
Abstract
Plants in the Nicotiana genus produce nicotine and related pyridine alkaloids as a part of their chemical defense against insect herbivores. These alkaloids are formed by condensation of a derivative of nicotinic acid, but the enzyme(s) involved in the final condensation step remains elusive. In Nicotiana tabacum, an orphan reductase A622 and its close homolog A622L are coordinately expressed in the root, upregulated by methyl jasmonate treatment, and controlled by the NIC regulatory loci specific to the biosynthesis of tobacco alkaloids. Conditional suppression of A622 and A622L by RNA interference inhibited cell growth, severely decreased the formation of all tobacco alkaloids, and concomitantly induced an accumulation of nicotinic acid β-N-glucoside, a probable detoxification metabolite of nicotinic acid, in both hairy roots and methyl jasmonate-elicited cultured cells of tobacco. N-methylpyrrolinium cation, a precursor of the pyrrolidine moiety of nicotine, also accumulated in the A622(L)-knockdown hairy roots. We propose that the tobacco A622-like reductases of the PIP family are involved in either the formation of a nicotinic acid-derived precursor or the final condensation reaction of tobacco alkaloids.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Recent rapid advances in genomics and DNA sequencing are steadily generating increasing numbers of new nucleotide sequences, which may encode polypeptides that show similar or partial homology to known metabolic enzymes. Large-scale transcriptional profiling may reveal that some are coordinately regulated by structural or regulatory genes involved in established plant metabolic pathways, thus providing the first clues as to their possible biochemical functions (Saito et al. 2008). Pinpointing catalytic functions of orphan enzymes, however, is not always straightforward, particularly for those involved in secondary metabolism in non-model plants, because enzymatic reactions may be ill-defined and candidate substrates may not be readily available.
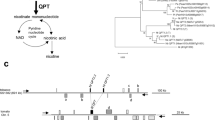
Previously we found a novel reductase gene while screening for genes whose expression was down-regulated in a low-nicotine mutant of tobacco (Nicotiana tabacum), and dubbed it A622, according to the clone number in a tobacco cDNA library (Hibi et al. 1994). A622 is a member of the PIP family of NADPH-dependent reductases, which were named after the three founding members; pinoresinol–lariciresinol reductase (Min et al. 2003), isoflavone reductase (Wang et al. 2006), and phenylcoumaran benzylic ether reductase (PCBER) (Min et al. 2003). Among the known members of this family (Fig. 1), PCBER and pterocarpan reductase are the most homologous to A622 (Akashi et al. 2006). Recently, two eugenol synthases were also reported to be included in this complex clade (Koeduka et al. 2008). However, tobacco has an orthologous PCBER, which is distinct from A622, and recombinant A622 does not show PCBER activity (Shoji et al. 2002). Since pterocarpans are phytoalexins found only in legumes, A622 does not appear to be a pterocarpan reductase either. Expression patterns of A622 (see below) suggest that A622 is unlikely to be a synthase which produces volatile phenylpropenes, such as eugenol and isoeugenol, in many herbs (Koeduka et al. 2008). A622 must be a reductase since the recombinant protein binds NADPH (Shoji et al. 2002), but its enzymatic reaction remains elusive.

Phylogenetic tree of the PIP-family reductases that include PLR, IFR, and PCBER. Shown is an unrooted neighbor-joining phylogenetic tree of tobacco A622 and A622L, and of related protein sequences from various plants. Sequence analysis was performed by using CLUSTAL X, and the nearest-joining method was used to create the phylogenetic tree. TREEVIEW was used to visualize the resulting tree. The scale indicates the average substitutions per site for each cladogram, and the numbers label the bootstrap value of each node (1,000 bootstrap trials). PCBER, phenylcoumaran bezylic ether reductase; IFR, isoflavone reductase; PTR, pterocarpan reductase; PLR, pinoresinol–lariciresinol reductase; EGS, eugenol reductase; IGS, isoeugenol reductase; and LAR, leucocyanidin reductase. NtA622 (Nicotiana tabacum, GenBank accession no. D28505), NtA622L (N. tabacum, AB445396), NtPCBER (N. tabacum, AB445398), BpPCBER (Betula pendula, AF282850), FiPCBER (Forsythia × intermedia, AF242491), PcPCBER (Pyrus communis, AF071477), PopPCBER (Populus trichocarpa, AJ005803), PtPCBER (Pinus taeda, AF081678), ThPCBER (Tsuga heterophylla, AF242495), LjPTR1 (Lotus japonicus, AB265589), LjPTR2 (L. japonicus, AB265590), LjPTR3 (L. japonicus, AB265591), LjPTR4 (L. japonicus, AB265592), CaIFR (Cicer arietinum, X60755), MsIFR (Medicago sativa, X58078), PsIFR (Pisum sativum, S72472), FiPLR (F. intermedia, U81158), LaPLR (Linum album, AJ849358), LuPLR (Linum usitatissimum, AJ849359), ThPLR (T. heterophylla, AF242501), TpPLR (Thuja plicata, AF242503), CbEGS1 (Clarkia breweri, EF467239), CbEGS2 (C. breweri, EF467240), CbIGS1 (C. breweri, EF467238), ObEGS1 (Ocimum basilicum, DQ372812), PhEGS1 (Petunia hybrida, EF467241), PhIGS1 (P. hybrida, DQ372813), DuLAR (Desmodium uncinatum, AJ550154), MdLAR (Malus domestica, DQ139836), VvLAR (Vitis vinifera, AJ865335)
Expression analyses indicated A622 to be closely associated with the biosynthesis of tobacco alkaloids. A622 is expressed specifically in the root, most strongly in the cortex of the root tip and in the outer cortex layer and endodermis in the differentiated region (Hibi et al. 1994; Shoji et al. 2002). The application of methyl jasmonate (MeJA) strongly up-regulates A622 expression, while the simultaneous addition of ethylene suppresses the MeJA-induced expression (Shoji et al. 2000a, 2002). Finally, the NIC regulatory loci for nicotine biosynthesis tightly control the expression of A622 (Hibi et al. 1994; Shoji et al. 2002). These expression profiles are very similar to those of the structural genes encoding enzymes involved in nicotine biosynthesis (Fig. 2), such as putrescine N-methyltransferase and N-methylputrescine oxidase (Hibi et al. 1994; Shoji et al. 2000b; Katoh et al. 2007), and are consistent with the spatial and temporal regulation of nicotine biosynthesis (Katoh et al. 2005).

