
Abstract
The Arabidopsis genome contains 10 genes belonging to the HD-Zip II family including ATHB2 and HAT2. Previous work has shown that ATHB2 is rapidly and strongly induced by light quality changes that provoke the shade avoidance response whereas HAT2 expression responds to auxin. Here, we present a genome-wide analysis of the HD-Zip II family. Phylogeny reconstruction revealed that almost all of the HD-Zip II genes can be subdivided into 4 clades (α–δ), each clade comprising 2–3 paralogs. Gene expression studies demonstrated that all the γ and δ genes are regulated by light quality changes. Kinetics of induction, low R/FR/high R/FR reversibility and auxin response analyses strongly suggested that HAT1, HAT3 and ATHB4, as ATHB2, are under the control of the phytochrome system whereas HAT2 is up-regulated by low R/FR as a consequence of the induction of the auxin signaling pathway provoked by FR-rich light. Root and shoot digital in situ revealed that γ and δ genes are also tightly regulated during plant development with both distinct and overlapping patterns. Phenotypes of gain of function and dominant negative lines demonstrated that one or more of the HD-Zip II γ genes negatively regulate cell proliferation during leaf development in a high R/FR light environment. Finally, target gene analysis using a chimeric transcription factor (HD-Zip2-V-G), known to activate ATHB2 target genes in a glucocorticoid-dependent manner, revealed that all the 10 HD-Zip II genes can be recognized by the HD-Zip 2 domain in vivo, implying an intricate negative feedback network.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The HD-Zip class of proteins is unique to plants, and characterized by the presence of a homeodomain closely linked to a leucine zipper motif (Ruberti et al. 1991). Several Arabidopsis HD-Zip protein encoding genes have been identified in the early 1990s and, on the basis of sequence homology in the HD-Zip domain, grouped into four families, HD-Zip I–IV (Sessa et al. 1994). Completion of the Arabidopsis genome sequence revealed that it encodes 48 HD-Zip proteins all belonging to the four families previously identified (Sessa et al. 1994; Morelli et al. 1998; Baima et al. 2001; Henriksson et al. 2005; Nakamura et al. 2006; data herein). Genome-wide analyses of the HD-Zip I (17 genes; Henriksson et al. 2005) and HD-Zip IV (16 genes; Nakamura et al. 2006) gene families have been recently reported. Furthermore, work from several laboratories has contributed to a detailed analysis of the 5 HD-Zip III genes establishing that they serve both to maintain the meristem and to pattern dorsoventrality in the initiating leaf (Bowman 2004 and references therein; Byrne 2006 and references therein). By contrast, no systematic analysis of the HD-Zip II gene family (10 genes) has yet been undertaken.
The molecular functions of ATHB2, a member of the HD-Zip II family, have been studied thoroughly. In vitro binding experiments showed that the HD-Zip domain of ATHB2 (HD-Zip2 domain) is sufficient for sequence-specific DNA binding. The HD-Zip2 domain interacts with DNA as a dimer and recognizes a 9 bp pseudopalindromic sequence [CAAT(G/C)ATTG] (Sessa et al. 1993). A mutational analysis of the HD-Zip2 domain revealed that conserved amino acid residues of helix 3, Val 47 and Asn 51, and Arg 55 are essential for the DNA binding activity of the HD-Zip2 domain. Furthermore, it was demonstrated that the preferential recognition of a G/C base pair at the central position by the HD-Zip2 domain is abolished either by the replacement of Arg55 with lysine or by the substitution of Glu46 and Thr56 with the corresponding residues of the HD-Zip domain of ATHB1, a member of the HD-Zip I family (Sessa et al. 1997). In transient expression experiments, ATHB2 repressed the expression of a reporter gene containing six copies of the HD-Zip2 binding site in its promoter, while an ATHB2 derivative with a strong transactivating domain (HD-Zip2-V-G) activated the same reporter gene (Steindler et al. 1999). Target gene analyses for ATHB2 has also been performed. In vivo and in vitro experiments indicated that ATHB2 constitutes a negative autoregulatory loop, and may be involved in a complicate regulatory network involving HD-Zip II genes, similar to the networks found in animal homeobox genes (Ohgishi et al. 2001).
Gene expression studies have identified ATHB2 as the first gene specifically and reversibly regulated by changes in the R/FR ratio in green plants that induce the shade avoidance response in most of the angiosperms (Carabelli et al. 1993, 1996; Steindler et al. 1997). The light regulation of the ATHB2 gene is quite complex. In etiolated seedlings, the gene is expressed at relatively high levels and is down-regulated by R or FR light. PHYA is responsible for the rapid down-regulation of ATHB2 by a FR pulse whilst a phytochrome other than A or B is responsible for the equally rapid down-regulation of ATHB2 by a R pulse. In young seedlings and mature plants, ATHB2 is expressed at low levels under high R/FR ratio light, but is rapidly and strongly induced by low R/FR. Returning the plants to high R/FR results in an equally rapid decrease in the ATHB2 mRNA levels. Kinetics of FR-rich light induction and its reversibility by R-rich light performed in phyB and phyA phyB plants revealed that ATHB2 is reversibly regulated by changes in the R/FR ratio largely through the action of a phytochrome other than A or B and secondarily by phytochrome B (Carabelli et al. 1996; Steindler et al. 1997). Further studies demonstrated that type II phytochrome D and E are indeed involved in the light regulation of the ATHB2 gene (Franklin et al. 2003).
Analysis of transgenic plants bearing constructs that alter ATHB2 expression revealed a series of interesting developmental phenotypes (Schena et al. 1993; Steindler et al. 1999). For example, seedlings overproducing ATHB2 had longer hypocotyls and petioles, smaller and fewer leaves. Moreover, these seedlings also had a thinner root mass, that is they produced less lateral roots than wild-type controls. The phenotypes of adult transgenic plants were similar to seedlings but more exaggerated. Altogether the phenotypes of plants overexpressing ATHB2 were reminiscent of those displayed by wild-type plants grown in low R/FR light, further suggesting a role for this HD-Zip protein in the regulation of the shade avoidance response (Steindler et al. 1999; Morelli and Ruberti 2000, 2002). This is also supported by the recent finding that ATHB2 is under the control of HFR1/SICS1, a negative controller of the shade avoidance response, ensuring that an exaggerated reaction does not occur when the plant is unsuccessful in escaping canopy shade (Sessa et al. 2005).
Very little is known about the other HD-Zip II genes except for HAT2 which was isolated as an auxin inducible gene by DNA microarray (Sawa et al. 2002). Analogously to seedlings overexpressing ATHB2, 35S::HAT2 transgenic plants produced long hypocotyls, epinastic cotyledons, long petioles and small leaves (Sawa et al. 2002).
Here, we present a comprehensive study of the HD-Zip II family. We report the results of phylogenesis reconstruction, expression patterns, and in vivo target gene analyses. Furthermore, a selection of three light-responsive HD-Zip II genes was further studied using gain of function and dominant negative lines.
