
Abstract
The hydrolytic products of glucosinolates in brassica crops are bioactive compounds. Some glucosinolate derivatives such as oxazolidine-2-thione from progoitrin in brassica oilseed meal are toxic and detrimental to animals, but some isothiocyanates such as sulforaphane are potent anti-carcinogens that have preventive effects on several human cancers. In most B. rapa, B. napus and B. juncea vegetables and oilseeds, there is no or only trace amount of glucoraphanin that is the precursor to sulforaphane. In this paper, RNA interference (RNAi) of the GSL-ALK gene family was used to down-regulate the expression of GSL-ALK genes in B. napus. The detrimental glucosinolate progoitrin was reduced by 65 %, and the beneficial glucosinolate glucoraphanin was increased to a relatively high concentration (42.6 μmol g−1 seed) in seeds of B. napus transgenic plants through silencing of the GSL-ALK gene family. Therefore, there is potential application of the new germplasm with reduced detrimental glucosinolates and increased beneficial glucosinolates for producing improved brassica vegetables.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Glucosinolates are secondary plant products that are synthesized in the order Capparales. The hydrolytic breakdown products of glucosinolates, especially isothiocyanates, are beneficial bioactive constituents that have cancer-preventive properties in humans, that contribute to flavors in brassica vegetables and condiments and that are used as biopesticides in biofumigation. In contrast, in rapeseed meal, the dominant glucosinolate progoitrin (2-hydroy-3-butenyl glucosinolate) is changed into an oxazolidine-2-thione, which causes goiter and has other detrimental effects on animal health. Therefore, enrichment of beneficial glucosinolate and reduction of detrimental glucosinolates are given great attention in Brassica crops’ breeding.
In canola, progoitrin has been dramatically reduced through conventional breeding although it might be reduced further through gene manipulation (Liu et al. 2010). In broccoli and a few other B. oleracea vegetables, glucoraphanin predominates and its hydrolysis product, sulforaphane, is identified as a potent anticarcinogenic compound. For this reason, sulforaphane has been extensively investigated due to its potent cancer-preventive effects (Fahey et al. 1997; Shapiro et al. 2001; Zhang et al. 1992). Sulforaphane also induces apoptosis in cancer cells through inhibition of histone deacetylase, which leads to cancer cell cycle arrest (Gamet-Payrastre et al. 2000). Increasing interest in glucosinolates and their degradation products is due to their potential as human cancer-prevention agents, crop-protection compounds and biofumigants in agriculture.
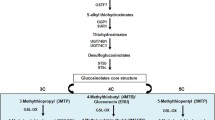
High concentrations of glucoraphanin are only found in broccoli and several other B. oleracea vegetables such as kale, Chinese kale, cabbage and purple cauliflower (Li et al. 2001). In B. oleracea, glucoraphanin concentration was enriched through conventional breeding (Mithen et al. 2003). However, in many brassica species such as B. napus, B. rapa, B. juncea, only undetectable or trace amounts of glucoraphanin occur (Rosa et al. 1997). It is desirable to develop other brassica vegetables with enriched glucoraphanin concentration and improved food quality. In Brassica species including vegetables such as Chinese cabbage, pak choi and turnip, gluconapin, the precursor of glucoraphanin according to the biosynthetic pathway of aliphatic glucosinolates, predominates (Fig. 1). Therefore, it is possible to block the side chain modification to produce glucoraphanin-enriched B. rapa vegetables and other brassica species.

A model of the biosynthetic pathways of aliphatic glucosinolates in B. napus depicted on the basis of genetic analysis (Magrath et al. 1993; Li et al. 2001; Liu et al. 2010). In this model, all aliphatic glucosinolates are derived from methionine and classified into C3, C4 and C5 glucosinolates based on the length of side chains. GSL-ELONG, GSL-PPO, GSL-OX, GSL-ALK and GSL-OH are the major genes in the pathways detected in Brassica species. GSL-PRO is involved in C3 side glucosinolates and GSL-ELONG, in C4 and C5 glucosinolates. GSL-OX and GSL-ALK modify all side chains, but GSL-OH, only C4 and C5 side chains. All gene function is inferred from genetic and transgenic analysis. Asterisks indicating that C3 glucosinolates were detected in new synthetic B. napus (Magrath et al. 1993) and induced in transgenic B. napus with RNAi construct of GSL-ELONG gene (Liu et al. 2010)
Glucoraphanin accumulation is associated with non-functional GSL-ALK genes in B. oleracea (Li and Quiros 2003), which is homologous to AOP genes in Arabidopsis (Neal et al. 2010). The corresponding homologs of AOP2 (GSL-ALK) have been identified in broccoli and purple cauliflower (B. oleracea). A two bp deletion of GSL-ALK gene ensures the non-functional GSL-ALK gene, which abolishes the conversion of methylsulfinyl glucosinolates into alkenyl glucosinolates so glucoraphanin is enriched in these B. oleracea vegetables (Li et al. 2003; Li and Quiros 2003). However, except for the deletion found in broccoli and purple cauliflower, there is no other mutation of the GSL-ALK gene family that is found in other B. oleracea varieties or other brassica species. Since there are multi-copy tandem repeats of the AOP homologs (Gao et al. 2004), it is not easy to change several functional genes into non-functional ones. Moreover, in amphidiploid brassica species such as B. napus and B. juncea, gene duplication and function redundancy is extensively increased. It is even more difficult to develop a mutant where multiple genes are knocked out simultaneously. RNA interference (RNAi) has been demonstrated to be an effective method to silence genes and gene families with sequence similarity to down-regulate GSL-ELONG gene expression in B. napus (Liu et al. 2010). In this research, RNAi was undertaken to silence the entire GSL-ALK gene family to reduce the detrimental glucosinolate progoitrin and increased the beneficial glucosinolate glucoraphanin in B. napus.