Biosynthesis of tobacco alkaloids. A salvage pathway of NAD provides nicotinic acid and nicotinic acid mononucleotide, the latter of which is also synthesized from quinolinate by way of the de novo pathway. Nicotinic acid or its metabolite is coupled with N-methylpyrrolinium cation (MP), Δ1-piperidine, and a nicotinic acid derivative to yield nicotine, anabasine, and anatabine, respectively. Nornicotine is derived from nicotine, whereas anatalline may be synthesized from anatabine. Nicotinic acid N-glucoside (NaNG) is reversibly formed from nicotinic acid. PMT, putrescine N-methyltransferase; MPO, N-methylputrescine oxidase
Our previous attempts to constitutively down-regulate A622 expression in tobacco plants yielded transgenic plants in which A622 expression was suppressed only partially, and the mild suppression phenotype was lost in the selfed progenies (our unpublished results). In this study, inducible suppression of A622 and its close homolog was tested in tobacco hairy roots and cultured cells, to circumvent possible growth inhibitory effects in the knockdown cells. We then carefully monitored metabolic changes in the pathway of tobacco alkaloids. Our results strongly suggest that A622 is a reductase involved in a late step of tobacco alkaloid biosynthesis.
Materials and methods
Plant materials and transformation
Binary vectors were introduced into Agrobacterium rhizogenes strain ATCC15834 or A. tumefaciens strain EHA105 by electroporation. To generate transgenic hairy roots, leaf discs prepared from sterile tobacco plants (Nicotiana tabacum cv. Petit Havana SR1) were inoculated with A. rhizogenes as described by Kanegae et al. (1994). After selection and disinfection on solid MS medium (Murashige and Skoog 1962) containing 50 mg l−1 kanamycin and 250 mg l−1 carbenicillin, hairy roots were subcultured in liquid MS medium with 3% sucrose every 2 weeks. Cultured tobacco BY-2 cells (N. tabacum cv. Bright Yellow-2) were grown in liquid MS medium supplemented with 20 mg l−1 KH2PO4, 0.5 g l−1 MES and 0.2 mg l−1 2,4-D. Transgenic tobacco BY-2 cells were generated with A. tumefaciens strain EHA105, as described by An (1985). Four-day-old BY-2 cells were elicited with 50 μM MeJA (Wako Chemical, Osaka, Japan) in an auxin-free medium (Shoji et al. 2008), whereas 1-month-old tobacco plants (cv. Burley 21) were treated with 100 μM MeJA for 1 day and the root tissues were analyzed for the up-regulated expression of nicotine biosynthesis-related genes (Shoji et al. 2002). Tobacco alkaloids and their metabolic precursors were analyzed after 2 days of MeJA treatment.
Vector construction
For the RNAi-mediated constitutive suppression of A622, a partial fragment of A622 cDNA (+323 to +522; the adenine in the translational start codon ATG as +1) was amplified by PCR, subcloned into pHANNIBAL (Wesley et al. 2001), and then transferred to the binary vector pBI121 (Clontech, Mountain View, CA) to provide pHANNIBAL-A622RNAi, which drives RNAi suppression by using the CaMV 35S promoter. For inducible RNAi suppression, an RNAi cassette containing an inverted repeat of the partial fragment of A622 and the pdk intron of pHANNIBAL-A622RNAi was transferred to a multi-cloning site downstream of the OLexA promoter in the vector pER8 (Zuo et al. 2000), to provide pXVE-A622RNAi.
Genomic PCR and RT-PCR
Genomic DNA was isolated from root tissues of N. tabacum (cv. Petit Havana SR1), N. sylvestris and N. tomentsiformis by using a PureLink Plant Total DNA Purification kit (Invitrogen, Carsbad, CA). The following PCR primers were used to amplify genomic DNA (10 ng) with Ex Taq DNA polymerase (TaKaRa Bio, Shiga, Japan) for 30 cycles of 94°C for 30 s, 55°C for 30 s, and 72°C for 30 s; 5′-GGACCTCTGCCATTAAGGACT and 5′-CGCAGATTGATATCGACAAAC for A622, 5′-TGCAGATTGATGTCGACAAC and 5′-GGACCTATGCCGTTAAGAGTC for A622L, and 5′-AGTTGGAGGAGGTGATGATG and 5′-TATGTGGGTCGCTCAATGTC for tobacco α-tubulin genes.
Total RNA was isolated by using an RNeasy Plant Mini kit (Qiagen, Hilden, Germany). cDNA was synthesized from 1 μg of the total RNA by Super Script II reverse transcriptase (Invitrogen), and amplified using Ex Taq DNA polymerase (TaKaRa Bio) under the following conditions: 24 cycles of 94°C for 30 s, 55°C for 30 s and 72°C for 30 s. The PCR primers used were; 5′-GCATATGGCCAAATTAGTC and 5′-TCAGAGAAAGCACACTCAA for A622, and 5′-GCATATGGCCAAATTGACT and 5′-TCAGAGAAAGCACACTCGT for A622L. For α-tubulin genes, the primers used in the genomic PCR were also used in the RT-PCR.