Materials and methods
Phylogenetic analysis
Available databases were searched by use of the WU-BLAST2 algorithm (BLASTP) to identify HD-Zip II sequences in the Arabidopsis genome. To generate the phylogenetic trees of the HD-Zip II family, the whole amino acid sequences were used. First, sequences were aligned using CLUSTALW 1.83 (Thompson et al. 1994) with default parameters (Gap Open Penalty = 10.0, Gap Extension Penalty = 0.2, Matrix = Gonnet 250), and manually refined. Afterwards, trees were constructed with PHYLIP 3.66 (Felsenstein 1989, 2005), using the neighbor-joining (NJ) method (Saitou and Nei 1987), randomizing input order of species. Internal branch support of the tree was estimated with 1000 bootstrap replicates. Showed tree includes all the groups which are present in more than 50% of the trees, plus the most frequent others that are compatible with these. A sequence belonging to HD-Zip I family was used as an outgroup (ATHB1).
Paralogs analysis
The search for putative paralogs which likely arose via segmental chromosome duplication events within the HD-Zip II family was carried out by the use of the “Paralogons in Arabidopsis thaliana” program (http://www.wolfe.gen.tcd.ie/athal/dup) as described by Blanc et al. (2003). Expectation values were obtained from SEG-filtered SSEARCH searches of Arabidopsis sequences against themselves (Blanc et al. 2003).
Promoter analyses and significance calculations
Putative promoter regions upstream of HD-Zip family I (17) and II (10) genes were analyzed to identify over-represented cis-elements, similarly to Nemhauser et al. (2004). 3000 nucleotide-long regions upstream of 5′-UTR (if known, otherwise from ATG) were analyzed, as downloaded from TAIR website (Release 7, TAIR7_upstream_1000_20070405). Then, a database was created containing all motifs to test. One thousand surrogates of HD-Zip I and II family promoter set were created by randomly sampling the list of genes represented on the Affymetrix ATH1 array and extracting their upstream sequences. For each set of n promoters, the null distribution for each motif was modeled by counting the number of occurrences for each word within each of the 1000 surrogate sets. The one-tailed P-value for each motif is based on the Z-score of the difference of the actual word count of the promoter set (Ctrue) minus the mean count from the 1000 surrogates (Crand) relative to the SD from the 1000 surrogates (SDrand) (i.e., Z = (Ctrue − Crand)/SDrand). By means of this Z-score we obtained the probability for each motif to be over-represented compared with random (Nemhauser et al. 2004). These calculations were implemented by using Perl scripts; analyses were repeated two times independently, with similar results.
Plant lines
Wild-type strain used was A. thaliana (L.) Heynh. var. Columbia (Col-0). 35S::HAT1, 35S::HAT2 and 35S::ATHB2N51A transgenic lines were obtained transforming Col-0 with the plant expression vector HAT1, HAT2 and ATHB2N51A, respectively. The HAT1 and HAT2 constructs were obtained with the GATEWAYTM technology (Gateway(R) system (Invitrogen; http://www.invitrogen.com). The HAT1 and HAT2 complete coding sequences, from ATG to terminator codon, were amplified by RT-PCR from poly(A)+ RNA extracted from 2 weeks-old plants, and cloned into the pDONR 201 vector (Invitrogen; http://www.invitrogen.com). Subsequently, the HAT1 and HAT2 coding sequences were cloned from the donor vector into the pBENDER binary vector (http://www2.mpiz-koeln.mpg.de/~weisshaa/BW-research/Vectors.html). ATHB2N51A was constructed by excising the HDZIP1-VP16 sequence from a derivative of pMON721 (Aoyama et al. 1995) using BglII and BamHI. The ATHB2N51A fragment was obtained using the procedure described by Sessa et al. (1997) with the following combination of primers: ATHB2 5′: 5′-CCCGAGATCTCTTCGAGAAAGACGAT-3′ ATHB2 3′: 5′-CCCGGGATCCTTAGGACCTAGGACGAA-3′. The expression plasmids were introduced into Agrobacterium tumefaciens strain GV3101pMP90R as previously described (Steindler et al. 1999). Col-0 was transformed using the vacuum infiltration method (Bechtold et al. 1993). Homozygous lines were selected as described by Steindler et al. (1999).
Growth and phenotypic analyses
Plants were grown as previously described (Steindler et al. 1999; Sessa et al. 2005). For hypocotyls images were taken with a MZ 12 binocular microscope (Leica) using a Spot Jr. digital camera (Diagnostic Instrument Inc.), and subsequently analyzed with the Scion Image software (Scion Corp.). Root growth, analysis and measurements were performed as previously described (Steindler et al. 1999). Leaves were cleared according to the protocol previously described (Weigel and Glazebrook 2002). Cleared samples were excised under a MZ8 binocular microscope, and then analyzed under dark-field optics or with differential interference contrast (DIC) optics, with an Axioskop 2 plus binocular microscope (Zeiss, Germany). Images were taken with the Coolpix 990 digital camera (Nikon Corp., Japan). To determine the mean leaf area, at least 30 samples were measured with the NIH Image Analysis Software (Research Services Branch (RSB) of the National Institute of Mental Health (NIMH), U.S.A., http://www.rsb.info.nih.gov/ij/). To determine the mean cell area, 100 adaxial subepidermal cells were measured in each distal, median and proximal region of 10 leaves (Horiguchi et al. 2005; Carabelli et al. 2007). T-test statistical analysis was performed using QuickCalcs Online Calculators for Scientists (GraphPad Software, Inc. http://www.graphpad.com/quickcalcs/).
Gene expression analysis
For gene expression analysis in high and low R/FR, 8-day-old seedlings grown as previously described (Sessa et al. 2005) were harvested after the designated light treatments for the indicated period of time. Light outputs in high R/FR were: 670 nm (Red) 96 μmol m−2 s−1; 735 nm (Far Red) 21 μmol m−2 s−1; 470 nm (Blue) 15 μmol m−2 s−1. Light outputs in low R/FR were: 670 nm, 12 μmol m−2 s−1; 735 nm, 105 μmol m−2 s−1; 470 nm, 15 μmol m−2 s−1. For gene response to exogenous IAA, seedlings were grown in liquid medium (1× MS; Murashige and Skoog 1962) for 7 days in a 16 h light/8 h dark cycle. IAA (Sigma-Aldrich, Germany) was dissolved in DMSO and then added to the medium to the indicated final concentration. An equivalent amount of DMSO was used as a control. For DEX (Sigma-Aldrich, Germany) and CHX (Sigma-Aldrich, Germany) treatments, conditions were as described by Ohgishi et al. (2001). Total RNA was extracted using RNeasy Plant Mini Kit (Qiagen, Germany) and polyA+ RNA was extracted from total RNA with the Dynabeads® Oligo (dT) (Dynal, Norway). Northern analysis was performed as previously described (Carabelli et al. 1996; Sessa et al. 2005). Details on the probes used are given in the Supplemental Material. For quantitative PCR experiments, total RNA was reverse-transcribed using the Superscript III™ First-Strand Synthesis System (Invitrogen) according to the manufacturer’s instructions. Quantitative PCR was performed with the LightCycler® 480 instrument (Roche), using LightCycler® 480 Probes Master (Roche) and Universal ProbeLibrary (UPL) Probes (Roche), 5′-labeled with fluorescein (FAM) and 3′-labeled with a dark quencher dye, according to the manufacturer’s instructions. Multiwell plates 384 were used and the qRT-PCR set-up was automated on the Tecan Freedom Evo® 75 platform (Tecan application note—Automated Real-Time PCR Set-up on a Tecan Freedom PCR Workstation, htp://www.tecan.com). Each amplification was performed in triplicate in a reaction volume of 10 μl using final UPL probe concentration of 100 nM and final primers concentration of 300 nM. The combination of UPL and primers used for each gene is detailed in the Supplemental Material. Each gene specific expression quantification assay was easily designed as an intron-spanning assay, using the web-based ProbeFinder software accessible at http://www.universalprobelibrary.com. Quantification of our target genes expression was expressed in comparison to the reference gene actin (ACT2) and relative expression ratio was calculated based on the qRT-PCR efficiency (E) for each gene and the crossing point (CP) deviation of our target genes versus a control (Pfaffl 2001).