Materials and methods
Preparation of GSL-ALK RNAi construct
The fragments from the B. oleracea GSL-ALK gene (NCBI Accession no. AY044424), the pyruvate orthophosphate dikinase (PDK) intron and modified USER binary vector pBI121U (Liu et al. 2010) were used to make an RNAi construct. Two pairs of primers UOD13A, UOD24A, and UOD13B, UOD24B, located in the middle exons of the broccoli GSL-ALK gene were used to amplify sense and anti-sense fragments (427 bp) with cDNA. An intron was amplified with two primers, INT5 and INT6, designed with the sequence of the PDK intron in pHellsgate 8 vector (Fig. 2a). These three amplified fragments GSL-ALK sense, PDK intron and GSL-ALK anti-sense, were inserted into the modified binary vector pBI121U to have an RNAi construct 35S::UOD (Fig. 2b). All primers were synthesized by Invitrogen, Toronto, Canada (Table 1).

Structure of GSL-ALK in B. oleracea and RNAi construct. a Gene structure of GSL-ALK in B. oleracea ((NCBI Accession no. AY044424). Forward primers UOD13A and UOD13B and reverse primers UOD24A and UOD24B used to amplify ALK gene fragments. Forward primers INT5 and reverse primers INT6 used to amplify PDK gene fragment from pHellsgate vector. b RNAi construct 35S::UOD contains the gene inserts amplified from the middle exon 2 of GSL-ALK, PDK as intron and 35S promoter. Two arrows are the locations of primers 35SB inside the 35S promoter and INUP1 inside the PDK intron part that were used to confirm transgenic individuals
A procedure similar to that described by Nour-Eldin et al. (2006) was used to assemble the RNAi construct. The 5′ universal uracil primer tails were “5′ −GGAGTTAAU+” and “5′ −GGTCTTAAU+” that were added to the forward primers UOD13A and UOD13B. The 3′ universal reverse primer tails “5′ −ACGAACAGGU+” and “5′ −ATCCTCTAGU+” in UOD24A and UOD24B were complementary to the tails of two uracil-containing primers INT5 and INT6. ‘GCTGAGGAGTTAATTAAGACCTCAGC’ was inserted into a binary vector pBI121 to produce the modified USER binary vector pBI121U. The vector was prepared by digesting with PacI and subsequent nicking by Nt.BbvCI (New England Biolabs, Toronto, Canada) according to the USER cloning method described by Nour-Eldin et al. (2006). Two GSL-ALK gene fragments and the intron fragment were digested with a USER™ enzyme (New England Biolabs, Toronto, Canada).The inserts and the linearized vector DNA were incubated for 20 min at 37 °C followed by 20 min at 25 °C and the hybridized product transformed into chemically competent E. coli DH10B cells (Invitrogen Canada). The positive clones were isolated and sequenced to confirm the accuracy of the sequence. The recombination RNAi construct, named 35S::UOD, was electroporated into Agrobacterium tumefaciens strain GV3101.