Immunoblot analysis
Root tissues and cultured cells were frozen in liquid nitrogen, homogenized by mortar and pestle, thawed, and then immediately mixed with CelLytic P Cell Lysis Reagent (Sigma, St. Louis, MO). After centrifugation of the homogenates, soluble proteins in the supernatant were separated by SDS-PAGE (10% T), and transferred onto a BioTrace PVDF membrane (Pall, East Hills, NY) using a Trans-blot SD Semi-Dry Electrophoretic Transfer Cell (Bio-Rad, Hercules, CA). The membrane was blocked in 1× TBS buffer containing 3% (w/v) bovine serum albumin for 1 h. Polyclonal anti-A622 mouse serum (Shoji et al. 2000a), polyclonal anti-phospho(enol)pyruvate carboxylase (PEPC) rabbit serum (Chemicon, Temecula, CA) and monoclonal anti-β-tubulin mouse serum (Sigma) were diluted 1:1,000 in 1× TBS buffer containing 0.05% Triton X-100, and incubated with the membranes for 1 h. After washing, HRP-conjugated anti-mouse IgG from sheep and HRP-conjugated anti-rabbit IgG from donkey (GE Healthcare, Buckinghamshire, UK) were used as secondary antibodies to detect A622, PEPC, and β-tubulin with the ECL-plus Western Blotting System (GE Healthcare).
Purification and identification of NaNG
Tobacco hairy roots (XA12 line) were cultured with 2 μM β-estradiol (Sigma) for 2 weeks and harvested. Fresh roots (10 g) were homogenized in 50 ml of 80% methanol with a mortar and pestle. The methanol extract was concentrated by an evaporator, dissolved in 1 ml of deionized water, and loaded onto an ODS column (YMC ODS-AM 120-S50; column volume of 35.3 ml). The target compound was eluted with 0.1% acetic acid at a flow rate of 2.5 ml min−1. The fraction which contained the compound in the highest purity was concentrated and dissolved in the acetone/water mixture for crystallization. Colorless needle-like crystals (3.1 mg) were obtained.
Chemical spectra of the purified compound were as follows. 1H-NMR (400 MHz, D2O): δ 3.64–3.91 (5H, m, H-2′-6′), 4.00 (1H, d, J = 12.4 Hz, H-6′), 5.84 (1H, d, J = 8.7 Hz, H-1′), 8.21 (1H, dd, J = 8.0 and 6.2 Hz, H-5), 9.00 (1H, dd, J = 8.0 and 1.1 Hz, H-4), 9.09 (1H, dd, J = 6.2 and 1.1 Hz, H-6), and 9.33 (1H, s, H-2). 13C-NMR (100 MHz, D2O): δ 60.3 (C-6′), 68.6 (C-4′), 74.0 (C-3′), 75.3 (C-2′), 79.5 (C-5′), 95.1 (C-1′), 127.8 (C-5), 137.1 (C-3), 142.6 (C-2), 142.8 (C-6), 147.6 (C-4), and 167.4 (3-CO2H). UVΛH2O nm (ε): 272 sh (1600), 265 (1830), and 208 sh (3660). Positive-mode FAB-MS: 286.0925 (calculated molecular mass of protonated NaNG; 286.0927 for C12H15NO7 + H). Hydrolysis by β-glucosidase was done as reported (Nishitani et al. 1995).
Metabolite analyses
To extract tobacco alkaloids, plant samples (50 mg dry weight) were lyophilized, homogenized and then soaked in 4 ml of 50 mM sulfuric acid. The homogenate was sonicated for 15 min and centrifuged at 3,100 rpm for 15 min. The supernatant was neutralized by adding 0.4 ml of 25% ammonia water. The mixture (1 ml) was loaded onto an Extrelut-1 column (bed volume, 3 ml; Merck, Whitehouse Station, NJ) and eluted with 6 ml of chloroform. The elutant was dried at 37°C. The dry residues were dissolved in ethanol containing 0.1% dodecane as an internal standard and analyzed by gas-liquid chromatography (GC-2010; Shimadzu, Kyoto, Japan) or gas-liquid chromatography mass spectrometry (Hewlett Packard 5890 SERIES II/JEOL MStation MS700 system; Agilent, Santa Clara, CA) with an Rtx-5 Amine capillary column (I.D. 0.25 mm, df 0.50 μm, 30 m; Restek, Buckinghamshire, UK). Nicotine, nornicotine, anatabine and anabasine were purchased from Wako or Sigma. The Anatalline standard was a gift from Dr. Oksman-Caldentey (Häkkinen et al. 2004).
For the analysis of NaNG and related pyridine metabolites, 1 g of fresh tissue was homogenized with 0.5 ml of 50 mM potassium phosphate buffer (pH7.0) and in a mortar and pestle. After incubation at 60°C for 30 min, the homogenate was centrifuged and the supernatant was passed through a disposable syringe filter unit (0.2 mm cellulose acetate; Iwaki, Chiba, Japan). Aliquots were analyzed by HPLC (LC-10 system; Shimadzu) with a Zorbax SB-Aq column (4.6 × 250 mm, 5 mm; Agilent). NaNG was eluted with a 0-70% gradient of methanol in 100 mM potassium phosphate (pH 7.0) at a flow rate of 1 ml min−1. The absorbance at 254 nm was monitored using a SPD-10A UV–Vis detector (Shimadzu). The retention times (min) of pyridine nucleotides were as follows: nicotinamide adenine dinucleotide (2.72), nicotinic acid mononucleotide (2.75), nicotinamide mononucleotide (3.14), NaNG (3.44), nicotinic acid (5.53), nicotinic acid adenine dinucleotide (10.7), and nicotinamide (17.6).
N-Methylpyrrolinium cation (MP) was measured by GLC after derivatization (Hashimoto et al. 1990). The aqueous extract (50 μl) in the potassium phosphate buffer described above was mixed with 10 μl of a 10% KCN solution and 100 μl of 0.05% (v/v) dodecane in chloroform. Aqueous KCN reacted quickly and quantitatively with MP to give 1-methyl-2-cyanopyrrolidine which, after being transferred to the chloroform phase by vigorous shaking, was analyzed by GLC with a Rtx-5 Amine capillary column (Shimadzu). Dodecane was used as an internal standard. The CN-derivatized product was identified by GC-MS (EI + mode, 70 eV). The authentic MP was synthesized by BioMol International (Plymouth Meeting, PA).
