencing respectively. The plant materials included young leaf, mature leaf, inflorescence, flower, young berry and mature berry. b Length distribution of small RNAs in grapevine six tissues above. c Categories and percentages of small RNAs in grapevine six tissues above. d Frequency of redundant and unique miRNAs from grapevine tissue sRNA libraries, YL, ML, IF, FL, YB and MB represent young leaves, mature leaves, inflorescence, flowers, young berries and mature berries, respectively
Materials and methods
Plant materials
Young leaves (10 days after leaf expansion), mature leaves (40 days after leaf expansion), inflorescences (10 days after emergence), fully open flowers, young berries (10 days after flowering), and large berries (70 days after flowering) were collected and used in deep sequencing in 2012, from 5-year-old table grapevine ‘Summer Black’ (hVV) grown at the grapevine resource garden of the Jiangsu Academy of Agricultural Sciences, China. Moreover, the inflorescences were treated twice with 50 mg/L gibberellic acid (GA) 10 days before flowering and at the full bloom stages, respectively; the berries were treated with 100 mg/L ethylene at 7 weeks after flowering (1 week before veraison). Meanwhile, the clear water used as control. Young berries (1 week after flowering, AF), moderate berries (5 weeks AF), and big berries (9 weeks AF) in GA treatments and controls were collected, and the “A” big berries (8 weeks AF), “B” big berries (9 weeks AF), “C” big berries (10 weeks AF), and “D” big berries (11 weeks AF) in the ethylene treatments and controls were also collected. These samples from treatments and controls were used in quantitative real time RT-PCR (qRT-PCR) assays. Each type of samples had three replicates during deep sequencing and qRT-PCR. After collection, all the samples were immediately frozen in liquid nitrogen and stored at −80 °C until use.
Preparation of sRNA and Degradome libraries for high-throughput sequencing
Small RNA library construction and sequencing was performed as described by Wang et al. (2011b). Briefly, total RNA was extracted from different ‘Summer Black’ grapevine tissues via phenol/chloroform extraction (Wang et al. 2011a). Then, 10 M LiCl was used to separate the low-molecular-weight RNA from the high molecular weight RNA following previously reported procedures (Wang et al. 2011a). The 15 nt–30 nt sRNA fractions were isolated from the low-molecular-weight RNA through excision from 15 % denaturing polyacrylamide gels. The sRNAs were then ligated to Illumina adaptors (Illumina, http://www.illumina.com) without dephosphorylation and rephosphorylation. The short RNAs were converted into cDNA via RT-PCR following the Illumina protocol. The libraries from the six tissues were sequenced via a Genome Analyzer II (Illumina). The larger molecular weight RNA samples from the six tissues were used to prepare libraries for Degradome sequencing following the procedures described by Addo-Quaye et al. (2008, 2009), and the amplified Degradome tag libraries were sequenced on an Illumina genome analyzer.
Bioinformatics analysis of Vv-miRNAs identified in grapevine small RNA libraries
Small RNA sequences were extracted from the raw reads that matched both the last 7 nt of the 5′-adaptor and the first 7 nt of the 3′-adaptor, as described by Moxon et al. (2008a, b). The adaptors and vector sequences of all sRNA sequences from 15 nt to 30 nt were removed, and the modified sequences were then mapped to the grapevine genome. The sequences that matched the genome were used to search the Rfam database (http://www.sanger.ac.uk/software/Rfam) and the GenBank noncoding RNA database (http://www.ncbi.nlm.nih.gov/) by BLASTN analysis to remove the rRNA, tRNA, small nuclear (snRNA), small nucleolar (snoRNA), other non-coding RNA, and all sequences that contained poly(A) tails. The remaining sequences were then compared against known plant miRNAs in the miRBase 18.0. Only matching sequences (0–3 base mismatches) were considered as known Vv-miRNAs, whereas other sequences similar to known Vv-miRNAs but have one single nucleotide polymorphism (SNP) with the known ones validated were considered potential new members of known miRNA families.
After identifying known Vv-miRNAs, the fold-back structure of the remaining sequences were predicted by miRCat (http://srna-tools.cmp.uea.ac.uk/) (Moxon et al. 2008a, b) using default parameters. The small RNA sequences that have stem loop precursors were regarded as putative novel Vv-miRNAs. We also searched the sRNA libraries for Vv-miRNA* sequences (complementary to Vv-miRNA in the precursor molecule). Only sequences with miRNA–miRNA* duplexes were regarded as novel Vv-miRNAs. The counts of all Vv-miRNAs identified from the various libraries were normalized according to the method reported by Pantaleo et al. (2010).
qRT-PCR validation of berry-specific miRNA expression
The miRNA-enriched libraries from grapevine berries treated with GA or ethylene were used as templates for qRT-PCR. To amplify the Vv-miRNAs from the reverse-transcribed cDNAs, we used the precise Vv-miRNA sequence as the forward primer and the mirRacer 3′ primer as the reverse primer (Wang et al. 2011a). qRT-PCR was conducted using a Rotor-Gene 3000 (Corbett Robotics, Australia) and Rotor-Gene software version 6.1 (Wang et al. 2004). For each reaction, 1 μL of diluted cDNA (equivalent to about 100 pg of total RNA) was mixed with 10 μL of 2× SYBR green reaction mix (SYBR® Green qRT-PCR Master Mix; Toyobo, Osaka, Japan), and 5 pmol each of the forward and reverse primers were added to a final volume of 20 μL. The conditions for the PCR amplification were as follows: initial denaturation at 95 °C for 2 min, followed by 50 cycles of denaturation at 95 °C for 30 s, annealing at 60 °C for 20 s, and extension at 72 °C for 20 s. The relative expression levels were determined via comparative quantification, as previously described (Wilson et al. 2005). The 5.8S rRNA was used as a reference gene in the qRT-PCR detection of miRNAs. All quantitative data were exported as Excel data in the Rotor-Gene 3000 (Corbett Robotics, Australia), and were further analyzed with an R 2 above 0.998 using the LinRegPCR program (Ramakers et al. 2003).
Degradome data analysis and identification of miRNA target mRNAs
The Degradome data was analyzed using the CLEAVELAND pipeline (Addo-Quaye et al. 2008, 2009). The predicted small RNA target was run against the transcriptome of interest, wherein we predicted the transcript set of V. vinifera mRNA v1.fa from the Genoscope FTP site (http://www.cns.fr/externe/Download/Projets/Projet_ML/data). The alignment scores were calculated for each hit up to a user-defined cutoff, the full RNA–RNA alignments were printed, and the cleavage site associated with each prediction was calculated. The cleavage site was simply the 10th nucleotide complementary to the aligned small RNA under a bioinformatics comparison among miRNAs, the target mRNAs, and target cleavage products. No alignments were retained for randomized queries. However, concise records of each predicted target for the random queries were retained, including the predicted cleavage sites.