Results and discussion
The Arabidopsis HD-Zip II family consists of 10 genes encoding highly related proteins
A search of Arabidopsis databases using the HD-Zip domain of ATHB2 identified 9 proteins with sequences highly similar to those of ATHB2 (E-value < 10−20), indicating that the HD-Zip II family actually consists of ten members.
An alignment of the amino acid sequences of the HD-Zip II proteins is shown in Fig. 1. Remarkably, all the ten HD-Zip domains where most of the positions are invariant or highly conserved align without any insertion or deletion. The high degree of homology within the HD-Zip II domains (Supplementary Table 1) and the conservation of amino acid residues (position 46 and 56 in the HD-Zip domain, Fig. 1) previously shown to contribute to the different DNA binding specificity of HD-Zip I and II transcription factors (Sessa et al. 1997) indicate that HD-Zip II proteins have the potential to recognize the same target genes.

Alignment of the full length amino acid sequences of the 10 HD-Zip II proteins. Gaps are indicated by dashes. For each column in the alignment, residues conserved in more than 60% of all sequences are highlighted. Following BLOSUM62 amino acid substitution scores, similar amino acids are depicted in grey, identical in black. The positions of introns outside of the HD-Zip domain are indicated by colored arrowheads. The amino acids at the corresponding positions are highlighted with the same colors. Magenta line boxes indicate amino acids at position 46 and 56 of the HD-Zip domain. All HD-Zip II domains have a glutamic residue at position 46 whereas eight out of the ten HD-Zip II domains have a threonine at position 56. At this position of ATHB18 and ATHB17 HD-Zip domains there is a conservative substitution (serine vs. threonine). ATHB18, At1G70920; ATHB17, At2G01430; HAT14, At5G06710; HAT22, At4G37790; HAT9, At2G22800; ATHB2, At4G16780; HAT1, At4G17460; HAT2, At5G47370; HAT3, At3G60390; ATHB4, At2G44910
The alignment in Fig. 1 also shows conserved motifs outside the HD-Zip domain. Immediately downstream of the HD-Zip domain, there is a seven amino acid motif (CPSCERV) of unknown function, conserved in five out of the ten HD-Zip II proteins. A highly similar sequence is also present in ATHB17 and ATHB18 (CPRCERV), HAT22 (CPSCERL), HAT9 (CPSCERI) and ATHB2 (CPSCEHV). At the N-terminal end, it is interesting the presence of a L×L×L motif in six out of the ten HD-Zip II proteins, important for conferring transcriptional repression function to the domain I of the AUX/IAA factors, involved in the regulation of auxin response (Tiwari et al. 2004). It is noteworthy the fact that ATHB2 and HAT2 have been previously shown to work as transcriptional repressors in vitro and in vivo (Steindler et al. 1999; Ohgishi et al. 2001; Sawa et al. 2002).
Over the N-terminal region, a major similarity has been previously found for five HD-Zip II factors; ATHB2, HAT1, HAT2, HAT3 and ATHB4 share the HD-Zip protein N terminus domain together with 24 proteins in other plants (PF04618, http://www.pfam.sanger.ac.uk). The average length of the domain is 104.7 amino acids with average identity equals 39%. Interestingly, 22 out of the 29 proteins show both the HD-Zip protein N terminus domain and the HD-Zip domain, as in the case of all the five Arabidopsis sequences. Out of the remaining Arabidopsis HD-Zip II proteins, four of them have a region N-terminal to the HD-Zip domain of similar length whereas ATHB18 has a much shorter amino acid sequence between the ATG and the DNA binding domain (Fig. 1). Alignment of the N-terminal regions of the ten HD-Zip II proteins from the first amino acid to the last conserved residue of the HD-Zip protein N terminus domain revealed that HAT9 and HAT22 share a significant homology in the N-terminus region with ATHB2, HAT1, HAT2, HAT3 and ATHB4. Several conserved residues of the HD-Zip protein N terminus domain are also present in the N-terminus region of HAT14 (Supplementary Fig. 1). The function of HD-Zip protein N terminus domain is not known. However, its conservation between monocot and dicot plants is suggestive of a key function.
The Arabidopsis HD-Zip II genes have the potential to form an intricate regulatory network
Previous work demonstrated that the ATHB2 protein interacts with the ATHB2 promoter directly recognizing the sequences 5′-TAATCATTA-3′ and 5′-TAATTATTA-3′, which are similar to the HD-Zip2 binding consensus sequence (Ohgishi et al. 2001; Sessa et al. 1993). Furthermore, a target-gene analysis using an ATHB2-derived transcription factor (HD-Zip2-V-G, Steindler et al. 1999), consisting of the HD-Zip domain of ATHB2, the transactivating domain of the herpex viral protein VP16 (Triezenberg et al. 1988) and the hormone binding domain of the rat glucocorticoid receptor GR (Picard et al. 1988), has been performed (Ohgishi et al. 2001). In transgenic Arabidopsis plants expressing HD-Zip2-V-G, glucocorticoid treatment activates the ATHB2 gene itself, independent of de novo protein synthesis, further confirming that the DNA binding domain of ATHB2 recognize its own promoter. Target-gene analysis using the HD-Zip2-V-G also suggested that the DNA binding domain of ATHB2 recognizes other HD-Zip II genes (HAT9, HAT22, HAT1, HAT2, HAT3, ATHB4; Ohgishi et al. 2001). Similar results were also observed in transgenic plants expressing a HAT2-derived transcription factor (H2-V-G, Sawa et al. 2002).
As a first step to investigate whether all the 10 HD-Zip II genes may potentially be recognized by the DNA binding domain of ATHB2, putative promoter regions of these genes were analyzed for the presence of HD-Zip II binding sites. To this end, 3000 nucleotide-long regions upstream of 5′-UTR (or from the translational start if the former was not known) were inspected for HD-Zip binding sites using the core sequences recognized by the HD-Zip domain of ATHB1 (BS1, NAATWATTN; Sessa et al. 1993) and ATHB2 (BS2, NAATSATTN; Sessa et al. 1993), respectively. Interestingly, all the 10 HD-Zip II genes do contain several HD-Zip binding sites (Fig. 2a). Moreover, a statistical analysis performed essentially as described by Nemhauser et al. (2004) (see “Material and methods” for details) indicated that the upstream regions of the HD-Zip II genes are significantly enriched for HD-Zip binding sequences (Supplementary Fig. 2). By contrast, the same analysis performed on all the 17 HD-Zip I genes showed no overrepresentation of HD-Zip binding sites in the putative promoter regions of these genes (Supplementary Fig. 2).