Results
A close homolog of A622 in N. tabacum originates from N. tomentosiformis
A search for A622-related sequences in the tobacco genome database (http://www.tobaccogenome.org/) identified a closely related homolog of A622, referred to hereafter as A622L. Predicted protein sequences of A622 and A622L are 95.5% identical and 98.4% similar (Supplementary Fig. S1), and constitute a closely related pair in the PIP family of reductases (Fig. 1). Both genes contain four less conserved introns which are inserted at the same positions (Supplementary Fig. S2). Genomic PCR using primers specific to either A622 or A622L amplified A622 genomic fragments from N. tabacum and N. sylvestris, and A622L genomic fragments from N. tabacum and N. tomentosiformis (Fig. 3a). Therefore, the A622 gene probably originates from N. sylvestris, as previously suggested (Hibi et al. 1994), whereas the A622L gene may be derived from N. tomentosiformis, the other probable progenitor of N. tabacum.

A622 and A622L are coordinately regulated. The α-tubulin gene was used as a control. a Genomic PCR analysis. Genomic DNA from N. tabacum (Nta) and its probable progenitors, N. sylvestris (Nsy) and N. tomentosiformis (Nto), was amplified using specific PCR primers. RT-PCR analysis: b Tissue-specific expression pattern. c Treatment of tobacco hairy roots with 100 μM MeJA for 24 h. d Treatment of cultured tobacco BY-2 cells with 50 μM MeJA for the indicated period. e Gene expression in the root of wild-type (WT) and nic1nic2 mutant (nic) tobacco plants
We examined the expression profiles of A622L by RT-PCR. The A622L transcript was abundant in the root but was undetectable in the stem and the leaf of tobacco plants (Fig. 3b). The application of MeJA upregulated the expression of A622L in the tobacco hairy roots (Fig. 3c), and induced its expression within 2 h in cultured tobacco BY-2 cells (Fig. 3d). Importantly, the expression of A622L in the tobacco root was under the control of the NIC regulatory loci (Fig. 3e). The expression patterns were highly similar to those of A622 (Fig. 3a–e), indicating that A622 and A622L are very probably involved in the same metabolic pathway.
A622 knockdown in tobacco hairy roots
We first tried to constitutively suppress the expression of A622 by RNA interference (RNAi) in transgenic tobacco plants and in tobacco hairy roots, by using the cauliflower mosaic virus 35S (CaMV35S) promoter. However, we could not obtain transgenic lines in which levels of the A622 transcript were highly reduced (data not shown). One possible explanation would be that efficient suppression of A622 in tobacco roots may perturb cellular metabolism and may be detrimental to cell survival or growth. We therefore used an inducible expression system (the XVE-β-estradiol system; Zuo et al. 2000) to drive the RNAi-based suppression of A622. Two stably transformed tobacco hairy root lines (XA12 and XA14) were obtained in which levels of the A622 and A622L transcripts were highly reduced when the roots were treated with 2 μM of the inducer β-estradiol at 3-day intervals for a total of 12 days (Fig. 4a). A polyclonal antiserum raised against A622 (Shoji et al. 2002), which recognizes A622L as well, was used to examine protein accumulation (Fig. 4b). No A622 protein was detectable in XA12 roots, whereas a trace amount was observed in XA14 roots. In wild-type roots (WT) and vector-transformed control roots (VC), the inducer treatment did not affect the abundance of A622 mRNA and protein.

Suppression of A622 and A622L in tobacco hairy roots retards root growth and inhibits alkaloid formation. Tobacco hairy roots of wild type (WT), a vector-transformed control (VC), and inducible A622 RNAi lines (XA12 and XA14) were treated with solvent DMSO (inducer; −) or with 2 μM β-estradiol (inducer; +) for 12 days. a RT-PCR analysis of A622 and A622L expression. b Immunoblot analysis using an antiserum against A622 and PEPC (loading control). c Root morphology of the XA14 line after addition of DMSO (above) or β-estradiol (below). d Growth of hairy roots (XA12 and XA14) after addition of DMSO (−) or β-estradiol (+). Data indicate the mean values ±SD, from five replicates. e Cellular contents of nicotine and nornicotine. Data indicate the mean values ±SD, from three replicates
The inducer treatment did not affect the growth of wild-type and vector control roots (data not shown). However, the growth of XA12 and XA14 roots was severely inhibited after the addition of β-estradiol (Fig. 4c). The root tip region including the root meristem and elongation zone was highly short-ended and compact in the inducer-treated XA12 and XA14 roots, compared to un-treated XA roots (Fig. 4d). This region of the root corresponds to the area with the highest level of A622 expression (Shoji et al. 2002). We next analyzed tobacco alkaloids, which consisted mostly of nicotine and a small amount of nornicotine in the hairy roots (Fig. 4e). Nicotine concentrations in wild-type and vector control roots were not significantly affected by the β-estradiol treatment, and ranged from 17 to 28 μmoles/g DW. In contrast, the inducer treatment strongly reduced the amount of nicotine to 2.4 μmoles/g DW in XA12 roots and to 7.5 μmoles/g DW in XA14 roots. Nornicotine content was moderately reduced after the inducer treatment in XA12 and XA14 roots, but not in wild-type roots. The treatment appeared to increase moderately the amount of nornicotine in the vector control roots, but this relative increase may be caused by the somewhat lower alkaloid content of un-treated control roots in this particular experiment.
When the cell extracts of hairy roots were analyzed by reverse-phase high-performance liquid chromatography (HPLC) with detection at 254 nm, a compound accumulating specifically in the inducer-treated XA12 and XA14 roots was found (Fig. 5a). This compound (retention time; 3.44 min) was purified by a series of ODS column chromatography and then crystallized. Its chemical spectra for 1H-NMR, 13C-NMR, UV, and FAB-MS (see “Materials and methods” for details) were consistent with those of nicotinic acid N-glucoside (NaNG; Nishitani et al. 1995). Enzymatic hydrolysis by β-glucosidase yielded free nicotinic acid (Fig. 5b), indicating a β-glucosidic conjugation. These structural analyses thus identified the accumulating compound as N-(β-d-glucopyranosyl)nicotinic acid (Fig. 5c). NaNG has previously been isolated from cultured tobacco cells supplied with nicotinic acid (Mizusaki et al. 1970; Nishitani et al. 1995).