Predicting the functions of Vv-miRNA target mRNAs
To determine the potential functions of Vv-miRNAs and their target genes and their possible involvement in biological processes, assignment of putative functions to target genes were validated for Vv-miRNAs, which was run locally to BLASTx against the NCBI database to search for similarity. The putative functions of these genes were determined based on their annotation in other plant species. The Gene Ontology and Kyoto Encyclopedia of Genes and Genomes ontology were analyzed to identify miRNAs and their target genes involved in biological processes and molecular functions, as previously described by Xie et al. (2011).
Results
Variations in sRNA populations in various sRNA libraries
Out of 70,034,868 raw reads sequenced from the six sRNA libraries from the young leaves, mature leaves, inflorescences, and young and mature berries of grapevines, 68,895,376 clean reads were obtained after removing the adaptor and low-quality sequences (Supplementary Table 1). Among the clean reads, 22,680,825 (32.92 %) matched the grapevine genome, and they are depicted as the overall grapevine sRNA population. The number of sRNAs differed among the six sRNA libraries (Supplementary Table 1), in that the young leaf, mature leaf, and young berry libraries had more sRNAs, whereas those of the inflorescence, flower, and mature berry libraries had fewer sRNAs (Fig. 1a). This observation reflects the spatiotemporal dynamic variations in the grapevine sRNA populations during growth and development. Comparative analysis of the size distribution of sRNAs in various grapevine tissues and stages revealed a conspicuous divergence among the six tissue sRNA libraries from three organs (Fig. 1b), which were characterized by two high peaks of sRNA length distributions in all the six tissue libraries. The maximum sRNA peaks in the young and mature leaf libraries were all at 21 nt, whereas the secondary peaks of these libraries were at 24 nt, with the difference in frequency of these two peaks in the mature leaf library greater than that in the young leaf library (Fig. 1b). In contrast to the two leaf sRNA libraries, the maximum and secondary size peaks in inflorescence and flower libraries were at 24 nt and 21 nt, respectively, and variation in the frequency of the two peaks in the flower (24 nt, 21 nt) was much larger compared with those in the inflorescence (Fig. 1b). Unlike the aforementioned four sRNA libraries, the sRNA size distribution peak of 24 nt in the mature berries was much lower compared with those of the young berries (Fig. 1b). Based on reports that most plant miRNAs are 21 nt, and small interfering RNAs (siRNAs) are 24 nt (Carra et al. 2009; Jones-Rhoades and Bartel 2004), the variations in length and frequency of sRNAs in libraries of various tissues may reflect the dynamic variation in the types and numbers of sRNAs over the span of grapevine development. This suggests the involvement of different sRNAs in the growth and development of grapevine organs or tissues.
The grapevine sRNAs were subsequently analyzed and classified into ten categories based on sRNA annotation principles (exon antisense, exon sense, intron antisense, intron sense, miRNA, rRNA, repeat, snRNA, snoRNA, and tRNA), with the unannotated sRNAs considered as unknowns. The comparative analysis revealed that the sRNA types in the six libraries had diverse proportions, and the percentages of the different sRNA categories differed over the periods of grapevine development (Fig. 1c). This is exemplified by the proportions of various sRNA categories in the leaves, flowers, and berries, where the percentages of the sRNAs in mature leaves and flowers exhibited more distinct variations than those in the other tissues. Meanwhile, the percentage of specific sRNA categories varied with diverse tissue development stages as demonstrated by tRNA and rRNA, which exhibited more significant percentage variations than those of the other sRNA categories in grapevine development (Fig. 1c). This confirms that spatiotemporal variations in sRNA compositions during grapevine growth and development, and these variations could reflect the spatiotemporal traits at the roles of the different types of sRNA during grapevine development.
Variation in overall miRNA abundance during grapevine development
The ratio of Vv-miRNAs to the overall sRNA population varied greatly and spatiotemporally, with miRNAs accounting for 35.5, 19.2, 22.3, 19.1, 20.5, and 21.2 % of the total sRNA component in the young leaves, mature leaves, inflorescences, flowers, young berries, and mature berries, respectively. Comparative analysis of the abundance of both redundant and unique Vv-miRNAs in the tissue sRNA libraries at different developmental stages (Fig. 1d) revealed that the abundance of redundant Vv-miRNAs were highest in the young leaf library, followed by the mature leaf, inflorescence, flower, young berry, and mature berry libraries. The abundance of unique miRNAs in the tissues was roughly similar, and only the unique miRNAs exhibited relatively high expression peaks compared with the redundant miRNAs in young berries, which suggest that the young berry library has the widest variety of miRNAs probably involved in physiologic activities. The analysis of the overall miRNA abundance reflects the dynamic variations in the overall abundance of the miRNAs during grapevine development. Moreover, the abundance of specific miRNA families in each library indicates the presence of wide variations among these miRNAs in grapevine tissues and organs at various stages, as exemplified by the miR166 and miR156 families, which were at levels of 81.3, 37.9, 43.4, 39.9, 50.5, and 59.3 % for miR166 and 4.0, 7.0, 9.4, 6.3, 2.9, and 10.0 % for miR156 in the total miRNA population in young leaf, mature leaf, inflorescence, flower, young berry, and mature berry libraries, respectively.
Spatiotemporal variation in populations of known miRNAs during grapevine development
The miRNA populations differed among various grapevine tissues and stages, which indicates dynamic variations in regulatory roles of miRNAs during grapevine development. We first matched the sequences from our sRNA libraries to known grapevine miRNAs deposited in miRBase 18.0 (http://www.mirbase.org/) to identify known Vv-miRNAs in grapevines. A total of 242 known miRNAs and 22 known miRNAs* were verified in the six sRNA libraries, including two types of miRNAs: 157 sRNA sequences with perfect or near perfect complementarity (0–3 base mismatches) to known Vv-miRNAs in miRBase 18.0, which were considered as known miRNAs (Table 1, bold words; Supplementary Table 2); 85 other sequences similar to known Vv-miRNAs identified in this study and those that have single nucleotide differences at the seed regions with their corresponding known miRNAs, which were considered as new members of known miRNA families, such as miR156a-1 and miR162-1 (Table 1, Italic; Supplementary Table 3). Compared with the Vv-miRNAs we identified in our earlier work (Wang et al. 2011b), we identified 110 known Vv-miRNAs in this work, including 25 known Vv-miRNAs (Vv-miR172a/b, miR2111, miR2950, miR3623-miR3640, miR477b, and miR845c/d) (Supplementary material Table 2: the regions with underlines) and 85 new members of known Vv-miRNA families (Supplementary material Table 3), which confirms that Illumina sequencing efficiently mines tissue- and stage-specific miRNAs, as well as low abundance miRNAs. Interestingly, the types and number of known miRNA populations identified in various libraries here had some differences (See Table 1; Supplementary Tables 2 and 3). This indicates spatiotemporal variations in the expression of different miRNAs during grapevine development.