Plant transformation and development of transgenic plants
The protocol of B. napus transformation described by Moloney et al. (1989) was used with modifications. Seeds of a canola cultivar ‘Westar’ were surface-sterilized for 15 min in 4 % sodium hypochlorite, with 0.1 % Tween 20 added as a surfactant. Then the seeds were washed thoroughly with sterile distilled water and germinated on 1/2 MS medium (2.2 g l−1 MS basal medium) (SIGMA, Canada) with 10 g l−1 sucrose. Hypocotyls were harvested from 5 day old seedlings, cut into 4–6 mm pieces and placed onto a pre-culture/co-culture MS medium (4.4 g l−1 MS basal medium) with 30 g l−1 sucrose, 1.0 mg l−1 2,4-D, 1.0 mg l−1 kinetin and 8 g l−1 agar. Incubation was performed for 3 days at 25 °C to prepare explants for transformation. Agrobacterium containing 35S::UOD was grown overnight on a shaker at 28 °C in 50 ml LB broth with 30 mg l−1 gentamicin and 50 mg l−1 kanamycin. The bacterial cells were harvested and re-suspended in the same volume of liquid hormone-free MS with 30 g l−1 sucrose. The explants were collected and mixed thoroughly with a tenfold dilution of Agrobacterium suspension of hormone-free MS with 30 g l−1 sucrose. The excess liquid medium was discarded and the explants were co-cultured with Agrobacterium containing 35S::UOD on the pre-culture/co-culture MS medium for 5 days. The explants were transferred to SIM medium (MS with 30 g l−1 sucrose, 8 g l−1 agar, 3 mg l−1 6-benzylaminopurine, 1 mg l−1 zeatin, 5 mg l−1 silver nitrate, 0.5 g l−1 MES and 300 mg l−1 timentin and 20 mg l−1 kanamycin) to perform selection of transformed cells. After a further 2 weeks of incubation at 25 °C, the explants were sub-cultured to fresh SIM medium. The first shoots were developed after 3–4 weeks. The proliferated green shoots were transferred to SEM medium (MS medium with 20 g l−1 sucrose, 8 g l−1 agar, 0.5 mg l−1 6-benzylaminopurine, 150 mg l−1 phloroglucinol, 0.03 mg l−1 GA3, 0.5 g l−1 MES) and the antibiotics as mentioned above for shoot elongation. The elongated shoots were transferred to RIM medium (1/2 MS medium containing 10 g l−1 sucrose, 0.5 mg l−1 IBA, 0.5 g l−1 MES and 300 mg l−1 timentin, 25 mg l−1 kanamycin). The rooted shoots were transferred to soil for acclimatization and plants were grown till maturity to harvest transgenic seeds (T1). Since the original transformed material was the canola cultivar ‘Westar’ with low concentration of glucosinolates (about 10 μmol g−1 seed). The T1 plants were crossed with a doubled haploid (DH) line DH241 in rapeseed with high concentration of glucosinolates (about 100 μmol g−1 seed) to test the effect of gene silencing in a genetic background of high glucosinolate concentration. The generated F1 transgenic plants and F2 seeds from F1 transgenic plants were analyzed to determine the effect of GSL-ALK gene silencing using semi-quantitative RT-PCR, HPLC and LC-MS analysis methods.
DNA extraction and PCR analysis
PCR was used to confirm the presence of the transgene in the positive T0, T1 and F1 transgenic plants. Plant genomic DNA was extracted using a CTAB (cety-trimethyl-ammonium bromide) method described by Li and Quiros (2003). The primer pair 35SB and INUP1 was used to identify 35S::UOD RNAi transgenic plants. PCR reactions were carried out in a 20 μl mixture at 94 °C for 3 min; 35 cycles of amplification at 94 °C for 1 min, 55 °C for 1 min, 72 °C for 1 min. PCR products were separated on 1 % agarose gel and sequenced to confirm that the inserts were correct.
Southern blot analysis
Copy numbers of inserts were determined by Southern blot analysis. Quantity and quality of genomic DNA was measured by NanoDrop 2000 (Thermo Scientific, DE, USA) with UV absorption at 260 nm. Seven micrograms of DNA was digested with EcoRI restriction enzyme and electrophoresed on 0.7 % agarose gel. DNA was transferred onto positively charged nylon membranes (Roche Applied Science, IN, USA) by overnight capillary transfer using 20× SSC buffer. The probe labelling was performed using PCR DIG probe synthesis kit (Roche Applied Science, IN) and primer pair INT5A and INT6A. Immobilization of DNA to nylon membrane was performed by UV radiation. Subsequent pre-hybridization, hybridization and immunological detection were performed following the procedure described by the DIG luminescent detection kit (Roche Applied Science, IN).
Semi-quantitative RT-PCR analysis of GSL-ALK gene expression
Semi-quantitative RT-PCR was used to analyze GSL-ALK gene expression in F1 transgenic plants. RNA was extracted from young leaves of F1 transgenic and non-transgenic plants using TRIZOL (Invitrogen, Toronto, Canada). Total RNA was used to synthesize single strand cDNA using Superscript III reverse transcriptase kit (Invitrogen, Toronto, Canada). Each 20 μl reaction mixture contained 50-100 ng RNA, oligo (dT18) primers and other components according to the manufacturer’s instructions. In semi-quantitative RT-PCR, primers ALKP1 and ALKP2 covering the partial coding region of the B. oleracea GSL-ALK gene were used to check the gene expression in transgenic and non-transgenic individuals and primers BRUBQ1 and BRUBQ2 were used to amplify the brassica ubiquitin (UBQ) gene as a check. A 50 μl RT-PCR reaction was set up and run at 94 °C for 3 min; 36 cycles for amplification of GSL-ALK cDNA and 26 cycles for UBQ at 94 °C for 1 min, 55 °C for 1 min, 72 °C for 1 min. PCR products were visualized by electrophoresis on 1 % agarose gel and sequenced to confirm that they were the correct ones. Quantification of RT-PCR products was scanned and measured by an Alpha Innotech digital gel documentation system and AlphaEase FC software (Fischer Scientific, Ottawa, Canada). Primers were listed in table 1.