Accumulation of NaNG in A622 knockdown tobacco hairy roots. a HPLC chromatograms of cell extracts from the XA12 line which had been treated with DMSO or 2 μM β-estradiol for 12 days. NaNG (retention time; 3.44 min) accumulated when A622 expression was suppressed. b Hydrolysis with β-glucosidase liberates nicotinic acid from NaNG. NaNG was dissolved in a citrate buffer (pH 5.0) and treated with commercial β-glucosidase. HPLC chromatograms are shown. c Chemical structure of NaNG
To study the metabolic consequences of the suppression of A622 in detail, the time course of the suppression and of the accumulation of metabolites were followed in two inducible RNAi hairy root lines. A622 protein levels began to decrease 2 days after the addition of β-estradiol in XA12, while the decline was already apparent after 1.5 days in XA14 (Fig. 6a). By the 4th day of suppression, A622 levels were very low in both lines. Treatment with the DMSO solvent did not affect the accumulation of A622 during 12 days in either hairy root lines (data not shown). We quantified the accumulation of NaNG and N-methylpyrrolinium cation (MP), a direct precursor for the pyrrolidine moiety of nicotine, in these roots. In wild-type roots and vector control roots, NaNG and MP did not accumulate during the 12-day culture period; their contents being lower than 2.4 μmoles/g DW and 6.5 μmoles/g DW, respectively, irrespective of the β-estradiol treatment (data not shown). The inducer, however, increased the accumulation of both compounds in XA12 and XA14 roots (Fig. 6b, c). In these root lines, NaNG started to accumulate 2 days after the addition of β-estradiol and steadily increased thereafter. The accumulation of MP could be observed even at the 1.5-day time point, slightly earlier than the onset of NaNG’s accumulation. During the period of 2–3 days, more MP accumulated than NaNG in XA roots. The final concentrations of NaNG and MP reached 100–140 μmoles/g DW and 70–85 μmoles/g DW, respectively, after 12 days of A622 suppression. These values were greater than the total alkaloid content of tobacco hairy roots (20–30 μmoles/g DW).

Time courses of A622 suppression and metabolite accumulation in tobacco hairy roots. a Immunoblot analyses of A622 and β-tubulin (control) accumulation in the XA12 and XA14 lines treated with β-estradiol. b Accumulation of MP and NaNG in the XA12 and XA14 lines
These results indicate the accumulation of NaNG and inhibition of root growth to be mechanistically linked. To address this relationship, we fed nicotinic acid to wild-type tobacco hairy roots at a final concentration of 8.12 μM (the concentration in the standard B5 medium) or 2.46 mM. In the high-nicotinate-containing medium, growth of tobacco hairy roots was severely inhibited (Fig. 7a). The elongation zone of the treated root was much shorter than that of the untreated root (Fig. 7b). We also found that NaNG accumulated in the roots after the addition of nicotinic acid to the medium (Fig. 7c). These results indicate that nicotinic acid or its immediate metabolite(s) is toxic to roots and that exogenously supplied nicotinic acid is converted to NaNG, a metabolite that appears to show low or negligible toxicity. The efficient conversion of exogenously supplied nicotinic acid to NaNG was also observed in un-elicited tobacco BY-2 cells which did not synthesize tobacco alkaloids (Fig. 7d).

Nicotinic acid supplied exogenously inhibits root growth and is converted to NaNG. a Wild-type tobacco hairy roots were cultured on the B5 agar medium containing nicotinic acid at 8.12 μM or 2.46 mM. The standard errors were calculated from five replicates. b Morphology of the roots cultured in the standard B5 medium (above) or in the high-nicotinic acid medium (below). Note that the region of root meristem and elongation is much reduced when cultured in the high concentration of nicotinic acid. c NaNG content of in wild-type tobacco hairy roots after eight days of incubation with 1 mM nicotinic acid in the culture medium. d NaNG content of in wild-type tobacco BY-2 cells after five days of incubation with 0.5 mM nicotinic acid in the culture medium
A622 knockdown in cultured tobacco cells
Although cultured tobacco BY-2 cells do not normally express structural genes for tobacco alkaloids and thus do not synthesize them, elicitation by MeJA induces transcription of alkaloid-synthesis-related genes and leads to alkaloid biosynthesis (Goossens et al. 2003). The MeJA-elicited BY-2 cells, however, mainly accumulate anatabine, a pyridine analog of nicotine, due to very low levels of the N-methylputrescine oxidase genes (Shoji and Hashimoto 2008). The metabolic impact of A622 suppression on MeJA-induced alkaloid biosynthesis was examined in cultured tobacco cells. Two control cell lines (VC1 and VC2) were transformed with an empty vector, whereas the A622 RNAi vector was introduced into four cell lines (A3, A21, A33, and A43). RT-PCR analysis in Fig. 8a shows that the application of 50 μM MeJA induced the expression of A622 and A622L in wild-type and control cells but not in the A622 RNAi cells. Immunoblot analysis also confirmed the results (Fig. 8b). The amount of anatabine in the four A622 RNAi cell lines was 1–10% of that in wild-type cells and the two control cell lines (Fig. 8c). Other tobacco alkaloids, nicotine, anabasine, and anatalline, also accumulated in highly reduced amounts in the A622 RNAi lines, compared to wild-type cells and control lines (Fig. 8d).

Suppression of A622 expression in cultured tobacco cells. Wild-type BY-2 cells (WT), two vector-transformed control cell lines (VC1 and VC2), and four A622 RNAi lines (A3, A21, A33, and A43) were treated with 50 μM MeJA for 48 h, and then analyzed for protein levels and metabolite contents. Wild-type BY-2 cells were also cultured in the absence of MeJA. a Immunoblot analyses of A622 and PEPC (control) accumulation. b Anatabine content. c Amounts of nicotine, anabasine, and anatalline. Note that the scale is different from that in (b). d NaNG content. ND; not detected
After MeJA treatment, NaNG was abundant in the A622 RNAi lines, but scarce in wild-type and control cells (Fig. 8e). We could not detect MP in wild-type, control, and A622 RNAi cells. Very low levels of MP in MeJA-elicited BY-2 cells are consistent with the finding that the elicited tobacco cells contain negligible levels of N-methylputrescine oxidase, which catalyzes the formation of this cation (Shoji and Hashimoto 2008).