Further analysis to compare known miRNAs in various sRNA libraries revealed that 191 of the 242 known miRNAs and miRNAs* mentioned above were expressed in all six libraries (Fig. 2a: blue words), 34 miRNAs and miRNAs* were coidentified in some of the six libraries (Fig. 2a: pink words), and 17 were specifically detected in single libraries (Fig. 2a: red words; Supplementary material Table 2 and 3, black grey regions). This finding suggests that most of the known miRNAs are highly conserved in diverse tissues or developmental stages of grapevine, and that they play important roles at various extents during grapevine development. Meanwhile, a small number of known miRNAs detected in some of the tissues had some tissue-specific characteristics, and a few were tissue-specific or stage-specific, expressed only in one tissue. These findings reflect the spatiotemporal roles of Vv-miRNAs and suggest dynamic variations in their regulatory roles during grapevine development.

Number of known (a) and novel (b) miRNA and miRNA* identified in six grapevine sRNA libraries. a, b represent known and novel miRNAs, respectively; kRs and nRs denote known RNAs and novel RNAs respectively; The words in blue denote the number of known and novel miRNA and miRNA* identified as common in all six grapevine sRNA libraries; The words in pink denote the number of known and novel miRNA and miRNA* identified in some of the six grapevine sRNA libraries; The words in red denote the number of tissue–specific known and novel miRNA and miRNA* identified only the individual grapevine sRNA libraries. The red dotted line circles denote the tissue–specific target genes for known and novel Vv-miRNAs
Dynamic expression of known miRNAs during grapevine development
The expression levels of known miRNAs in various tissues and stages of grapevine development were comparatively analyzed to gain insight into regulatory roles of miRNAs during grapevine development. The results indicate that the Vv-miRNA expression patterns (modes) varied widely within and between various Vv-miRNA families during grapevine development. All 242 known miRNAs validated belonged to 50 miRNA families, of which 29 families had one member with diverse expression levels in various tissues. The remaining 21 families had 2–25 members and they were categorized into two groups according to the expression modes of each family. The first group, which includes the miR156, miR159, miR160, miR166, miR167, miR169, miR171, miR172, miR319, miR3633, miR396, miR398, and miR399 families, exhibited diverse expression modes during grapevine development, and each family was clustered into several subgroups based on the expression modes of members per miRNA family during grapevine development (Supplementary material Fig. 1). This difference in expression modes among different members of the same families may have caused the diversity in the roles of various members within these miRNA families during grapevine development. In the second group, the remaining eight families (miR164, miR3629, miR3631, miR393, miR394, miR395, miR403, and miR535) exhibited identical or similar expression modes within each family. This aspect is exemplified by the miR395, miR164, miR3631, and miR3629 families, exhibiting “V,” “M,” “stepwise,” and “flowerpot” shaped modes, respectively (Supplementary material Fig. 2). All these findings confirm the variations and complexities in the regulatory roles of various miRNAs or miRNA families on their target genes during grapevine development.
Although our results largely agree with earlier reports that miRNAs* accumulate at lower levels than their miRNA partners, the counts of the miRNA* for miR2950, miR3623, miR3627, and miR477b in most tissues or developmental stages of grapevine exceeded their corresponding miRNA counts. These miRNAs* in six tissues have the following ratios: 1:3.0 for miR3623; 1:1.4 for miR3637; 1:5.7 for miR2950 and 1:7.5 for miR477b (Fig. 3). This finding demonstrates the complex regulation modes of these four miRNAs and miRNAs*. The four Vv-miRNAs* possessed relatively higher expression levels than their partners, which suggests that grapevine miRNAs* could have some independent regulatory roles, similar to those observed in silkworms (Jagadeeswaran et al. 2010). The expression modes of these miRNAs*, like their miRNAs, also exhibited some variations during grapevine development, which further suggests that these miRNAs* play different regulatory roles on corresponding target genes.

Temporal characterization of miRNAs along with their miRNAs*. These bar graphs represent the expression levels of miR2950 and miR2950*, miR3623 and miR3623, miR3637 and miR3637* in grapevine young leaves, mature leaves, inflorescences, flowers, young berries and mature berries, respectively. Moreover, these four miRNAs’ stars possess the higher expression than themselves in most of six tissues researched here
Spatiotemporal variations in novel miRNA populations in grapevine sRNA libraries
High-throughput sequencing of the six sRNA libraries could be used to discover more novel tissue- or stage-specific miRNAs, which is important for comprehensively understanding the regulatory functions of miRNAs on their target genes during grapevine development. Secondary structure analysis of the annotated flanking regions of the candidate miRNAs indicated that 132 novel miRNAs that belong to 115 unique sequences met the criteria for miRNA annotation by Meyers et al. (2008) and they were thus considered potential Vv-miRNAs in the six sRNA libraries (Supplementary material Table 4). Analysis of the sequences showed that about 50 % of the novel miRNAs in each sRNA library started with ‘U,’ which is an important trait of miRNAs, and 17, 18, 22, 22, 19, and 22 miRNAs met the criteria in the corresponding six tissues, respectively. This finding provides evidence that these potential miRNAs might be actual miRNAs. Compared to our earlier studies (Wang et al. 2011a, b, 2013), although the cultivars used were the same, the samples were derived from different tissues and stages of hVV grapevine. Of the 132 predicted novel miRNAs in the present work, 127 were first uncovered (Supplementary material Table 4) and the remaining 5 miRNAs were also identified as the novel ones in our previous study (Wang et al. 2011b). These results confirm that grapevines have numerous novel specific miRNAs, and demonstrate that Illumina sequencing is an effective technology for mining novel miRNAs.
Comparison of the novel miRNAs and miRNAs* in the six sRNA libraries revealed that only eight miRNAs, namely, miR006, miR012a, miR012b, miR014, miR025, miR006*, miR014*, and miR025* (Fig. 2b; blue words), are common to all six libraries. The other 102 (novel miRNA/miRNA*: 86/16) were only detected in a single tissue (Table 2; Fig. 2b, red words), and only about 15.2 % of the novel miRNAs and their corresponding miRNAs* were detected in some of all tissues (Fig. 2b, pink words). The majority of the novel miRNAs and miRNA* in grapevine possessed much more conspicuous tissue- or stage-specific characteristics, which suggests acute variations in their regulatory roles during grapevine development. On the other hand, the number of the tissue- and stage-specific Vv-miRNAs identified in various libraries also exhibited huge differences (See Supplementary material Fig. 3), which further supports the wide variations in the regulatory functions of these novel miRNAs during grapevine development. Up to 55.4 % of the novel tissue-specific miRNAs were found exclusively in the berries, which suggest that numerous miRNAs are involved in grapevine berry development.
Multiple novel miRNAs usually formed clusters on the same chromosomes, such as the 15 novel miRNAs in chromosomes 19, 14 on chromosome 14, 13 on chromosome 8, and 11 on chromosome 17. This aspect is consistent with previous reports on V. vinifera (Pantaleo et al. 2010) and other plant species (Lee et al. 2002; Baskerville and Bartel 2005). Some novel miRNAs with the same mature sequences have multiple precursors in various locations on the same chromosome. For example, miR012a has five potential precursors on chromosome 14, whereas miR064b has two multiples on chromosome 19. The relationship of these special Vv-miRNA structures with the accumulation levels and processing of miRNAs needs to be evaluated.