The DNA binding domain of ATHB2 directly recognizes all the HD-Zip II genes in vivo. (a) Schematic representation of HD-Zip binding sites on the upstream regions of the HD-Zip II genes. 3000 nucleotide-long regions upstream of 5′-UTR (or from the translational start if the former was not known) were inspected for HD-Zip binding sites using the core sequences recognized by the HD-Zip domain of ATHB1 (BS1, NAATWATTN; Sessa et al. 1993) and ATHB2 (BS2, NAATSATTN; Sessa et al. 1993), respectively. The distance between the HD-Zip II genes and the next upstream ORFs is more than 3000 bp in all cases except for HAT1 and ATHB18 (−2346 bp and −1315 bp, respectively). Red and blue lines indicate BS1 and BS2 binding sites, respectively. S = G or C, W = A or T, N = A, T, C or G. The asterisk indicates that a 3000 bp region upstream of the translational start has been considered. (b) In vivo target gene analysis of all HD-Zip II genes. Transgenic Arabidopsis plants carrying the HD-Zip2-V-G gene were non-treated or treated with DEX and/or CHX. Total RNAs were extracted and subjected to qRT-PCR using UPL probes and primers specific to the genes indicated in the left of the histograms. −DEX −CHX, +DEX −CHX, −DEX +CHX and +DEX +CHX indicate RNA samples prepared from non-treated plants, or those treated with DEX, CHX, and DEX and CHX, respectively
Next, the expression of all the 10 HD-Zip II genes was analyzed in transgenic plants expressing HD-Zip2-V-G in the absence and in the presence of DEX and/or CHX by means of qRT-PCR. As shown in Fig. 2b, DEX increased the transcript levels from all the 10 HD-Zip II genes, both in the absence and in the presence of CHX. On the other hand, the DEX treatment did not change the transcript levels of ATHB1, a gene of the HD-Zip I family (Fig. 2b). Together, the data indicate that the DNA binding domain of ATHB2 directly recognizes all the 10 HD-Zip II genes in vivo.
Our target-gene analyses also revealed a major effect of CHX on the transcript levels of ATHB17 and ATHB18. Translational inhibition by CHX has been shown to influence mRNA stability, although this depends on the gene in question (Green 1993; Sullivan and Green 1993). Its effect could be to stabilize transcripts of the ATHB17 and ATHB18 genes. Minor effects of CHX were also observed on the transcript levels of HAT2 whereas no significant change in the expression of any other HD-Zip II gene occurred in the presence of the protein synthesis inhibitor cycloheximide (Fig. 2b). However, DEX up-regulated most of these HD-Zip II genes whose expression is not affected upon CHX treatment more in the presence of cycloheximide than in its absence (Fig. 2b). This suggests that HDZip2-V-G competes for target genes with the repressors molecules ATHB2, HAT2 and possibly other HD-Zip II proteins, whose amount is increased by the action of HD-Zip2-V-G. This competition does not occur in the presence of CHX since protein synthesis is inhibited, and thus the magnitude of the transcriptional activation is larger in the presence of cycloheximide.
The finding that HD-Zip2-V-G can recognize all the 10 HD-Zip II genes in vivo indicates that the HD-Zip II proteins have the potential to form an intricate regulatory network. The high degree of conservation of the HD-Zip domain within the HD-Zip II family strongly suggests that any of the HD-Zip II proteins may regulate its own expression as well as that of any HD-Zip II gene co-expressed in time and/or space. It is also worth noting that ATHB2 and HAT2 act as repressors, thus implying a negative feedback regulation within the HD-Zip II family. Future work will have to investigate the regulatory properties of the other HD-Zip II transcription factors.
The Arabidopsis HD-Zip II family comprises several paralogous genes
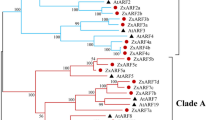
To assess the phylogeny of the HD-Zip II proteins, full length amino acid sequences were aligned to construct a phylogenetic tree by using the neighbor-joining (NJ) method (Saitou and Nei 1987). A sequence belonging to HD-Zip I family was used as an outgroup (ATHB1) and resulting tree was supported by bootstrap values over 50% (Fig. 3a). Hence, different subfamilies can be recognized: α (ATHB17 and ATHB18), β (HAT9 and HAT22), γ (HAT1, HAT2 and ATHB2) and δ (HAT3 and ATHB4), whereas HAT14 is sister to the β, γ and δ subfamilies (Fig. 3a).

Phylogenetic analyses of HD-Zip II proteins. (a) The tree based on the full length amino acid sequences of all HD-Zip II proteins was constructed by using the neighbor-joining method, and bootstrap support is indicated near the nodes of HD-Zip II sequences. ATHB1, a HD-Zip I protein, was used as an outgroup. The subfamilies within the HD-Zip II family are denoted α, β, γ and δ. (b) Schematic representation of the intron/exon organization within the HD-Zip domain encoding region in HD-Zip I and II genes. Red, orange and light orange rectangles indicate helix I, II and III of the homeodomain, respectively; a green rectangle indicates the leucine zipper motif. Black arrowheads indicate intron positions. The asterisk indicates that all the HD-Zip I α, β and γ encoding regions are characterized by the intron/exon organization schematically represented with the exception of ATHB1. (c) Intron/exon junctions within the HD-Zip domain encoding region in all HD-Zip II genes
Remarkably, all the HD-Zip II genes have two introns within the HD-Zip encoding region, and their position is conserved at the nucleotide level even in the less related genes (Fig. 3b, c). The first one splits the homeodomain, while the second one essentially separates the HD from the dimerization domain. This exon/intron organization of the HD-Zip II domain is not found in any of the HD-Zip I subfamilies (Henriksson et al. 2005, Fig. 3b). Furthermore, it is noteworthy the fact that the exon/intron organization outside the HD-Zip domain is conserved within the HD-Zip II subfamilies: the β subfamily does not have introns besides the ones in the HD-Zip region; the α, γ and δ subfamilies have one more intron in the N-terminal region whose position is conserved within each subfamily (Fig. 1). HAT14 also has an intron in the N-terminal region, but its position is distinct from that of HD-Zip II α, γ and δ genes.
The phylogeny of the HD-Zip II proteins as well as the conservation of the exon/intron organization outside the region encoding the HD-Zip domain in the genes belonging to the same subfamily strongly suggest the presence of paralogous genes within the HD-Zip II family. Thus, the chromosomal location of the HD-Zip II genes was analyzed in relation to the segmental duplication history of these regions, as described by Blanc et al. (2003). These analyses revealed that HAT9/HAT22 (Exp = 3.9 × 10−76, see “Materials and methods” for details), HAT1/HAT2 (Exp = 7.7 × 10−65) and HAT3/ATHB4 (Exp = 3.6 × 10−57) constitute very likely pairs of paralogous genes (Fig. 4). Moreover, the ATHB2 gene very close to the HAT1 locus on chromosome 4 seems also to be paralog to HAT2, even if with a lower statistical significance (Exp = 4.6 × 10−41) compared with HAT1. This suggests that ATHB2 and HAT1 arose from a tandem duplication event, which occurred before the whole genome duplication. The absence of a HD-Zip II gene close to HAT2 on chromosome 5 is likely due to the high frequency of gene loss associated with genome duplication events (Wagner 2001).

Chromosomal location and duplication events for HD-Zip family II genes in the Arabidopsis genome. Black circles indicate centromeres. Colored boxes symbolize recent duplicated segments in chromosomes II–V. Boxes linked by lines symbolize sister regions containing HD-Zip II paralogous genes
Together, the data on the duplication history of the Arabidopsis genes further support the phylogeny of the HD-Zip II proteins.