Analysis of desulfoglucosinolates by LC-MS
Desulfoglucosinolates in seeds of an F1 individual of transgenic line (DH241 × UOD-20)-4 was analyzed on a Varian 212-LC binary gradient LC-MS equipped with Polaris™ C18 A column (100 mm × 2.0 mm, 3 μM) (Varian Inc., CA, USA). The binary mobile phase system consisted of solvent A, distilled water with 0.1 % formic acid and solvent B, acetonitrile with 0.1 % formic acid at a flow rate of 300 μl/min. For better separation of non-polar and polar compounds, the following gradient elution program was used: 0.01 min, 98:2 A/B; 8 min, 93:7 A/B; 12 min 85:15 A/B; 30 min, 45:55 A/B; 35 min, 8:92 A/B; 40 min, 8:92 A/B; 45 min, 98.5:1.5 A/B; 48 min, 98.5:1.5 A/B and 52 min, 100:0 A/B. The system was equilibrated by running the above mobile phase for 15 min before loading samples. Twenty microliter of desulphoglucosinolates samples were injected using an HTS Pal autosampler (Varian Inc, CA, USA). For MS analysis, the flow was diverted to a Varian 500 mass spectrometer LC Ion Trap (Varian Inc., CA, USA) equipped with an electro spray ionization (ESI) interface and configured for positive ionization mode. The interface and MSD detector parameters were set as follows: positive polarity capillary voltage 80 V, needle voltage 5,000 V, electron multiplier voltage 1,325 V, trap damping gas flow rate 0.8 ml/min, infusion flow rate 20.0 μl/min, purge flow rate 200 μl/min, nebulizer gas pressure 50 psi, drying gas pressure 30 psi, drying gas temperature 350 °C, scan rage 110–700 m/z and scan time 3.34 s/scan. Nitrogen was used as nebulizing and drying gas. Data were processed and analyzed by Varian MS workstation software.
Determination of profile and concentration of glucosinolates in seeds
HPLC was used to identify the profile and concentration of glucosinolates in seeds from T0 and F1 transgenic and non-transgenic plants. Two hundred mg of seeds were ground in liquid nitrogen. Total desulfoglucosinolates were extracted and purified using Sephadex/sulfatase protocols as previously described (Kliebenstein et al. 2001) with some modifications. Fifty microliters of desulfoglucosinolate extract was injected into a 5-mm column (Lichrocart 250–4 RP18e, Fisher Scientific, Ottawa, Canada) on a Waters HPLC. Desulfoglocosinolates were detected at 229 nm. The running program was an 8-min gradient from 1.5 to 7.0 % (v/v) methanol, a 4-min gradient from 7 to 15 % (v/v) methanol, a 18-min gradient from 15 to 55 % (v/v) methanol, a 5-min gradient from 55 to 92 % (v/v) methanol, 5 min at 92 % (v/v) methanol, a 5-min gradient from 92 to 1.5 % (v/v) methanol, 3 min at 1.5 % (v/v) methanol and final 4 min at 0 %(v/v) methanol. All desulfoglucosinolate absorbance data (measured at 229 nm) were converted to micromoles glucosinolates per gram dry weight seeds using response factors determined from the purified standards for each of the glucosinolates (Vinjamoori et al. 2004).
Results
Development of transgenic plants and altered seed glucosinolate concentration in transgenic canola
A canola cultivar ‘Westar’ was transformed with an RNAi construct through Agrobacterium-mediated method. Over 40 T0 transgenic plants were identified using 35SB and INUP1 primers. Most transgenic plants did not show morphological changes and set seeds, except for a few plant. The T1 seeds were harvested from 28 independent transformants and seed glucosinolates were analysed using HPLC. Among these T1 seeds, eighteen transgenic lines contained detectable glucoraphanin concentration, ranging from 0.1 to 0.98 μmol g−1 (Supplementary material S1). The ratio of glucoraphanin to progoitrin, however, varied considerably. The highest ratio of glucoraphanin to progoitrin in the T1 seeds of transgenic line UOD-20 was 0.31 and others ranged from zero to 0.23. Therefore, the different concentration of a new detectable glucosinolate component glucoraphanin and ratio of glucoraphanin to progoitron in T1 seeds of transformants implied different effect of GAL-ALK gene silencing. The B. napus canola cultivar contains low concentrations of glucosinolates, making it difficult to detect glucosinolate changes. The copy numbers in eight transgenic lines were determined through PCR amplification of gene inserts with the primer pair 35SB and INUP1 (Supplementary material S2). Transgenic and non-transgenic individuals in six transgenic lines fitted a ratio of 3:1, suggesting that there was one T-DNA insert in each line, while in other two lines, they fitted a ratio of 15:1, suggesting that there were two copies of T-DNA inserts. To determine the effect of GSL-ALK gene silencing on the genetic background of high glucosinolate concentration, one T1 line UOD-21 with two copies of T-DNA inserts and two T1 lines UOD-18 and UOD-20 with a single copy of T-DNA insert were used. These three T1 lines displayed different concentrations of glucoraphanin and the ratio of glucoraphanin to progoitrin were selected to cross with the DH 241, a DH line with high concentration of glucosinolates (approximately 100 μmol g−1 seed). The generated F1 transgenic lines, UOD-21 × DH241, UOD-18 × DH241 and DH241 × UOD-20 and the history of development of transgenic plants was presented in Supplementary material S3. Seeds from those F1 transgenic plants were analyzed to further determine the effect of GSL-ALK gene silencing. The copy numbers of inserts in these three crosses were confirmed by Southern blot analysis (Fig. 3). The results were the same as those obtained by analysis of segregation ratio through amplification of inserts. Two transgenic lines UOD-18 and UOD-20 contained one copy of insert while two copies of inserts in the transgenic line UOD-21 was confirmed.