Discussion
Two A622-related genes in tobacco
Nicotiana tabacum is a natural allotetraploid species that was formed relatively recently by combining two ancestral genomes closely related to the modern N. sylvestris and N. tomentosiformis species (Clarkson et al. 2005, and references therein). The majority, but not all, of tobacco genes are thought to be pairs of orthologous genes derived from the two progenitor species (Okamuro and Goldberg 1985). Molecular and cytogenetic analyses also revealed rapid elimination of repeated DNA sequences from the N. tomentosiformis genome donor of a synthetic, allotetraploid tobacco (Skalická et al. 2005). Our previous DNA blot analysis of N. tabacum and N. sylvestris genomic DNA under high stringency conditions, using the tobacco A622 cDNA as a hybridization probe, failed to detect an A622-like DNA sequence of possible N. tomentosiformis origin in the tobacco genome (Hibi et al. 1994). This led us to speculate that A622 in tobacco is a single-copy gene of N. sylvestris origin (Hibi et al. 1994). If true, the putative A622 gene of N. tomentosiformis origin may have been eliminated from the tobacco genome after allotetraploidization. Alternatively, an A622 ortholog may not be present in the modern N. tomentosiformis species. In this case, A622 should not be involved in the metabolic pathway present in this species. In this study, we identified a tobacco A622L gene which is highly homologous to A622, and showed that this gene is derived from N. tomentosiformis. Therefore, the tobacco genome contains two closely related A622 genes that are derived from the two progenitor species of tobacco. Our previous failure to detect A622 homologs in the tobacco genome might be due to the highly stringent hybridization conditions used (Hibi et al. 1994).
Expression of A622 and A622L shows a very similar pattern; predominantly occurring in the root, up-regulated or induced in hairy roots and cultured cells by MeJA elicitation, and positively regulated by the NIC regulatory loci which specifically control the accumulation of tobacco alkaloids. The expression profiles are largely conserved in tobacco genes encoding enzymes for the biosynthesis of nicotine (Hibi et al. 1994; Shoji et al. 2000b; Shoji et al. 2002; Reed and Jelesko 2004; Cane et al. 2005; Heim et al. 2007; Katoh et al. 2007; Shoji et al. 2008). These results suggest that A622 and A622L are orthologous genes of N. sylvestris and N. tomentosiformis origins, and might be involved in the formation of tobacco alkaloids in these Nicotiana species.
Down-regulation of A622 causes profound changes in alkaloid metabolism
In this study, we used a 0.2-kb cDNA fragment of A622 to trigger RNAi-mediated gene suppression in tobacco hairy roots and tobacco cultured BY-2 cells. In the transformants, the expression of both A622 and A622L was effectively downregulated since these two genes showed extensive sequence identity in the region used. Although we did not test the individual suppression of these homologous genes, we believe that the strong perturbation of alkaloid metabolism requires both genes to be simultaneously suppressed.
When A622 and A622L were suppressed in the tobacco cells that synthesize pyridine alkaloids, biosynthesis of all the tobacco alkaloids was severely inhibited. In the tobacco roots that synthesize nicotine and nornicotine, MP rapidly accumulated in compensation for the decrease of these pyrrolidine-containing alkaloids, in which MP serves as the direct precursor of the pyrrolidine ring (Mizusaki et al. 1968). In the MeJA-elicited BY-2 cells, MP synthesis is highly limiting due to the very weak expression of N-methylputrescine oxidase, and non-pyrrolidine-type alkaloids (e.g., anatabine, anabasine, and anatalline) are consequently synthesized (Shoji and Hashimoto 2008). In such cells, suppression of A622 inhibited synthesis of the non-pyrrolidine-type alkaloids but did not lead to accumulation of MP. These results indicate that A622 is required for the coupling reactions between a pyrrolidine precursor and MP (to give nicotine), and between two pyrrolidine precursors (to yield anatabine).
If A622 is involved in the ring-coupling reactions, its suppression should also result in accumulation of the hypothetical pyrrolidine precursor of tobacco alkaloids. Indeed, we observed high levels of NaNG, a glucose conjugate of nicotinic acid, in A622-suppressed cells. However, we conclude that NaNG is a product of the detoxification of nicotinic acid, not a direct precursor for the pyrrolidine moiety of tobacco alkaloids, for the following reasons. First, unlabeled nicotinic acid efficiently inhibited the incorporation of radio-labeled NaNG into nicotine when administered to tobacco roots (Mizusaki et al. 1970), indicating that NaNG is initially deglucosylated to nicotinic acid before being incorporated into nicotine. Second, we observed that when nicotinic acid was supplied at a milli-molar level to the culture medium, it strongly inhibited root growth and was converted to NaNG in tobacco cells. Conversion of exogenously supplied nicotinic acid to NaNG in tobacco cells has also been reported by others (Mizusaki et al. 1970; Nishitani et al. 1995). Importantly, efficient N-glucosylation of nicotinic acid was also observed in un-elicited tobacco BY-2 cells (this study) and tobacco leaf (Mizusaki et al. 1970), neither of which synthesize tobacco alkaloids. Therefore, the formation of NaNG can be uncoupled from the biosynthesis of alkaloids and may be a basic cellular detoxification mechanism against undesired accumulation of toxic intermediates in the biosynthetic and salvage pathways of NAD. Third, the initial increase of NaNG in A622-suppressed tobacco roots occurred about 12 h after the onset of MP accumulation. This time lag suggests that an unidentified pyrrolidine precursor of tobacco alkaloids first accumulated, and then was rapidly metabolized to NaNG, by way of nicotinic acid. Fourth, we have been unable to detect any reduction of NaNG when recombinant A622 was used in the in vitro assays (our unpublished results).