Novel berry-specific miRNAs and their potential roles in grapevine berries
Considering the roles of GA in fruit development and ethylene in fruit ripening, we studied the response of novel berry-specific miRNAs to exogenous GA and ethylene. The results show that the novel miRNAs miR014, miR051, miR053, miR100, and miR104, were responsive to the GA and/or ethylene treatments (Fig. 4). Compared with the control, miR051 and miR100 were down-regulated in the GA treatments; miR014, miR085, and miR104 were down-regulated in the ethylene treatments; and miR053 was up-regulated in the GA and ethylene treatments (Fig. 4); interestingly, the expression levels of miR034, miR061 and miR100 all were regulated conspicuously in GA treatments of the 5 weeks after flowering (Fig. 4), while those of miR014, miR034, miR085 and miR104 all were mediated distinctly mostly in ethylene treatments of the 10 and 11 weeks after flowering (Fig. 4), indicating that the response of these miRNAs to GA and/or ethylene had some spatiotemporal traits too. Target analysis revealed that the identified target genes (GSVIVT01008148001, GSVIVT01009517001, GSVIVT01029055001, GSVIVT0101030500 1, GSVIVT01014221001, and GSVIVT01025159001) for the foregoing novel miRNAs participated in plant hormone signal transduction, which supports our experimental results. These results suggest that the novel miRNAs from berries are involved in the GA and/or ethylene signal pathways; thus, they regulate grapevine berry development and ripening.

Expression patterns of miRNAs in grapevine berries under Control and GA/ethylene treatments. The line graphs (a, b, f) denote the expression profiles of miR061, miR100 and miR034 of novel berry-specific miRNAs in the berry tissues of 1, 5 and 9 weeks after flowering in control and treatments with GA, respectively; other line graphs (c, d, e, g) represent those of miR014, miR085, miR104 and miR034 of novel berry-specific miRNAs in the berry tissues of 4, 5, 6 and 7 weeks after flowering in control and treatments with ethylene. N weeks AF denotes n weeks after flowering. Each reaction was repeated three times and standard error was pointed with line graphs. The template amount was corrected by 5.8S rRNA
Dynamic expression of novel Vv-miRNAs during grapevine development
We compared the expression frequency of all identified novel Vv-miRNA in different grapevine tissues and/or stages to determine the regulation of target genes by Vv-miRNAs during grapevine development. It was found that the expression of the majority of the novel Vv-miRNAs accumulate at lower levels, which is consistent with previous reports (Moxon et al. 2008a, b; Song et al. 2010; Wang et al. 2011b), and could be clustered into five modes. These expression modes include an instance where 65.1 % (86) of the novel Vv-miRNAs were expressed in only one of the tissues used to construct the sRNA libraries (Supplementary material Table 4), which indicates that many novel miRNAs may be tissue-specific in grapevines. Another group of miRNAs, namely, miR007, miR013, miR023, and miR040, was ubiquitously expressed in all except one grapevine tissue and absent in mature berries, young berries, and young leaves. Some novel Vv-miRNAs also exhibited organ-specific expression pattern, such as leaves (young and mature leaves), flowers (inflorescences and flowers), or berries (young and mature berries). This aspect is best exemplified by miR019, miR029, and miR052 (in leaves), miR054 and miR057 (in flowers), as well as miR088 and miR096 (in berries). Also group of novel Vv-miRNAs was only ubiquitously expressed at diverse levels in two of the three organs. This group includes miR020, miR022, and miR026 (not detected in berries), as well as miR051, miR056, and miR060 (absent in the leaves). Only five novel miRNAs were ubiquitously expressed at various levels in every tissue and stage. These results demonstrate the clear spatiotemporal specificity of novel miRNAs, with drastic variations in their action on target genes during grapevine development.
Analysis of Vv-miRNA target genes using Degradome sequencing
Systematically studying the target mRNAs of Vv-miRNAs and their modes of action is necessary for understanding the functions of miRNAs during grapevine development. The six tissue mRNA libraries from grapevines were subjected to Degradome sequencing. A total of 102 target genes for 22 known miRNAs families and 90 targets for the 34 novel miRNAs were validated (Supplementary material Tables 5 and 6). Among the 192 target genes, 31 were validated and their functions were analyzed in our earlier report (Wang et al. 2013), but more targets need to be identified. It was revealed that the kinds and numbers of target genes identified here in various tissues were of conspicuous differences, where the numbers of target for known miRNAs were 42, 46, 51, 43, 71 and 12, and while those for novel miRNAs were 11, 4, 10, 30, 22 and 18 in young leaf, mature leaf, inflorescence, flower, young berry and mature berry libraries, respectively. These differences reflect the spatiotemporal variations in the regulatory patterns of Vv-miRNAs. Furthermore, the types and numbers of the target genes within the same miRNA families also differed among various tissues. In the Vv-miR396 family, five target genes were identified in the young leaf library, five were identified in the mature leaf library, seven were identified in the inflorescence library, five were identified in the flower library, seven were identified in the young berry library, and two were identified in the mature berry library. Although the same number of target mRNA was identified in some tissues as described above, these targets had some discrepancies (Fig. 5), similar to that observed in the Vv-miR156 family (Fig. 5). This finding suggests differences in the targeting patterns of the same Vv-miRNA family in various tissues.

Target genes identified for miR396 and miR156 family in various tissue sRNA libraries. miR396 family were totally validated 14 target genes in grapevine young leaves, mature leaves, inflorescences, flowers, young berries and mature berries, but the kinds and number of targets detected in each tissue possessed obvious discrepancy; miR156 family were totally detected 10 targets in six grapevine tissues above, the types and number of target mRNAs identified in each tissue showed the similar case to those of miR396 family
All target genes were subjected to GO and KEGG analyses. The results showed that the target genes of the Vv-miRNAs were involved in more than 100 pathways, which could be mainly classified into several groups: the first group, which were implicated in metabolic processes, such as riboflavin, galactose, alpha-linolenic acid, cyanoamino acid, ether lipid, and pyruvate metabolism; the second group with being involved in biosynthesis processes, like flavonoid, phenylpropanoid, diterpenoid, aminoacyl-tRNA, and benzoxazinoid biosynthesis, etc.; the third group, which were involved in signal transduction processes, such as the Wnt, Jak-STAT, TGF-beta, adipocytokine, plant hormone, and p53 signaling pathways; the genes of the fourth group were related to disease resistance like Chagas disease (American trypanosomiasis), plant-pathogen interaction, Huntington’s disease, and Alzheimer’s disease; the last group with being related to degradation processes, such as polycyclic aromatic hydrocarbon, aminobenzoate, bisphenol, limonene, pinene, RNA degradation. Interestingly, we found that the functions of some target genes during grapevine development varied in terms of conservation. For example, GSVIVT01015074001, which is involved in flavone and flavonol biosynthesis, was only identified in young and mature berries. This differential expression might be due to the function of this gene in the biosynthesis of flavor compounds.