HD-Zip II γ and δ genes are up-regulated by light quality changes that induce the shade avoidance response
It has been previously shown that the expression of ATHB2 and ATHB4 is strongly induced by low R/FR (Carabelli et al. 1993, 1996). Moreover, it has been demonstrated that the up-regulation of the ATHB2 gene induced by low R/FR is mediated by the phytochrome system (Carabelli et al. 1996; Steindler et al. 1997). In addition, Affymetrix Arabidopsis Genome GeneChip array (ATH1) analyses on seedlings exposed to low R/FR for 1 h revealed that the HAT1, HAT2 and HAT3 genes are also responsive to light quality changes (ArrayExpress database, accession no. E-MEXP-443; Sessa et al. 2005). To investigate whether low R/FR light affects the expression of the remaining HD-Zip II genes, total and polyA+ RNAs were extracted from 7-day-old seedlings grown in high R/FR and then exposed to low R/FR for different times. Northern analyses confirmed that HAT1, HAT2 and HAT3 genes are significantly induced by low R/FR, and revealed that HAT22, HAT9, HAT14 and ATHB17 are essentially unaffected by light quality changes (Supplementary Fig. 3). No specific signal was detected for ATHB18 (data not shown). However, qRT-PCR revealed no significant change in the expression of this gene in seedlings exposed to low R/FR (Supplementary Fig. 4).
To further characterize the light-regulation of the HAT1, HAT2, HAT3 and ATHB4 genes, their early response to low R/FR light was analyzed by Northern or qRT-PCR (Fig. 5a, b). The kinetics of HAT1, HAT3 and ATHB4 induction, as that of ATHB2, is very rapid and transient; the transcript level of these genes reaches its maximum at 30 min and then slowly decreases. By contrast, the HAT2 transcript level increases slowly and reaches its maximum at 1 h. This observation suggested that the expression of HAT1, HAT3 and ATHB4, but not that of HAT2, may be directly regulated by the phytochrome system. Previous work has demonstrated that HAT2 expression is induced by auxin (Sawa et al. 2002), and thus, it seems likely that the HAT2 gene is up-regulated by low R/FR as a consequence of the induction of the auxin signaling pathway provoked by FR-rich light (Devlin et al. 2003; Carabelli et al. 2007). Consistent with this hypothesis, the kinetics of induction by low R/FR of auxin-responsive genes such as IAA19 and IAA29, is indistinguishable from that of HAT2 (Supplementary Fig. 5). Furthermore, the expression of ATHB2, HAT1, HAT3 and ATHB4 is essentially unaffected by exogenous auxin whereas, as expected, HAT2 transcript levels are dramatically increased in IAA-treated seedlings (Supplementary Fig. 6).

HAT1, ATHB2, HAT3 and ATHB4 genes are rapidly and reversibly regulated by light quality changes. (a) Northern analysis of HAT1, HAT2, ATHB2 and HAT3 in Col-0 seedlings grown in high R/FR (0) and then exposed to low R/FR for the indicated times. ATL18 was used to monitor equal loading. (b) qRT-PCR analysis of ATHB4 in Col-0 seedlings treated as described in (a). The histogram shows the relative expression levels of ATHB4 in high and low R/FR light. Each value is the mean of three separate quantitative PCR reactions normalized to actin 2. Relative transcript abundance of ATHB4 at each time point is normalized to its relative level in Col-0 seedlings in high R/FR (0). (C) Northern analysis of HAT1, HAT2, ATHB2 and HAT3 in Col-0 seedlings grown in high R/FR (0) and then exposed for 60 min to low R/FR (60), or exposed for 60 min to low R/FR and then returned to high R/FR for 15 min (75), 30 min (90), and 60 min (120). ATL18 was used to monitor equal loading. (d) qRT-PCR analysis of ATHB4 in Col-0 seedlings treated as described in (c). The histogram shows the relative expression levels of ATHB4 in the light conditions indicated. Each value is the mean of three separate quantitative PCR reactions normalized to actin 2. Relative transcript abundance of ATHB4 under each light condition is normalized to its relative level in Col-0 seedlings in high R/FR (0)
To investigate whether the induction of HAT1, HAT3 and ATHB4 is indeed directly mediated by the phytochrome system, an experiment of light reversion was conducted (Fig. 5c, d). Total RNA was extracted from 7-day-old seedlings grown in high R/FR and then exposed to low R/FR for 60 min or exposed for 60 min to low R/FR and then returned to high R/FR for 15 min (75), 30 min (90) and 60 min (120). Northern and qRT-PCR analyses showed that the induction of HAT1, HAT3 and ATHB4, as that of ATHB2, is rapidly reverted by light-switching with a two-fold reduction in transcript levels after 15 min of exposure to high R/FR (Fig. 5c, d). Interestingly, the HAT2 mRNA level decreases slowly showing a two-fold decrease after 30 min of the same light treatment (Fig. 5c), further supporting the hypothesis that HAT2 induction is mediated by auxin changes caused by low R/FR.
Together the data demonstrate that five out of the ten HD-Zip II genes are regulated by light quality changes. Among them, ATHB2, HAT1, HAT3 and ATHB4 seem to be directly regulated by the phytchrome system whereas HAT2 appears to respond to the auxin changes provoked by low R/FR light. This is particularly interesting considering that HAT1 and HAT2 are paralogous genes, thus implying that genes belonging to the same subfamily evolved distinct regulatory properties.
HD-Zip II γ and δ genes are tightly regulated during plant development in a high R/FR light environment
To investigate whether the expression of HD-Zip II genes is spatially and temporally regulated during plant development in a high R/FR light environment, we took advantage of Arabidopsis microarray databases available on line (Zimmermann et al. 2004, GENEVESTIGATOR, https://www.genevestigator.ethz.ch/index.php; AREX, http://www.arexdb.org/index.jsp; AtGenExpress Visualization Tool (AVT), http://jsp.weigelworld.org/expviz/expviz.jsp; Winter et al. 2007, Arabidopsis eFB Browser, http://bar.utoronto.ca/efp/cgi-bin/efpWeb.cgi).
HD-Zip II gene expression patterns were analyzed in the root using the digital in situ approach which measures gene expression among three developmental stages along the longitudinal axis and five radial cell layers (Birnbaum et al. 2003). The analyses revealed that several HD-Zip II genes are regulated during root development. All the three HD-Zip II γ genes are expressed at both stage I and II; however, their expression pattern is quite distinct during development. ATHB2 transcript levels decrease whereas HAT2 increase from stage I to stage II and III. On the other hand, HAT1 is expressed at comparable levels at stage I and II, and significantly less at stage III. The expression pattern of the three genes is also distinct along the radial axis. ATHB2 is expressed essentially at the same level in the five radial cell layers whereas HAT1 and HAT2 display tissue-specificity. HAT22 and HAT9 β genes are expressed exclusively at stage III with distinct patterns along the radial axis whereas the ATHB17 α transcript is present at low levels throughout root development mainly in stele cells. HD-Zip II δ genes are not expressed in the root (Supplementary Fig. 7).