A622 as a PIP-family reductase
Five PIP family proteins, pinoresinol–lariciresinol reductase (Min et al. 2003), isoflavone reductase (Wang et al. 2006), PCBER (Min et al. 2003), and eugenol synthases (Louie et al. 2007; Koeduka et al. 2008), have been structurally characterized and shown to have a very similar polypeptide-chain fold, consisting of an N-terminal Rossman-fold domain that binds NADPH, and of a C-terminal segment that largely contributes to substrate binding. Because A622 has high sequence identity with loblolly pine PCBER (57.7%) and sweet basil eugenol synthase (40.4%), for which the structure of a ternary complex bound to NADPH and a substrate-like inhibitor is known (Louie et al. 2007), we modeled the 3D structure of A622 based on these crystal structures. A superimposed model bound to the cofactor (Supplementary Fig. S3) indicates that A622, PCBER, and eugenol synthase are very similar in overall structure. Conservation of the Rossman-fold domain appears particularly strong; most of the amino acid residues interacting with the NADP + cofactor (Louie et al. 2007) are invariant in the A622 sequence. Structural modeling of A622 thus implies that A622 would abstract the 4-pro-R-hydride from the re-side of the dehydronicotinamide ring of NADPH, as in other PIP-catalyzed reactions (Gang et al. 1999; Chu et al. 1993; Schlieper et al. 1990).
Since suppression of A622 strongly inhibits condensation reactions to form several pyridine alkaloids and induces extensive accumulation of NaNG, this reductase might yield a coupling-competent intermediate from nicotinic acid or might be involved directly in the coupling reactions. As nicotinic acid itself is not a substrate for A622 (our unpublished results), a derivative of nicotinic acid may serve as its substrate. In all the known PIP-catalyzed reactions, the NADPH-derived hydride is transferred to a phenyl-ring substituent positioned para to the 4-hydroxyl group of the substrates. In the proposed reaction model, deprotonation of the 4-hydroxyl group of the substrate facilitates the formation of a quinone–methide intermediate prior to reduction (Louie et al. 2007). 6-Hydroxynicotinic acid and its C-3 derivatives have a hydroxyl group at analogous positions. Although 6-hydroxynicotinic acid was identified as a metabolite of nicotinic acid in tobacco roots, it was not incorporated into nicotine when administered to the tobacco roots (Dawson et al. 1960; Mizusaki et al. 1970). The identity of the true A622 substrate is a matter of speculation but its structure may be suited to the formation of a presumed quinone–methide intermediate.
References
Akashi T, Koshimizu S, Aoki T, Ayabe S (2006) Identification of cDNAs encoding pterocarpan reductase involved in isoflavan phytoalexin biosynthesis in Lotus japonicus by EST mining. FEBS Lett 580:5666–5670. doi:10.1016/j.febslet.2006.09.016
An G (1985) High efficiency transformation of cultured tobacco cells. Plant Physiol 79:568–570
Cane KA, Mayer M, Lidgett AJ, Michael AJ, Hamill JD (2005) Molecular analysis of alkaloid metabolism in AABB v. aabb genotype Nicotiana tabacum in response to wounding of aerial tissues and methyl jasmonate treatment of cultured roots. Funct Plant Biol 32:305–320. doi:10.1071/FP04008
Chu A, Dikova A, Davin LB, Bedgar DL, Lewis NG (1993) Stereospecificity of (+)-pinoresinol and (+)-lariciresinol reductases from Forsythia intermedia. J Biol Chem 268:27026–27033
Clarkson JJ, Lim KY, Kovarik A, Chase MW, Knapp S, Leitch AR (2005) Long-term genome diploidization in allopolyploid Nicotiana section Repandae (Solanaceae). New Phytol 168:241–252. doi:10.1111/j.1469-8137.2005.01480.x
Dawson RF, Christman DR, D’Adamo A, Solt ML, Wolf AP (1960) The biosynthesis of nicotine from isotopically labeled nicotinic acids. J Am Chem Soc 82:2628–2633. doi:10.1021/ja01495a059
Gang DR, Kasahara H, Xia Z-Q, van Mijnsbrugge K, Bauw G, Boerjan W, Van Montagu M, Davin LB, Lewis NG (1999) Evolution of plant defense mechanisms: relationships of phenylcoumaran benzylic ether reductases to pinoresinol-lariciresinol and isoflavone reductases. J Biol Chem 274:7516–7527. doi:10.1074/jbc.274.11.7516
Goossens A, Haekkinen ST, Laakso I, Seppaenen-Laakso T, Biondi S, Sutter VD, Lammertyn F, Nuutila AM, Soederlund H, Zabeau M, Inze D, Oksman-Caldentey K-M (2003) A functional genomics approach toward the understanding of secondary metabolism in plant cells. Proc Natl Acad Sci USA 100:8595–8600. doi:10.1073/pnas.1032967100
Häkkinen ST, Rischer H, Laakso I, Maaheimo H, Seppänen-Laakso T, Oksman-Caldentey KM (2004) Anatalline and other methyl jasmonate-inducible nicotine alkaloids from Nicotiana tabacum cv. BY-2 cell cultures. Planta Med 70:936–941. doi:10.1055/s-2004-832620
Hashimoto T, Mitani A, Yamada Y (1990) Diamine oxidase from cultured roots of Hyoscyamus niger. Its function in tropane alkaloid biosynthesis. Plant Physiol 93:16–221
Heim WG, Sykes KA, Hildreth SB, Sun J, Lu RH, Jelesko JG (2007) Cloning and characterization of a Nicotiana tabacum methylputrescine oxidase transcript. Phytochemistry 68:454–463. doi:10.1016/j.phytochem.2006.11.003
Hibi N, Higashiguchi S, Hashimoto T, Yamada Y (1994) Gene expression in tobacco low-nicotine mutants. Plant Cell 6:723–735