Southern blot analysis of F1 plants of three tranformant and non-tranformant crosses. Genomic DNA was digested with EcoRI and probed with PDK intron fragment obtained by PCR DIG labeling reaction. Lane 1 UOD-18 × DH241 (single copy); 2 DH241 × UOD-20 (no insert); 3 DH241 × UOD-20 (single copy); 4 UOD-21 × DH241 (single copy); 5 UOD-21 × DH241 (two copies); 6 DH241 (control); 7 UOD-18 × DH241 (no insert); 8 DH241 × UOD-20 (single copy)
Semi-quantitative RT-PCR analysis of GSL-ALK gene expression
To determine the effect of GSL-ALK gene silencing, thirteen F1 individuals, including three transgenic individuals as biological repeats within each F1 transgenic lines (UOD-21 × DH241, UOD-18 × DH241 and DH241 × UOD-20) and four non-transgenic individuals as negative control, were analyzed. The F1 transgenic individuals were confirmed through amplification of the T-DNA inserts. The expression of the GSL-ALK genes in nine transgenic individuals of three transgenic lines and four non-transgenic individuals was analyzed using semi-quantitative RT-PCR. The results showed that the expression levels of the GSL-ALK genes in non-transgenic plants was relatively high, and in the transgenic plants, dramatically reduced, suggesting that the gene silencing was very effective (Fig. 4 and Supplementary material S4).

A Semi-quantitative RT-PCR analysis of GSL-ALK gene expression in F1 RNAi transgenic and non-transgenic plants. ALK gene expression showed down-regulated expression in the RNAi transgenic lines. a Panel 1 PCR amplification products of genomic DNA of RNAi inserts in crosses of rapeseed and RNAi transgenic canola lines with primers S35B and INDOWN1; panel 2 semi-RT-PCR products of GSL-ALK gene cDNA in the same set of samples as in panel 1 with primers ALKP1 and ALKP2; panel 3 RT-PCR products of ubiquitin cDNA as a check. Lanes 1–4 four negative controls of non-transgenic F1 individuals; lanes 5–13, three lanes within each transgenic F1 individuals, UOD-21 × DH241 (lanes 5–7), UOD-18 × DH241 (lanes 8–10), DH241 × UOD-20 (lanes 11–13). B Quantification of GSL-ALK gene transcripts in the RNAi transgenic lines and non-transgenic controls was scanned in AlphaImager HP and measured AlphaImager EP Software. The error bars indicating SE from three or four repeats. Double asterisk indicates changes that are highly significantly difference (p < 0.01) compared to the negative controls
HPLC and LC-MS analysis for identification of glucosinolates
To identify the glucosinolate components, the (F2) seeds of F1 transgenic individual (OUD20 × DH241)-D and non-transgenic individual were analyzed using HPLC. Compared with non-transgenic individuals, the (F2) seeds of the F1 transgenic plant produced a new glucosinolate peak while other peaks were common between transgenic and non-transgenic individuals (Fig. 5). To accurately analyze the changes in glucosinolates, extracted desulfoglucosinolates were analyzed with LC-MS. Individual glucosinolates were identified on the basis of their [M + H]+ and [M + Na]+ ions. In LC-MS spectra of desulfoglucosinolates in seeds of an F1 transgenic individual (DH241 × UOD-20)-D, the [M + Na]+ ions were m/z 332 desulfoprogoitrin, m/z 380 desulfoglucoraphanin, m/z 394 desulfoglucoalyssin, m/z 316 desulfogluconapin, m/z 364 desulfoglucoerucin, and the [M + H]+ ion was m/z 385 4-hydroxy-desulfoglucobrassicin (Fig. 5c). Besides these major peaks, a few other peaks detected such as desulfogluconapoleiferin, desulfoglucobrassicanapin and desulfoglucobrassicin. The results showed that the new component of glucosinolate desulfoglucoraphanin was dramatically induced in seeds of the transgenic F1 plant compared to non-transgenic F1 plants, which confirmed the effect of the GSL-ALK gene silencing.