HD-Zip II gene expression was also investigated in the aerial part of the plant (Schmid et al. 2005). Interestingly, all the three HD-Zip II γ genes are co-expressed during leaf development and their transcript levels significantly increase with leaf age. On the other hand, the expression of HAT3 δ gene decreases during leaf development whereas its paralog ATHB4 is essentially not expressed at any of the developmental stages analyzed (Supplementary Fig. 8a). Most of the γ and δ genes are also expressed in the shoot apical meristem. The transcript levels of HAT2 and HAT1 γ genes increase, respectively, upon phase change and at the reproductive phase whereas those of ATHB2 γ gene are undetectable at all three phases. The expression of ATHB4 δ gene increases upon phase change whereas HAT3 δ transcript levels are comparable at vegetative and transition phases and slightly decrease at the reproductive phase (Supplementary Fig. 8b). Remarkably, the expression of several HD-Zip II genes is also dynamically regulated during flower organ development. ATHB18 α gene is expressed in stamens at stage 12 but not at stage 15. HAT22 β gene expression increases significantly during flower development in sepals, petals and stamens and decreases in carpels. ATHB2 γ gene is expressed in petals at stage 12 more than at stage 15 whereas HAT2 γ transcript levels increase during flower development in petals and stamens. HAT3 δ gene is expressed exclusively in carpels and its transcript levels decrease during flower development (Supplementary Fig. 9).
Together, digital in situ analyses indicated that the expression of the HD-Zip II genes, including those induced by light quality changes, is tightly regulated during plant development in a high R/FR light environment. Furthermore, these analyses revealed both overlapping and distinct expression patterns of paralogous genes, thus suggesting the existence of redundancy and divergence within the HD-Zip II protein family.
Functional redundancy within the HD-Zip II γ subfamily
The finding that the HD-Zip II γ and δ genes, all induced by light quality changes (low R/FR) which provoke the shade avoidance response, are tightly regulated during organ formation strongly suggests that they may play a major role in plant development under a high R/FR light environment as well.
To test this hypothesis, the HD-Zip II γ subfamily was selected for further analyses. Previous work has shown that seedlings overexpressing ATHB2 have elongated hypocotyls, smaller cotyledons and leaves whereas 35S::HD-Zip2-V-G seedlings in the presence of dexamethasone display reciprocal phenotypes (Steindler et al. 1999). It has also been shown that ectopic expression of HAT2 results in phenotypes similar to those caused by elevated levels of ATHB2 (Sawa et al. 2002; Supplementary Table 2). As observed in plants with elevated levels of ATHB2, the expression of several HD-Zip II genes is down-regulated in plants overexpressing HAT2. Moreover, plants expressing a derivative of HAT2 with opposite regulatory properties (35S::HAT2-V-G) have higher transcript levels of the same HD-Zip II genes (Sawa et al. 2002).
To investigate whether all the HD-Zip II proteins of the γ subfamily, which share a DNA binding domain highly homologous (identity ≥ 83%, Supplementary Table 1), act as negative regulators of gene expression, we generated and characterized transgenic plants ectopically expressing HAT1. At the seedling stage, the ectopic expression of HAT1, as that of ATHB2 and HAT2, causes longer hypocotyls and smaller leaves in high R/FR (Supplementary Table 2). Roots are also strongly affected, since the length of the main root is reduced (cm ± s.e.m.: Col-0, 3.14 ± 0.10; 35S::HAT1#6-3, 2.22 ± 0.09*; 35S::HAT1#1-3, 1.82 ± 0.05*; *P ≤ 0.01 transgenic line versus wild type) and produce almost no lateral roots in HAT1 transgenic plants (n° ± s.e.m.: Col-0, 6.55 ± 0.56; 35S::HAT1#6-3, 0.19 ± 0.08*; 35S::HAT1#1-3, 0.04 ± 0.04*; *P ≤ 0.01 transgenic line versus wild type), as previously observed in 35S::ATHB2 and 35S::HAT2 seedlings (Steindler et al. 1999; Sawa et al. 2002). The phenotype of 35S::HAT1 plants is therefore consistent with a molecular function of HAT1 identical to that of ATHB2 and HAT2. This is further supported by the finding that, as observed in plants with elevated levels of ATHB2 and HAT2 (Ohgishi et al. 2001; Sawa et al. 2002; Supplementary Fig. 10), the expression of several HD-Zip II genes is significantly down-regulated in plants overexpressing HAT1 (Supplementary Fig. 10).
As a first step to investigate how HD-Zip II γ proteins affect plant growth, the phenotype of plants overexpressing each of the three γ proteins was further analyzed. Among the plant organs, we selected the leaf for a two-fold reason. First, leaves are particularly interesting since they constitute most of the aboveground portion of the plant and are derived from determinate growth. Second, digital in situ analysis revealed that all the HD-Zip II γ genes are expressed during leaf development (Supplementary Fig. 8a).
To evaluate the morphological changes provoked by elevated levels of ATHB2, HAT1 and HAT2, first leaves of different age but same area (1.5 mm2) in wild-type and transgenic seedlings were selected, and the mean cell area of adaxial subepidermal cells was measured in three leaf regions: distal (D), median (M) and proximal (P). Remarkably, the mean cell area was significantly increased in all the transgenic lines overexpressing each of the three HD-Zip II γ genes (Table 1, Fig. 6), thus indicating that cell number rather than cell size contribute to the reduced leaf size of seedlings overexpressing ATHB2, HAT1 and HAT2.

DIC optics analysis of the adaxial subepidermal cell layer in the median region of the first leaves of seedlings with altered levels of HD-Zip II γ proteins. Plants were grown in a light/dark cycle for 8.5 days (Col-0, 35S::ATHB2N51A #4-3), 10 days (35S::HAT1 #1-3, 35S::HAT2 #8-4) or 11.5 days (35S::ATHB2 #3-2), and first leaves with an equal area (1.5 mm2) were selected for DIC optics analysis of the adaxial subepidermal cell layer. Images represent first leaf median regions of wild type and transgenic lines. Selected cells are colored green to highlight differences among the lines. Scale bar: 50 μm
To get further insight into the function of HD-Zip II γ proteins, we devised a strategy which takes into account both the DNA binding properties of the HD-Zip proteins (Sessa et al. 1993, 1997) and the high conservation of the HD-Zip domain within the γ subfamily. Previous work has demonstrated that the HD-Zip domain recognizes DNA exclusively in the dimeric form (Sessa et al. 1993). Furthermore, it has been shown that a single amino acid substitution at position 51 of the HD-Zip2 domain (N51A) abolishes DNA binding in vitro and in vivo even if present in only one of the two subunits forming the HD-Zip dimeric complex (Sessa et al. 1997; Steindler et al. 1999). Finally, several evidence indicate that HD-Zip proteins are able to form heterodimers with members of the same family whereas heterodimerization across distinct HD-Zip families apparently dos not occur (Meijer et al. 2000).
A derivative of ATHB2 containing a single amino acid substitution at position 51 of the homeodomain was thus constructed (ATHB2N51A), and utilized to generate transgenic plants (Supplementary Table 2). Overexpression of ATHB2N51A in Arabidopsis plants should sequester the endogenous ATHB2 protein in heterodimeric complexes functionally inactive, and thus prevent the formation of functional ATHB2 homodimers. Because of the high homology between the three HD-Zip II γ proteins, ATHB2N51A should sequester HAT1 and HAT2 as well.