Kanegae T, Kajiya H, Amano Y, Hashimoto T, Yamada Y (1994) Species-dependent expression of the hyoscyamine 6β-hydroxylase gene in the pericycle. Plant Physiol 105:483–490. doi:10.1104/pp.105.2.483
Katoh A, Ohki H, Inai K, Hashimoto T (2005) Molecular regulation of nicotine biosynthesis. Plant Biotechnol 22:389–392
Katoh A, Shoji T, Hashimoto T (2007) Molecular cloning of N-methylputrescine oxidase from tobacco. Plant Cell Physiol 48:550–554. doi:10.1093/pcp/pcm018
Koeduka T, Louie GV, Orlova I, Kish CM, Ibdah M, Wilkerson CG, Bowman ME, Baiga TJ, Noel JP, Dudareva N, Pichersky E (2008) The multiple phenylpropene synthases in both Clarkia breweri and Petunia hybrida represent two distinct protein lineages. Plant J 54:362–374. doi:10.1111/j.1365-313X.2008.03412.x
Louie GV, Baiga TJ, Bowman ME, Koeduka T, Taylor JH, Spassova SM, Pichersky E, Noel JP (2007) Structure and reaction mechanism of basil eugenol synthase. PLoS One 2:e993. doi:10.1371/journal.pone.0000993
Min T, Kasahara H, Bedgar DL, Youn B, Lawrence PK, Gang DR, Halls SC, Park H, Hilsenbeck JL, Davin LB, Lewis NG, Kang C (2003) Crystal structures of pinoresinol-lariciresinol and phenylcoumaran benzylic ether reductases and their relationship to isoflavone reductases. J Biol Chem 278:50714–50723. doi:10.1074/jbc.M308493200
Mizusaki S, Kisaki T, Tamaki E (1968) Phytochemical studies on the tobacco alkaloids. XII. Identification of γ-methylaminobutyraldehyde and its precursor role in nicotine biosynthesis. Plant Physiol 43:93–98
Mizusaki S, Tanabe Y, Kisaki T, Tamaki E (1970) Metabolism of nicotinic acid in tobacco plants. Phytochemistry 9:549–554. doi:10.1016/S0031-9422(00)85688-5
Murashige T, Skoog F (1962) A revised medium for rapid growth and bioassays with tobacco tissue cultures. Physiol Plant 15:473–479. doi:10.1111/j.1399-3054.1962.tb08052.x
Nishitani H, Yamada Y, Ohshima N, Okumura K, Taguchi H (1995) Identification of N-(beta-D-glucopyranosyl)nicotinic acid as a major metabolite from niacin in cultured tobacco cells. Biosci Biotechnol Biochem 59:1336–1338
Okamuro JK, Goldberg RB (1985) Tobacco single-copy DNA is highly homologous to sequences present in the genomes of its diploid progenitors. Mol Gen Genet 198:290–298. doi:10.1007/BF00383009
Reed DG, Jelesko JG (2004) The A and B loci of Nicotiana tabacum have non-equivalent effects on the mRNA levels of four alkaloid biosynthetic genes. Plant Sci 167:1123–1130. doi:10.1016/j.plantsci.2004.06.006
Saito K, Hirai MY, Yonekura-Sakakibara K (2008) Decoding genes with coexpression networks and metabolomics—‘majority report by precogs’. Trends Plant Sci 13:36–43. doi:10.1016/j.tplants.2007.10.006
Schlieper D, Tiemann K, Barz W (1990) Stereospecificity of hydrogen transfer by fungal and plant NADPH: isoflavone oxidoreductases. Phytochemistry 29:1519–1524. doi:10.1016/0031-9422(90)80112-T
Shoji T, Hashimoto T (2008) Why does anatabine, but not nicotine, accumulate in jasmonate-elicited cultured tobacco BY-2 cells? Plant Cell Physiol 49:1209–1216. doi:10.1093/pcp/pcn096
Shoji T, Nakajima K, Hashimoto T (2000a) Ethylene suppresses jasmonate-induced gene expression in nicotine biosynthesis. Plant Cell Physiol 41:1072–1076. doi:10.1093/pcp/pcd027
Shoji T, Yamada Y, Hashimoto T (2000b) Jasmonate induction of putrescine N-methyltransferase genes in the roots of Nicotiana sylvestris. Plant Cell Physiol 41:831–839. doi:10.1093/pcp/pcd001
Shoji T, Winz R, Iwase T, Nakajima K, Yamada Y, Hashimoto T (2002) Expression patterns of two tobacco isoflavone reductase-like genes and their possible roles in secondary metabolism in tobacco. Plant Mol Biol 50:427–440. doi:10.1023/A:1019867732278
Shoji T, Ogawa T, Hashimoto T (2008) Jasmonate-induced nicotine formation in tobacco is mediated by tobacco COI1 and JAZ genes. Plant Cell Physiol 49:1003–1012. doi:10.1093/pcp/pcn077
Skalická K, Lim KY, Matyasek R, Matzke M, Leitch AR, Kovarik A (2005) Preferential elimination of repeated DNA sequences from the parental, Nicotiana tomentosiformis genome donor of a synthetic, allotetraploid tobacco. New Phytol 166:291–303. doi:10.1111/j.1469-8137.2004.01297.x
Wang X, He X, Lin J, Shao H, Chang Z, Dixon RA (2006) Crystal structure of isoflavon reductase from alfalfa (Medicago sativa L.). J Mol Biol 385:1341–1352. doi:10.1016/j.jmb.2006.03.022
Wesley SV, Helliwell CA, Smith NA, Wang MB, Rouse DT, Liu Q, Gooding PS, Singh SP, Abbott D, Stoutjesdijk PA (2001) Construct design for efficient, effective and high-throughput gene silencing in plants. Plant J 27:581–590. doi:10.1046/j.1365-313X.2001.01105.x
Zuo J, Niu Q-W, Chua N-H (2000) An estrogen receptor-based transactivator XVE mediates highly inducible gene expression in transgenic plants. Plant J 24:265–273. doi:10.1046/j.1365-313x.2000.00868.x
Acknowledgements
We thank Chojiro Kojima for molecular modeling of A622, Kirsi-Marja Oksman-Caldentey for providing anatalline, and Junko Tsukamoto for MS analysis. This study was supported in part by the Global COE Program (Frontier Biosciences: strategies for survival and adaptation in a changing global environment), MEXT, Japan.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Kajikawa, M., Hirai, N. & Hashimoto, T. A PIP-family protein is required for biosynthesis of tobacco alkaloids. Plant Mol Biol 69, 287–298 (2009). https://doi.org/10.1007/s11103-008-9424-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11103-008-9424-3