HPLC chromatograms of desulfoglucosinolates in seeds of F1 non-transgenic and transgenic individuals and MS spectra of desulfoglucosinolates in seeds of an F1 transgenic individual (DH241 × UOD-20) in B. napus. a HPLC profile in an F1 individual (B. napus rapeseed DH241 × canola ‘Westar’); b HPLC profile in an F1 transgenic individual (DH241 × UOD-20); peak 1 Desulfoprogoitrin (C4); peak 2 desulfoglucoraphanin (C4); peak 3 desulfogluconapoleiferin (C5); peak 4 desulfoglucoalyssin (C5); peak 5 desulfogluconapin (C4); peak 6 hydroxy-desulfoglucobrassicin; peak 7 desulfoglucobrassicanapin (C5); peak 8 Desulfoglucoerucin (C4); peak 9 desulfoglucobrassicin. c MS spectra of desulfoglucosinolates in seeds of an F1 transgenic individual (DH241 × UOD-20) in B. napus. Dominant ions at m/z 332, 380, 346, 394, 316, 385, 330, 364 and 391 in MS spectra 1–9 correspond to desulfoglucosinolates with the same numbers in HPLC profiles of F1 transgenic individual
Determination of seed glucosinolate concentration in transgenic rapeseed
Seed glucosinolates of F1 transgenic plants were analyzed by HPLC. The seeds (F2) harvested from three F1 transgenic lines UOD-21 × DH241, UOD-18 × DH241 and DH241 × UOD-20, 4-6 individuals within each transgenic line were considered replicates and three non-transgenic F1 individuals were used as negative controls. ANOVA was performed and the results showed that the concentration of seed glucosinolates of three F1 transgenic lines was significantly different to the negative control (Fig. 6). Glucoraphanin was produced in all F1 transgenic individuals. The average concentration of glucoraphanin in the seeds from F1 transgenic lines UOD-21 × DH241, UOD-18 × DH241 and DH241 × UOD-20 was 15.1, 19.2 and 42.6 μmol g−1 seed, respectively. The highest glucoraphanin concentration (57.4 μmol g−1 seed) was detected in one F1 individual (DH241 × UOD-20)-D (Table 2). In the F1 transgenic line DH241 × UOD-20, the average ratio of progoitrin to glucoraphanin was about 0.65, indicating that the concentration of glucoraphanin was higher than that of progoitrin. Moreover, in these F1 individuals derived from the transgenic line UOD-20, the reduction of progoitrin and the increase of glucoraphanin, glucoalyssin and glucoerucin concentration were statistically significantly different to the other two transgenic lines UOD-18 and UOD-21. It was suggested that RNAi effects in transgenic line UOD-20 were stronger than in the other two transgenic lines. The concentrations of progoitrin, gluconapoleiferin and glucobrassicanapin were dramatically reduced, whereas glucoraphanin, glucoalyssin and glucoerucin increased dramatically in all transgenic rapeseed lines (Fig. 6). However, gluconapin was reduced only in the UOD-20 derived individuals, and the average concentrations of gluconapin in transgenic UOD-21 × DH241, UOD-18 × DH241 lines were similar to that of negative controls, suggesting reduction of gluconapin occurred only when gene silencing was stronger. The levels of gene silencing effects are considered to be insertion position effect. The results indicated that the glucosinolates in up-stream were enriched, most glucosinolates, especially progoitrin in down-stream were reduced when the biosynthetic pathway of aliphatic glucosinolates was partially blocked by silencing the GSL-ALK genes. It further confirmed that silencing the GSL-ALK genes was achieved very effectively.

Glucosinolate concentrations in seeds of F1 transgenic lines UOD21 × DH241, UOD18 × DH241, DH241 × UOD20 and F1 non-transgenic negative control. Asterisks (1) progoitrin; (2) glucoraphanin; (3) gluconapoleiferin; (4) glucoalyssin; (5) gluconapin; (6) hydroxy-glucobrassicin; (7) glucobrassicanapin; (8) glucoerucin; (9) glucobrassicin. Data were subjected to statistical analysis by analysis of variance (ANOVA) with Duncan’s multiple range test (p = 0.05 or 0.01) in software SPSS v13.0. Bars represent SE (n > 3). Different letters in a column show significant differences (p < 0.01)
Discussion
All brassica species have relatively high levels of gene duplication and gene functional redundancy. Therefore, it is difficult to knock out all members of a gene family to create a non-functional mutant with desired traits. To enrich glucoraphanin concentration, it is necessary to block the chain modification of aliphatic glucosinolate biosynthesis, that is, to knock out or knock down all GSL-ALK genes. According to the published literature, there are two to four members of the AOP gene family in B. oleracea (Gao et al. 2004) and two mapped loci in the A genome of B. rapa (Lou et al. 2008). Actually, when using the whole genome sequence of B. rapa available at the website http://brassicadb.org/brad/, three loci and five members of GSL-ALK homologs were identified after blasting the whole B. rapa genome sequence with the B. oleracea GSL-ALK gene (NCBI Accession no. AY044424). Two loci on R3 and R9 and each containing two members might correspond to the two loci mapped previously (Lou et al. 2008). One locus on R5 with single copy was also identified (unpublished data). Therefore, there are five members of the AOP gene family in B. rapa, two to four members of the AOP gene family in B. oleracea (Gao et al. 2004), and nine members of the AOP gene family in B. napus. Since there is no mutant of the GSL-ALK gene family that has ever been found in other brassica species except in broccoli and purple cauliflower of B. oleracea, glucoraphanin has not been detectable in most brassica species including B. rapa, B. napus and B. juncea, from which dozens of vegetables are commonly cultivated globally. B. napus vegetables such as rutabaga and swede are mainly consumed in Japan and China, whereas B. rapa vegetables are more popular worldwide. Since B. rapa is recalcitrant to Agrobacterium-mediated transformation, it is feasible to have transgenes in B. napus and then to introduce transgenes easily from B. napus to B. rapa through interspecific hybridization and gene introgression. In this report, RNAi of the GSL-ALK gene family in B. napus leads to the reduction of progoitrin and increase of glucoraphanin, that is, the reduction of a toxic glucosinolate and the enrichment of a cancer-preventive compound in the RNAi transgenes. Glucoraphanin was increased to a relatively high level in some transgenic individuals and progoitrin was reduced by about 60 %. Thus, the GSL-ALK RNAi transgenic rapeseed with high concentration of glucoraphanin might be grown as sprouts (Shapiro et al. 2006) or made into health supplements for human consumption to capture the cancer preventive effect.