Conservation and tissue specificity of Vv-miRNA regulatory roles on target genes
Grapevine miRNA:mRNA pairs detected via Degradome sequencing validated Vv-miRNAs target genes and their functions. We compared the miRNA:mRNA pairs in various mRNA libraries and analyzed the conservation of the target modes of various miRNAs on their targets. The results show that the conservation of cleavage roles of the known and the novel miRNAs on their corresponding target genes varied among different grapevine tissues and stages, with 2, 2, 4, 3, 25 and 6 known tissue-specific miRNA:mRNA pairs, and 10, 3, 8, 28, 21 and 17 novel tissue-specific ones in the young leaves, mature leaves, inflorescences, flowers, young berries and mature berries, respectively. About 96.1 % (99/103) of the target genes of the novel miRNAs (Supplementary material Table 6) and about 46.7 % (42/90) of the targets of the known miRNAs (Supplementary material Table 5) were tissue-specific, which demonstrate that the cleavage roles of novel Vv-miRNAs on target genes are much more varied than those of known miRNAs during grapevine development.
Interestingly, the tissue-specific Vv-miRNAs were only expressed in a single tissue or stage. This aspect is exemplified by tissue-specific miRNAs, such as miR005, miR015, miR043, miR061, miR068, miR074a/b, miR089, miR092, miR097, miR098a, miR100, miR101, miR102, and miR103, regulate their target genes only in the corresponding tissues where they are found (Supplementary material Fig. 3; Supplementary material Table 6). By contrast, some non-tissue-specific or non-stage-specific miRNAs cleaved their target gene in multiple tissues or stages. For example, the miRNAs such as miR020, miR026, miR029, miR060 and miR088 are non-tissue-specific and they target their corresponding mRNAs in various tissues (Supplementary material Tables 4 and 6).
Spatiotemporal variations in the cleavage activity of miRNAs on their target genes during grapevine development
Plant miRNAs regulate the expression of their target mRNAs mainly through cleavage; thus, the accumulation of the cleavage products can be used to estimate the miRNA cleavage activity on their target genes. Analyzing the accumulation of the target cleavage products of the same miRNA in various tissues revealed dynamic variations during grapevine development. For example, miR172 has four target genes (GSVIVT01022081001, GSVIVT01025100001, GSVIVT01030611001, and GSVIVT01016352001) in different grapevine tissues, and the abundance of the cleavage products of these four target mRNAs exhibited the diverse cleavage modes (Fig. 6). Similarly, the miR156, miR159, miR160, miR166, miR167, miR171, miR319, miR396, and miR828 families cleaved multiple target genes in several tissues and the abundance of the resulting cleavage products exhibited different expression patterns during grapevine development.

Number of all target mRNAs and tissue-specific ones for known and novel miRNAs in diverse grapevine tissue mRNA libraries. The biggest division section in purple pie diagram is the number of all target mRNAs for known miRNAs in grapevine young leaves, mature leaves, inflorescences, flowers, young berries and mature berries, while the other 6 small division sections in this purple one represent the number of tissue-specific target genes in each tissue, respectively; similarly, the biggest division section in blue pie diagram is the number of all targets for novel miRNAs in six grapevine tissues above, while the remaining division sections in the blue diagram denote the number of tissue-specific targets in per tissue, respectively
Extent of target gene cleavage sites for novel Vv-miRNAs in grapevine flower tissue
The cleavage sites of the novel miRNAs on their target genes were extensive in the grapevine flower tissues. In particular, miR074a/b had the highest number (22) of identified target genes exclusive to the flower tissue, but most (81.8 %) of these target genes had 3–12 miRNA-targeted cleavage sites. Majority of the cleavage sites were in the 12th–16th nucleotides at the 5′-end of the miRNAs (Fig. 7), which differs from the previous reports wherein such cleavage sites were mainly in the target sequence that pairs with the 10th nucleotide at the 5′-end of the miRNAs (Moxon et al. 2008a, b; Carra et al. 2009; Pantaleo et al. 2010; Mica et al. 2010; Mallory et al. 2005). Moreover, the cleavage sites on miR074a/b target genes were comprehensively distributed on the target sequences complementary to almost all of the base sites (1–21) (except for the 5th and 20th base of mature miRNAs; Fig. 7), as well as found upstream and downstream of the target regions for mature miRNAs, which is similar to those reported in tomato (Moxon et al. 2008a, b). We further analyzed the functions of target genes regulated by miR074a/b based on functional annotation of orthologous genes in other plants through GO and KEGG analyses, and found that these target genes were related to the pollen tube development, embryo development, flower development, fertility restoration, embryo-specific genes, cellular dedifferentiation and redifferentiation, plant growth, disease resistance, signal transduction processes, which were exemplified by pentatricopeptide repeat-containing protein, embryo-specific protein 3, F-box/kelch-repeat protein SKIP6-like, disease resistance response protein 206-like, leucine-rich repeat receptor-like protein kinase, and phytosulfokines 6-like. The potential functions of the target genes suggest that miR074a/b is closely related to grapevine flower development, but the correlation between the cleavage activity of miR074a/b on its target genes and the grapevine flower development process, and whether the extensive cleavage is due to miRNA involvement in a variety of complicated metabolic processes during flower development are unclear.

Extent of target gene cleavage sites for novel miR074a/b in grapevine flower tissue. The novel miR074a/b had 22 target genes, of which 18 targets possessed more than 3 cleavage sites, and the most cleavage sites of most target genes for novel miR074a/b mainly located in the 12th–16th nucleotide of 5′-end of miRNAs, while GSVIVT01030074001, GSVIVT01008850001, GSVIVT01022227001 and GSVIVT01004867001 of 4 targets (the color regions) had much more extent cleavage sites (13, 12, 11 and 10), and these sites comprehensively distributed the target sequences complementary to miR1074a/b and their upstreams and downstreams. C sites denotes Cleavage sites;  represents target cleavage site
represents target cleavage site
Modes of miRNA action and the factors that affect them
The dynamic variations in the modes by which the Vv-miRNAs regulate their target mRNAs were systematically analyzed and the probable factors affecting these modes were elucidated to characterize the functions of Vv-miRNA during grapevine development. Based on the abundance of the identified Vv-miRNAs, their families, and their corresponding target cleavage products in the six grapevine tissues/stages, the patterns of Vv-miRNA activity could be grouped into five main categories (as outlined below), and the factors that affect their cleavage modes were also analyzed.
The first mode involves all members of the miRNA family co-participating in the regulation of their target genes, and the variations in the accumulation of target gene cleavage products in most tissues were consistent with expression levels of the corresponding miRNA family in the same tissues. As shown in Fig. 8a, the accumulation of cleavage products of all four genes targeted by Vv-miR156 in most tissues is consistent with the Vv-miR156 expression pattern. However, in young berries, the high abundance of the cleavage products of three of the four miR156 target genes was contradictory to those of the Vv-miR156 family (Fig. 8a). These three targets are SPB-like and are highly expressed in young grapevine berries (Cao et al. 2011).