To investigate whether overexpression of ATHB2N51A does indeed produce a dominant-negative phenotype, first leaves of same area (1.5 mm2) in wild-type and transgenic seedlings were selected, and the mean cell area of adaxial subepidermal cells was measured in distal, median and proximal leaf regions. Remarkably, the mean cell area was significantly decreased in both of the transgenic lines overexpressing ATHB2N51A (Table 1, Fig. 6), thus demonstrating that one or more HD-Zip II γ proteins negatively regulate cell proliferation during leaf development in a high R/FR light environment. Future work will have to investigate the contribution of each of the three HD-Zip II γ transcription factors to leaf cell proliferation regulation by means of single, double and triple loss-of-function mutant analysis.
Conclusions
In this study, we performed a genome-wide analysis of the Arabidopsis HD-Zip II family. The results provide evidence for a complex pattern of expression and regulation of this gene family, and they strongly suggest that HD-Zip II genes act as members of highly integrated networks in controlling organ development as well as plant responses to light quality changes. Clearly, a future challenge will be to unravel the HD-Zip II regulatory networks active during organ formation in high and/or low R/FR, and characterize their dynamics. The extraordinary genetic resources developed for Arabidopsis together with the recent progresses in proteomics to identify target genes and monitor protein interactions at subcellular resolution will allow us to tackle this challenge.
Abbreviations
- BS1:
-
Binding site 1
- BS2:
-
Binding site 2
- CHX:
-
Cycloheximide
- DEX:
-
Dexamethasone
- DIC:
-
Differential Interference Contrast
- DMSO:
-
Dimethyl sulfoxide
- HD-Zip:
-
Homeodomain-Leucine Zipper
- IAA:
-
Indole Acetic Acid
- MS:
-
Murashige and Skoog medium
- ORF:
-
Open Reading Frame
- qRT-PCR:
-
quantitative Reverse Transcription-Polymerase Chain Reaction
- RT-PCR:
-
Reverse Transcription-Polymerase Chain Reaction
- R/FR:
-
Red/Far-Red
- UPL:
-
Universal Probe Library
- UTR:
-
Untranslated Region
References
Aoyama T, Dong CH, Wu Y, Carabelli M, Sessa G, Ruberti I et al (1995) Ectopic expression of the Arabidopsis transcriptional activator Athb-1 alters leaf cell fate in tobacco. Plant Cell 7:1773–1785
Baima S, Possenti M, Matteucci A, Wisman E, Altamura M, Ruberti I et al (2001) The Arabidopsis ATHB8 HD-Zip protein acts as a differentiation-promoting transcription factor of the vascular meristems. Plant Physiol 126:643–6555. doi:10.1104/pp.126.2.643
Bechtold N, Ellis JE, Pellettier G (1993) In planta Agrobacterium mediated gene transfer by infiltration of adult Arabidopsis thaliana plants. C R Acad Sci Paris 316:1194–1199
Birnbaum K, Shasha DE, Wang JY, Jung JW, Lambert GM, Galbraith DW et al (2003) A gene expression map of the Arabidopsis root. Science 302:1956–1960. doi:10.1126/science.1090022
Blanc G, Hokamp K, Wolfe KH (2003) A recent polyploidy superimposed on older large-scale duplications in the Arabidopsis genome. Genome Res 13:137–144. doi:10.1101/gr.751803
Bowman JL (2004) Class III HD-Zip gene regulation, the golden fleece of ARGONAUTE activity? Bioessays 26:934–938. doi:10.1002/bies.20103
Byrne ME (2006) Shoot meristem function and leaf polarity: the role of class III HD-ZIP genes. PLoS Genet 2:e89. doi:10.1371/journal.pgen.0020089
Carabelli M, Sessa G, Baima S, Morelli G, Ruberti I (1993) The Arabidopsis Athb-2 and -4 genes are strongly induced by far-red-rich light. Plant J 4:469–479. doi:10.1046/j.1365-313X.1993.04030469.x
Carabelli M, Morelli G, Whitelam G, Ruberti I (1996) Twilight-zone and canopy shade induction of the Athb-2 homeobox gene in green plants. Proc Natl Acad Sci USA 93:3530–3535. doi:10.1073/pnas.93.8.3530
Carabelli M, Possenti M, Sessa G, Ciolfi A, Sassi M, Morelli G et al (2007) Canopy shade causes a rapid and transient arrest in leaf development through auxin-induced cytokinin oxidase activity. Genes Dev 21:1863–1868. doi:10.1101/gad.432607
Devlin PF, Yanovsky MJ, Kay SA (2003) A genomic analysis of the shade avoidance response in Arabidopsis. Plant Physiol 133:1617–1629. doi:10.1104/pp.103.034397
Felsenstein J (1989) PHYLIP: phylogeny inference package version 3.2. Cladistics 5:164–166
Felsenstein J (2005) PHYLIP (Phylogeny Inference Package) version 3.6. Distributed by the author. Department of Genome Sciences, University of Washington, Seattle
Franklin KA, Praekelt U, Stoddart WM, Billingham OE, Halliday KJ, Whitelam GC (2003) Phytochromes B, D, and E act redundantly to control multiple physiological responses in Arabidopsis. Plant Physiol 131:1340–1346. doi:10.1104/pp.102.015487
Green PJ (1993) Control of mRNA stability in higher plants. Plant Physiol 102:1065–1070
Henriksson E, Olsson AS, Johannesson H, Johansson H, Hanson J, Engstrom P et al (2005) Homeodomain leucine zipper class I genes in Arabidopsis. Expression patterns and phylogenetic relationships. Plant Physiol 139:509–518. doi:10.1104/pp.105.063461
Horiguchi G, Kim GT, Tsukaya H (2005) The transcription factor AtGRF5 and the transcription co-activator AN3 regulate cell proliferation in leaf primordia of Arabidopsis thaliana. Plant J 43:68–78. doi:10.1111/j.1365-313X.2005.02429.x
Meijer AH, de Kam RJ, d’Erfurth I, Shen W, Hoge JH (2000) HD-Zip proteins of families I and II from rice: interactions and functional properties. Mol Gen Genet 263:12–21. doi:10.1007/PL00008671
Morelli G, Ruberti I (2000) Shade avoidance responses. Driving auxin along lateral routes. Plant Physiol 122:621–626. doi:10.1104/pp.122.3.621
Morelli G, Ruberti I (2002) Light and shade in the photocontrol of Arabidopsis growth. Trends Plant Sci 7:399–404. doi:10.1016/S1360-1385(02)02314-2
Morelli G, Baima S, Carabelli M, Di Cristina M, Lucchetti S, Sessa G et al (1998) Homeodomain-leucine zipper proteins in the control of plant growth and development. In: Last R, Lo Schiavo F, Morelli G, Raikel N (eds) Cellular integration of signaling pathways in plant development, vol H 104, pp 251–262. Springer Verlag, NATO-ASI series
Murashige T, Skoog F (1962) A revised medium for rapid growth and bioassays with tobacco tissue coltures. Physiol Plant 15:473–497. doi:10.1111/j.1399-3054.1962.tb08052.x
Nakamura M, Katsumata H, Abe M, Yabe N, Komeda Y, Yamamoto KT et al (2006) Characterization of the class IV homeodomain-Leucine Zipper gene family in Arabidopsis. Plant Physiol 141:1363–1375. doi:10.1104/pp.106.077388