The RNAi effects varied in different transgenic plants. The positional effects of trangenes might be important and relatively large transgenic populations are essential to identify the best transgenic plants that have the beneficial trait for practical use. The copy numbers in transgenic plants might have less effect than positional effect according to the results of seed glucosinolates in T1 and F1 generations (Table 2, Supplementary material S1 and S2) since the transgenic line UOD20 with single copy T-DNA insert produced highest concentration of glucoraphanin in seeds of the F1 plants. From the twenty-eight transgenic individuals that were generated, the best transgenic line that was identified contained approximately 60 % reduction of progoitrin. Therefore, there is potential to reduce progoitrin concentration and increase glucoraphanin in B. napus through production of more transgenic plants. The sequence of different genes in the same family and their expression levels and functionality are diverse. Another strategy is to have double RNAi transgenic plants since one construct might not be enough to silence all related gene members.
The current rapeseed DH line contains approximately 100 μmol g−1 seed of total glucosinolates and transgenic plants with about 40 μmol g−1 seed glucoraphanin were produced. In broccoli, glucoraphanin concentration is about 20 μmol g−1 DW of head tissue (Kushad et al. 1999) and 70 μmol g−1 seed (Velasco and Becker 2000). Through selection for high concentration glucosinolates, it may be possible to increase glucoraphanin concentration further if the selected rapeseed contains higher glucosinolates than the DH line used in this study. Therefore, it is possible to produce B. napus that contains higher concentration of glucoraphanin as health vegetables than broccoli. Additionally, when seeds and sprouts are manufactured as health-supplementary food, it is much easier to produce large quantities of B. napus seeds than broccoli.
RNAi of the GSL-ALK gene family in B. napus did not show much change in total glucosinolates’ concentration in either the low glucosinolate background of canola or the high glucosinolate background of rapeseed. This is advantageous for the enrichment of glucoraphanin through silencing of GSL-ALK gene family. According to the literature, knock-out and knock-down of transcription factors MYB28, MYB29 and MYB76 in the biosynthetic pathways lead to reduced aliphatic glucosinolates (Gigolashvili et al. 2007; Sønderby et al. 2007) and in the double mutants of MYB28/MYB29, the gene expression of MAM1 and MAM3 controlling side chain elongation are significantly down-regulated (Beekwilder et al. 2008). Moreover, cytochrome P450 monooxygenases coded for by CYP79F1, CYP79F2 and CYP83A1 are involved in the formation of oldoxime and knock out mutant of CYP79F1 nearly eliminated all short chain aliphatic glucosinolates in Arabidopsis (Reintanz et al. 2001). Therefore, manipulation of these transcription factors, side chain elongation and cytochrome P450 genes might change the total concentration of aliphatic glucosinolates. However, since the GSL-ALK genes function in the modification step of alkenyl to alkyl glucosinolates (Fig. 1), their silencing might not affect the expression of B. napus genes homologous to those Arabidopsis transcription factors, side chain elongation and cytochrome P450 genes mentioned previously. Interestingly, gene silencing of the GSL-ELONG homologous to Arabidopsis MAM1 resulted in dramatic reduction of 4C and 5C aliphatic glucosinolates and induction of 3C side chain aliphatic glucosinolates (Liu et al. 2010), suggesting that the regulation of aliphatic glucosinolates’ biosynthesis in B. napus is quite complex.