Regulatory modes of various Vv-miRNA families on their target mRNAs during grapevine development. a Temporal regulation mode of miR156 family in grapevine young leaves, mature leaves, inflorescences, flowers, young berries and mature berries, all members of this miRNA family co-participated in the regulation of their target genes, b regulatory modes of miR171 family on corresponding target genes, miR171b of this family might be the main factor contributing to cleavage of the target gene, c co-regulatory mode of miR319 and miR159 families on their common target genes, all members of both miR319 and miR159 families co-regulated their target GSVIVT0102447001, d regulatory patterns of miRNA families having single members on corresponding target genes, the miRNA families with only one member play conspicuous regulatory roles mainly in the tissues where corresponding miRNAs were highly expressed. YL, ML, IF, FL, YB and MB represent young leaves, mature leaves, inflorescences, flowers, young berries, mature berries, respectively
The second mode of action of miRNA on their target genes involves one of the members of the same miRNA family regulating their target genes. For example, the miR171 family has eight members, but only the expression of miR171b in various tissues was consistent with the accumulation of the cleavage products of its target gene (GSVIVT01019006001) products, whereas the other members and the miR171 family (total) did not follow this rule (Fig. 8b). Similarly, miR166b from the miR166 family also exhibited the second mode of action.
The third mode of Vv-miRNA regulation on target genes involves several miRNA families co-regulating the same target genes. The best examples of this pattern are miR159 and miR319 families, which co-regulate GSVIVT0102447001. As shown in Fig. 8c, the abundance of GSVIVT0102447001 cleavage product is consistent with the total expression levels of the miR159 and miR319 families during grapevine development.
The fourth mode of action involves miRNA families with only one member that conspicuously regulates corresponding target mRNAs in tissues where they are highly expressed. These miRNAs include miR162, miR168, and miR482, which and whose target cleavage products all had the highest accumulations in young berries compared with the other tissues (Fig. 8d).
The last mode of action involves tissue-specific miRNAs with the corresponding tissue-specific cleavage activity against their target genes as described above. Most tissue-specific miRNAs were novel miRNAs, among which 34 novel miRNAs with the target genes we identified and 24 miRNAs with only one target gene (namely, miR015, miR031, miR043, miR068, miR089, miR092, miR097, miR098a/b, miR100-103, miR110-113, and miR114a/b/c/d/e/f/g) each showed tissue-specific cleavage of its target in corresponding single tissues. The remaining five miRNAs (miR005, miR061, miR074a/b, miR099, and miR115) had the similar regulatory modes, but they had multiple targets in the corresponding single tissues, unlike the aforementioned 24 miRNAs (Supplementary material Fig. 3; Supplementary material Table 6).
Discussion
Although several studies have reported on miRNAs from V. vinifera and hybrids of V. vinifera, V. labrusca, and Vitis amurensis Rupr. (Jaillon et al. 2007; Carra et al. 2009; Pantaleo et al. 2010; Wang et al. 2011a, b; Mica et al. 2010; Lazzari et al. 2009; Wang et al. 2012, 2013), they mainly focused on the bioinformatics prediction and identification of Vv-miRNAs, including their precise sequences, as well as predicting target genes and analyzing homologous/orthologous function. The spatiotemporal variations in the regulatory roles of Vv-miRNAs on their target genes have not been systematically studied during grapevine development at the transcriptome level except for one on Degradome sequencing of a single leaf mRNA library from V. vinifera (Pantaleo et al. 2010) and another on validating several target genes using RLM-RACE and PPM-RACE (Sun et al. 2012; Wang et al. 2013). Illumina and Degradome sequencing were employed in the present study to elucidate the spatiotemporal regulation of various Vv-miRNAs on their targets to understand the transcriptome-wide functions of global Vv-miRNAs during grapevine growth and development.
Characterization of mRNA libraries in different grapevine tissues
Sequencing of the six grapevine tissue sRNA libraries revealed that the expression of miRNAs with important regulatory functions varied widely, which indicates that the regulatory roles of miRNAs varied significantly during grapevine development. In addition, the percentage of sRNAs with different lengths in the six libraries differed significantly, which may reflect the differences in the types of sRNA and their contents in various grapevine tissues and/or stages. These findings possibly explain the variations in the modes by which miRNAs act on their target genes during grapevine development. Further study revealed that the percentage of sRNAs with different lengths in different tissue libraries from hVV differed from those reported in the V. vinifera sRNA libraries used by Pantaleo et al. (2010). They found that the leaf, tendril, inflorescence, and berry sRNA libraries from the wine grapevine V. vinifera ‘Pinot Noir’ have only one peak of 21 nt sRNA, whereas the percentage of sRNAs with other lengths were much lower. By contrast, 21 nt and 24 nt sRNA peaks were observed in each of the six sRNA libraries of ‘Summer Black’ in the present study. The 21 nt peak was highest in the young and mature leaf libraries, whereas the 24 nt peak was the highest in inflorescences and flowers. Interestingly, the peaks of these two sRNA sizes in young and mature berries behaved differently compared with those in the other organs, with the 24 nt sRNA exhibiting the highest peak in the young berries and the 21 nt sRNA exhibiting the highest peak in the mature berries. This significant discrepancy in sRNA size distribution between the grapevine cultivars in the two studies might be attributed to their different lineages. The differences in sRNA size distribution between the various grapevine tissues and/or stages, especially during berry development, are due to the variations in the types of sRNAs during grapevine development. These variations are much more conspicuous during berry development, which further confirms that multiple types of sRNAs are involved in grapevine berry development.
Tissue- and stage-specific miRNAs in the different grapevine tissue libraries
Deep sequencing of the sRNA libraries from various grapevine tissues and/or stages revealed numerous known miRNAs and many novel miRNAs, as well as the discovery of more tissue- or stage-specific miRNAs. In contrast to findings from earlier related studies (Carra et al. 2009; Pantaleo et al. 2010; Wang et al. 2011a, b, 2012), we report the most types (50 families) and the highest number of known (157) and novel (132) miRNAs, as well as the highest number of tissue- or stage-specific (98) miRNAs. Therefore, deep sequencing of sRNA libraries from many different tissues and/or stages is vital for systematically identifying the populations and roles of miRNAs in grapevines.
Most of the known miRNAs have highly conserved functions during grapevine development. This view is further supported by the presence of only 12 tissue-specific miRNAs among the 242 known miRNAs. Unlike the known miRNAs in this study, 86 novel tissue- or stage-specific miRNAs were uncovered, 7.5 times more novel tissue-specific miRNAs than the known tissue-specific miRNAs. This finding indicates that many novel miRNAs are involved in specific regulation during grapevine development. About 46 (55.4 %) novel tissue-specific miRNAs were only found in the berries, which suggest that many novel miRNAs might be involved in the complicated physiology during grapevine berry development. Furthermore, most of the novel miRNAs were also specific for grapevines, and the larger number of novel Vv-miRNAs could be useful in characterizing and understanding grapevine species.
Spatiotemporal characterization of miRNAs cleaving on target mRNAs
Degradome sequencing of mRNA libraries from diverse tissues/stages can facilitate the large-scale identification of target mRNAs and enables exact validation of the spatiotemporal characteristics of miRNA cleavage. Our datasets show that the types of targets and the abundance of miRNA target cleavage products differ among various grapevine tissues and stages. Some miRNAs do not have targets in some tissues. However, some targets could be found in other tissues. For instance, no target genes were identified for Vv-miR164 family in young leaves, mature leaves, and inflorescences, whereas multiple targets for this family were detected in flowers, young berries, and mature berries. Vv-miR394, miR477, and miR482 had similar regulatory modes. These observations indicate that Vv-miRNA cleavage on their target genes has conspicuous spatiotemporal traits during grapevine development. In addition, multiple target mRNAs were verified for three miRNA families (miR156, miR396, and miR828) in various grapevine tissues, but the abundance of the target gene cleavage products significantly varied among different tissues (Supplementary Table 6). Compared with the known miRNAs, the target genes of novel miRNAs in various tissues were tissue- or stage-specific, such as miR074a/b, which has 22 targets in the single flower tissue. These results present further evidence of the spatiotemporal aspects of the cleavage of their target genes during grapevine development.
Specificity of miRNA target gene cleavage sites during grapevine development
Identification of the miRNA target cleavage sites confirmed the presence of target genes and the modes by which miRNAs interact with these targets. Degradome sequencing accurately identified the cleavage sites in miRNA targets. Our data revealed that about 85.2 % of the known miRNAs had single cleavage sites on their target genes, whereas the remaining 14.8 % of the known miRNAs had two to three cleavage sites. These cleavage sites were mainly mapped on the mRNA nucleotides complementary to the 9th, 10th, or 11th nucleotides from the 5′-ends of the corresponding miRNAs, all of which were most likely the exact locations of the cleavage sites of the miRNAs with their target genes (Kasschau et al. 2003; Llave et al. 2002). Novel miRNAs, on the other hand, had more target gene cleavage sites compared with known miRNAs. About 24.9 % of the novel miRNAs had multiple cleavage sites, with some miRNAs having up to 13 cleavage sites (e.g. miR074a/b) (Moxon et al. 2008a, b). These observations on both known and novel miRNAs suggest the universality of target gene cleavage by miRNAs, similar to siRNAs (Llave et al. 2002). Most of the novel flower-specific miRNAs had multiple cleavage sites on their target genes, with 76.7 % having 3–13 cleavage sites. In particular, miR074 had all of its 22 targets identified exclusively in the flowers, and 81.8 % (18) of these targets had multiple cleavage sites. By contrast, some non-flower-specific novel miRNAs usually have only one mRNA cleavage site, with several miRNAs having two to three sites. These findings indicate that many Vv-miRNAs have complicated cleavage roles during grapevine flower development, and tell the truth that some comprehensive involvement of these novel miRNAs happen during grapevine flower development.
References
Addo-Quaye C, Eshoo TW, Bartel DP, Axtell MJ (2008) Endogenous siRNA and miRNA targets identified by sequencing of the Arabidopsis degradome. Curr Biol 18:758–762
Addo-Quaye C, Miller W, Axtell MJ (2009) CleaveL and A pipeline for using degradome data to find cleaved small RNA targets. Bioinformatics 25:130–131
Allen RS, Li JY, Alonso-Peral MM, White RG, Gubler F, Millar AA (2010) MicroR159 regulation of most conserved targets in Arabidopsis has negligible phenotypic effects. Silence 1:18
Bartel DP (2004) MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 116:281–297
Baskerville S, Bartel DP (2005) Microarray profiling of microRNAs reveals frequent co-expression with neighboring miRNAs and host genes. RNA 11:241–247
Cao X, Wang C, Fang JG, Yang G, Yu HP, Song CN (2011) Cloning, subcellular localization and expression analysis of SPL9 and SPL10 genes from grapevine. Acta Horticulturae Sinica 38(2):240–250 (in Chinese)
Carra A, Mica E, Gambino G, Pindo M, Moser C, Pè ME, Schubert A (2009) Cloning and characterization of small non-coding RNAs from grapevine. Plant J 59:750–763
Carrington JC, Ambros V (2003) Role of microRNAs in plant and animal development. Science 301:336–338
Fahlgren N, Howell MD, Kasschau KD et al (2007) High-throughput sequencing of Arabidopsis microRNAs: evidence for frequent birth and death of MIRNA genes. PLoS ONE 2:e219
Jagadeeswaran GR, Zheng Y, Sumathipala N, Jiang HB, Arrese EL, Soulages JL, Zhang W, Sunkar R (2010) Deep sequencing of small RNA libraries reveals dynamic regulation of conserved and novel microRNAs and microRNA-stars during silkworm development. BMC Genomics 11:52
Jaillon O, Aury JM, Noel B, Policriti A, Clepet C, Casagrande A, Choisne N, Aubourg S, Vitulo N, Jubin C, Vezzi A, Legeai F, Hugueney P, Dasilva C, Horner D, Mica E, Jublot D, Poulain J, Bruyère C, Billault A, Segurens B, Gouyvenoux M, Ugarte E, Cattonaro F, Anthouard V, Vico V, Fabbro CD, Alaux M, Gaspero GD, Dumas V, Felice N, Paillard S, Juman I, Moroldo M, Scalabrin S, Canaguier A, Clainche IL, Malacrida G, Durand E, Pesole G, Laucou V, Chatelet P, Merdinoglu D, Delledonne M, Pezzotti M, Lecharny A, Scarpelli C, Artiguenave PèG, Valle FME, Morgante M, Caboche M, Adam-Blondon AF, Weissenbach J, Quétier F, Wincker P, French-Italian public consortium for grapevine genome characterization (2007) The grapevine genome sequence suggests ancestral hexaploidization in major angiosperm phyla. Nature 449:463–467
Jones-Rhoades MW, Bartel DP (2004) Computational identification of plant microRNAs and their targets, including a stress induced miRNA. Mol Cell 14:787–799
Kasschau KD, Xie Z, Allen E, Llave C, Chapman EJ, Krizan KA, Carrington JC (2003) P1/HC-Pro, a viral suppressor of RNA silencing, interferes with Arabidopsis development and miRNA function. Dev Cell 4:205–217