References
Beekwilder J, van Leeuwen W, van Dam NM, Bertossi M, Grandi V, Mizzi L, Soloviev M, Szabados L, Molthoff JW, Schipper B, Verbocht H, de Vos RC, Morandini P, Aarts MG, Bovy A (2008) The impact of the absence of aliphatic glucosinolates on insect herbivory in Arabidopsis. PLoS ONE 3:e2068
Fahey JW, Zhang Y, Talalay P (1997) Broccoli sprouts: an exceptionally rich source of inducers of enzymes that protect against chemical carcinogens. Proc Natl Acad Sci USA 94:10367–10372
Gamet-Payrastre L, Li P, Lumeau S, Cassar G, Dupont MA, Chevolleau S, Gasc N, Tulliez J, Terce F (2000) Sulforaphane, a naturally occurring isothiocyanate, induces cell cycle arrest and apoptosis in Ht29 human colon cancer cells. Cancer Res 60:1426–1433
Gao M, Li G, Yang B, McCombie RW, Quiros CF (2004) Comparative analysis of a Brassica BAC clone containing several major aliphatic glucosinolate genes with its corresponding Arabidopsis sequence. Genome 47:666–679
Gigolashvili T, Yatusevich R, Berger B, Muller C, Flugge UI (2007) The R2R3-MYB transcription factor HAG1/MYB28 is a regulator of methionine-derived glucosinolate biosynthesis in Arabidopsis thaliana. Plant J 51:247–261
Kliebenstein DJ, Kroymann J, Brown P, Figuth A, Pedersen D, Gershenzon J, Mitchell-Olds T (2001) Genetic control of natural variation in Arabidopsis thaliana glucosinolate accumulation. Plant Physiol 126:811–825
Kushad MM, Brown AF, Kurilich AC, Juvik JA, Klein BP, Wallig MA, Jeffery EH (1999) Variation of glucosinolates in vegetable crops of Brassica oleracea. J Agric Food Chem 47:1541–1548
Li G, Quiros CF (2003) The Brassica oleracea gene BoGSL- ALK regulates in planta alkenyl aliphatic glucosinolates in Arabidopsis thaliana. Theor Appl Genet 106:1116–1121
Li G, Riaz A, Goyal S, Abel S, Quiros CF (2001) Inheritance of three major genes involved in the synthesis of aliphatic glucosinolates in Brassica oleracea. J Am Soc Hortic Sci 126:427–431
Li G, Gao M, Yang B, Quiros CF (2003) Gene to gene alignment between the Arabidopsis and Brassica oleracea genomes. Theor Appl Genet 107:168–180
Liu Z, Hammerlindl J, Keller W, McVetty PBE, Daayf F, Quiros CF, Li G (2010) MAM gene silencing leads to the induction of C3 and reduction of C4 and C5 side chain aliphatic glucosinolates in Brassica napus. Mol Breed 27:467–478
Lou P, Zhao J, He H, Hanhart C, Del Carpio DP, Verkerk R, Custers J, Koornneef M, Bonnema G (2008) Quantitative trait loci for glucosinolate accumulation in Brassica rapa leaves. New Phytol 179:1017–1032
Magrath R, Herron C, Giamoustaris A, Mithen R (1993) The inheritance of aliphatic glucosinolates in Brassica napus. Plant Breed 111:55–72
Mithen R, Faulkner K, Magrath R, Rose P, Williamson G, Marquez J (2003) Development of isothiocyanate-enriched broccoli, and its enhanced ability to induce phase 2 detoxification enzymes in mammalian cells. Theor Appl Genet 106:727–734
Moloney MM, Walker JM, Sharma KK (1989) High efficiency transformation of Brassica napus using Agrobacterium vectors. Plant Cell Rep 8:238–242
Neal CS, Fredericks DP, Griffiths CA, Neale AD (2010) The characterization of AOP2: a gene associated with biosynthesis of aliphatic alkenyl glucosinolates in Arabidopsis thaliana. BMC Plant Biol 10:170–186
Nour-Eldin HH, Hansen BG, Norholm MHH, Jensen JK, Halkier BA (2006) Advancing uracil-excision based cloning towards an ideal technique for cloning PCR fragments. Nucleic Acids Res 34:e122
Reintanz B, Lehnen M, Reichelt M, Gershenzon J, Kowalczyk M, Sandberg G, Godde M, Uhl R, Palme K (2001) Bus, a bushy Arabidopsis CYP79F1 knockout mutant with abolished synthesis of short-chain aliphatic glucosinolates. Plant Cell 13:351–367
Rosa EAS, Heaney RK, Fenwick GR, Portas CAM (1997) Glucosinolates in crop plants. Hortic Rev 19:99–215
Shapiro TA, Fahey JW, Wade KL, Stephenson KK, Talalay P (2001) Chemoprotective glucosinolates and isothiocyanates of broccoli sprouts: metabolism and excretion in humans. Cancer Epidemiol Biomarkers Prev 10:501–508
Shapiro TA, Fahey JW, Dinkova-Kostova AT et al (2006) Safety, tolerance, and metabolism of broccoli sprout glucosinolates and isothiocyanates: a clinical phase I study. Nutr Cancer 55:53–62
Sønderby IE, Hansen BG, Bjarnholt N, Ticconi C, Halkier BA et al (2007) A systems biology approach Identifies a R2R3 MYB gene subfamily with distinct and overlapping functions in regulation of aliphatic glucosinolates. PLoS ONE 2:e1322
Velasco L, Becker HC (2000) Variability for seed glucosinolates in a germplasm collection of the genus Brassica. Genet Resour Crop Evol 47:231–238
Vinjamoori V, Byrum JR, Hayes T, Das PK (2004) Challenges and opportunities in the analysis of raffinose oligosaccharides, pentosans. J Anim Sci 82:319–328
Zhang GY, Talalay P, Cho C, Posner GH (1992) A major inducer of anticarcinogenic protective enzymes from broccoli: isolation and elucidation of structure. Proc Natl Acad Sci USA 89:2399–2403
Acknowledgments
The research was supported by the Genome Canada/Genome Alberta and Genome Prairie and Manitoba Provincial Government, and by NSERC discovery grant.
Author information
Authors and Affiliations
Corresponding author
Additional information
Zheng Liu and Arvind H. Hirani: contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Liu, Z., Hirani, A.H., McVetty, P.B.E. et al. Reducing progoitrin and enriching glucoraphanin in Braasica napus seeds through silencing of the GSL-ALK gene family. Plant Mol Biol 79, 179–189 (2012). https://doi.org/10.1007/s11103-012-9905-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11103-012-9905-2