Lazzari B, Caprera A, Cestaro A, Merelli I, DelCorvol M, Fontana P, Milanesi L, Velasco R, Stella A (2009) Ontology-oriented retrieval of putative microRNAs in Vitis vinifera via GrapeMiRNA: a web database of de novo predicted grape microRNAs. BMC Plant Biol 9:82
Lee Y, Jeon K, Lee JT, Kim S, Kim VN (2002) MicroRNA maturation: stepwise processing and subcellular localization. EMBO J 21:4663–4670
Leung AKL, Sharp PA (2010) MicroRNA functions in stress responses. Mol Cell 40(2):205–215
Llave C, Xie Z, Kasschau KD, Carrington JC (2002) Cleavage of Scarecrow-like mRNA targets directed by a class of Arabidopsis miRNA. Science 297:2053–2056
Lu YD, Gan QH, Chi XY, Qin S (2008) Identification and characteristization of microRNAs and their targets in grapevine (Vitis Vinifera). Agric Sci China 7(8):929–943
Mallory AC, Vaucheret H (2004) MicroRNAs: something important between the genes. Curr Opin Plant Biol 7:120–125
Mallory AC, Bartel DP, Bartel B (2005) MicroRNA-directed regulation of Arabidopsis AUXIN RESPONSE FACTOR17 is essential for proper development and modulates expression of early auxin response genes. Plant Cell 17:1360–1375
Meyers BC, Axtell MJ, Bartel B, Bartel DP, Baulcombe D, Bowman JL, Cao XF, Carrington JC, Chen XM, Green PJ, Griffiths-Jones S, Jacobsen SE, Mallory AC, Martienssen RA, Poethig RS, Qi YJ, Vaucheret H, Voinnet O, Watanabe Y, Weigel D, Zhu JK (2008) Criteria for annotation of plant MicroRNAs. Plant Cell 20:3186–3190
Mica E, Piccolo V, Delledonne M, Ferrarini A, Pezzotti M, Casati C, Del Fabbro C, Valle G, Policriti A, Morgante M, Pesole G, Enrico Pè M, Horner DS (2010) Correction: high-throughput approaches reveal splicing of primary microRNA transcripts and tissue specific expression of mature microRNAs in Vitis vinifera. BMC Genomics 11:9
Moxon S, Jing R, Szittya G, Schwach F, Pilcher RL, Moulton V, Dalmay T (2008a) Deep sequencing of tomato short RNAs identifies microRNAs targeting genes involved in fruit ripening. Genome Res 18:1602–1609
Moxon S, Schwach F, Maclean D, Dalmay T, Studholme DJ, Moulton V (2008b) A tool kit for analysing large-scale plant small RNA datasets. Bioinformatics 24:2252–2253
Pantaleo V, Szittya G, Moxon S, Miozzi L, Moulton V, Dalmay T, Burgyan J (2010) Identification of grapevine microRNAs and their targets using high-throughput sequencing and degradome analysis. Plant J 62:960–976
Ramakers C, Ruijter JM, Deprez RH, Moorman AF (2003) Assumption-free analysis of quantitative real-time polymerase chain reaction (PCR) data. Neurosci Lett 339:62–66
Song CN, Wang C, Zhang CQ, Nicholas KK, Yu HP, Ma ZQ, Fang JG (2010) Deep sequencing discovery of novel and conserved microRNAs in trifoliate orange (Citrus trifoliata). BMC Genomics 11:431
Sun X, Nicholas KK, Han J, Leng XP, Fang JG (2012) Characterization of grapevine microR164 and its target genes. Mol Biol Rep 39(10):9463–9472
Sunkar R, Zhou X, Zheng Y, Zhang W, Zhu JK (2008) Identification of novel and candidate Vv-miRNAs in rice by high throughput sequencing. BMC Plant Biol 8:25
Szittya G, Moxon S, Santos DM, Jing R, Fevereiro MP, Moulton V, Dalmay T (2008) High-throughput sequencing of Medicago truncatula short RNAs identifies eight new miRNA families. BMC Genomics 9:593
Thomson DW, Bracken CP, Goodal GJ (2011) Experimental strategies for microRNA target identification. Nucleic Acids Res 39(16):6845–6853
Velasco R, Zharkikh A, Troggio M, Cartwright DA, Cestaro A, Pruss D, Pindo M, FitzGerald LM, Vezzulli S, Reid J, Malacame G, Iliev D, Coppola G, Wardell B, Micheletti D, Macalma T, Facci M, Mitchell JT, Perazzolli M, Eldredge G, Gatto P, Cyzerski R, Moretto M, Gutin N, Stefanin M, Chen Y, Segala C, Kavenport C, Demattè L, Mraz A, Battilana J, Stormo K, Costa F, Tao QZ, Si-Ammour A, Harkins T, Lackey A, Perbost C, Taillon B, Stella A, Solovyev V, Fawcett JA, Sterck L, Vandepolele K, Grando SM, Toppo S, Moser C, Lanchbury J, Bogden R, Skolnick M, Sgaramella V, Bhatnagar SK, Fontana P, Gutin A, Ven de Peer Y, Salamini F, Viola R (2007) A high quality draft consensus sequence of the genome of a heterozygous grapevine variety. PLoS ONE 2:e1326
Wang XJ, Reyes JL, Chua NH, Gaasterland T (2004) Prediction and identification of Arabidopsis thaliana microRNAs and their mRNA targets. Genome Biol 5:R65
Wang C, Shangguan LF, Nicholas KK, Wang XC, Han J, Song CN, Fang JG (2011a) Characterization of microRNAs identified in a table grapevine cultivar with validation of computationally predicated grapevine miRNAs by miR-RACE. PLoS ONE 6(7):e21259
Wang C, Wang XC, Nicholas KK, Song CN, Zhang CQ, Li XY, Han J, Fang JG (2011b) Deep sequencing of grapevine flower and berry short RNA library for discovery of novel microRNAs and validation of precise sequences of grapevine microRNAs deposited in miRBase. Physiol Plant 143:64–81
Wang C, Han J, Liu C, Nicholas K, Kayesh E, Shangguan LF, Li XY, Fang JG (2012) Identification of microRNAs from Amur grapes (Vitis amurensis Rupr.) by deep sequencing and analysis of microRNA variations with bioinformatics. BMC Genomics 13:122
Wang C, Han J, Nicholas KK, Wang XC, Liu H, Li XY, Leng XP, Fang JG (2013) The characterization of target mRNAs for table grapevines miRNAs with an integrated strategy of modified RLM RACE, PPM RACE and qRT-PCRs of cleavage products. J Plant Physiol 170(10):943–957
Wilson DN, Chung H, Elliott RC, Bremer E, George D, Koh S (2005) Microarray analysis of postictal transcriptional regulation of neuropeptides. J Mol Neurosci 25:285–298
Xie FL, Frazier TP, Zhang BH (2011) Identification, characterization and expression analysis of MicroRNAs and their targets in the potato (Solanum tuberosum). Gene 473:8–22. doi:10.1016/j.gene.2010.09.007
Acknowledgments
This work was supported by the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD), and the Graduate innovative training project in Jiangsu Province CXZZ11_0665 and CXZZ12_0284.
Conflict of interests
The authors declare that they have no conflict of interests.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Wang, C., Leng, X., Zhang, Y. et al. Transcriptome-wide analysis of dynamic variations in regulation modes of grapevine microRNAs on their target genes during grapevine development. Plant Mol Biol 84, 269–285 (2014). https://doi.org/10.1007/s11103-013-0132-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11103-013-0132-2