Nemhauser JL, Mockler TC, Chory J (2004) Interdependency of brassinosteroid and auxin signaling in Arabidopsis. PLoS Biol 2:e258. doi:10.1371/journal.pbio.0020258
Ohgishi M, Oka A, Morelli G, Ruberti I, Aoyama T (2001) Negative autoregulation of the Arabidopsis homeobox gene ATHB-2. Plant J 25:389–398. doi:10.1046/j.1365-313x.2001.00966.x
Pfaffl MW (2001) A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res 29:2002–2007. doi:10.1093/nar/29.9.e45
Picard D, Salser SJ, Yamamoto KR (1988) A movable and regulable inactivation function within the steroid binding domain of the glucocorticoid receptor. Cell 54:1073–1080. doi:10.1016/0092-8674(88)90122-5
Ruberti I, Sessa G, Lucchetti S, Morelli G (1991) A novel class of plant proteins containing a homeodomain with a closely linked leucine zipper motif. EMBO J 10:1787–1791
Saitou N, Nei M (1987) The neighbor-joining method: a new method for reconstructing phylogenetic tree. Mol Biol Evol 4:406–425
Sawa S, Ohgishi M, Goda H, Higuchi K, Shimada Y, Yoshida S et al (2002) The HAT2 gene, a member of the HD-Zip gene family, isolated as an auxin inducible gene by DNA microarray screening, affects auxin response in Arabidopsis. Plant J 32:1011–1022. doi:10.1046/j.1365-313X.2002.01488.x
Schena M, Lloyd AM, Davis RW (1993) The HAT4 gene of Arabidopsis encodes a developmental regulator. Genes Dev 7:367–379
Schmid M, Davison TS, Henz SR, Pape UJ, Demar M, Vingron M et al (2005) A gene expression map of Arabidopsis thaliana development. Nat Genet 37:501–506. doi:10.1038/ng1543
Sessa G, Morelli G, Ruberti I (1993) The Athb-1 and -2 HD-Zip domains homodimerize forming complexes of different DNA binding specificities. EMBO J 12:3507–3517
Sessa G, Carabelli M, Ruberti I, Baima S, Lucchetti S, Morelli G (1994) Identification of distinct families of HD-Zip proteins in Arabidopsis thaliana. In: Puigdomenech P, Coruzzi G (eds) Molecular-genetic analysis of plant development and metabolism. NATO-ASI series, vol H 81, pp 411–426. Springer Verlag
Sessa G, Morelli G, Ruberti I (1997) DNA-binding specificity of the homeodomain-leucine zipper domain. J Mol Biol 274:303–309. doi:10.1006/jmbi.1997.1408
Sessa G, Carabelli M, Sassi M, Ciolfi A, Possenti M, Mittempergher F et al (2005) A dynamic balance between gene activation and repression regulates the shade avoidance response in Arabidopsis. Genes Dev 19:2811–2815. doi:10.1101/gad.364005
Steindler C, Borello U, Carabelli M, Morelli G, Ruberti I (1997) Phytochome A, phytochome B and other phytochome(s) regulate ATHB-2 gene expression in etiolated and green Arabidopsis plants. Plant Cell Environ 20:759–763. doi:10.1046/j.1365-3040.1997.d01-123.x
Steindler C, Matteucci A, Sessa G, Weimar T, Ohgishi M, Aoyama T et al (1999) Shade avoidance responses are mediated by the ATHB-2 HD-zip protein, a negative regulator of gene expression. Development 126:4235–4245
Sullivan ML, Green PJ (1993) Post-transcriptional regulation of nuclear-encoded genes in higher plants: the roles of mRNA stability and translation. Plant Mol Biol 23:1091–1104. doi:10.1007/BF00042344
Thompson JD, Higgins DG, Gibson TJ (1994) CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res 22:4673–4680. doi:10.1093/nar/22.22.4673
Tiwari SB, Hagen G, Guilfoyle TJ (2004) Aux/IAA proteins contain a potent transcriptional repression domain. Plant Cell 16:533–543. doi:10.1105/tpc.017384
Triezenberg SJ, Kingsbury RC, McKnight SL (1988) Functional dissection of VP16, the transactivator of herpes simplex virus immediate early gene expression. Genes Dev 2:718–729. doi:10.1101/gad.2.6.718
Wagner A (2001) Birth and death of duplicated genes in completely sequenced eukaryotes. Trends Genet 17:237–239. doi:10.1016/S0168-9525(01)02243-0
Weigel D, Glazebrook J (2002) Arabidopsis: a laboratory manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, New York
Winter D, Vinegar B, Nahal H, Ammar R, Wilson GV, Provart NJ (2007) An “electronic fluorescent pictograph” browser for exploring and analyzing large-scale biological data sets. PLoS One 2:e718. doi:10.1371/journal.pone.0000718
Zimmermann P, Hirsch-Hoffmann M, Hennig L, Gruissem W (2004) GENEVESTIGATOR: Arabidopsis microarray database and analysis toolbox. Plant Physiol 136:2621–2632. doi:10.1104/pp.104.046367
Acknowledgements
We thank Bernd Weisshaar for the pBENDER vector. We also thank Takashi Aoyama and Massimiliano Sassi for helpful discussion, and Daniela Bongiorno for skillful technical assistance. This work was supported, in part, by grants from the EU 5th framework project REGIA (QLG2-1999-00876), MUR Strategic Program FIRB 2003 and FIRB ERA-PG, and ASI Biotechnology Program.
Author information
Authors and Affiliations
Corresponding author
Additional information
A. R. Ciarbelli and A. Ciolfi are contributed equally to this work.
Electronic supplemental material
Below are the link to the electronic supplementary material.
Supplementary Fig. 1
Alignment of the N-terminal regions of the 10 HD-Zip II proteins. For each column in the alignment, residues conserved in more than 40% of all sequences are highlighted in different colors using BLOSUM62 scores. Blue = K, R; Cyan = F, W, Y; Dark Green = I, L, M, V; Dark Grey = N, S, T; Green = A; Grey = Q; Magenta = H; Purple = P; Red = D, E; Yellow = G (TIFF 2296 kb)
Supplementary Fig. 2
The upstream regions of the HD-Zip II genes are significantly enriched for BS1 and BS2 sequences. S = G or C; W = A or T; N = A, T, C or G. Promoters refer to the 3000 bp-long upstream regions analyzed. The distance between the HD-Zip I genes and the next upstream ORFs is more than 3000 bp in 11 out of 17 genes. For the remaining HD-Zip I genes, the distance is as follows: ATHB5 = 2554 bp, ATHB6 = 1079 bp, ATHB20 = 260 bp, ATHB23 = 1595 bp, ATHB52 = 1267 bp, ATHB54 = 1847 bp. a = Total number of sites identified; b = Expected number of sites based on 1000 randomly sampled groups of promoters; c = Standard deviation of Expected Number; d = Number of promoters containing at least one site; e = Percentage of promoters containing at least one site. >, value is greater than 0.25 (TIFF 521 kb)
Supplementary Fig. 3
HD-Zip II γ and δ genes are induced by low R/FR light. Northern analysis of all the HD-Zip II genes with the exception of ATHB18 in Col-0 seedlings grown in high R/FR (0) and then exposed to low R/FR for the indicated times. Total RNA (A) was used for the analysis of HAT22, HAT1, HAT2, ATHB2, HAT3 transcripts and polyA+ RNA (B) for that of ATHB17, HAT14, HAT9, ATHB4 mRNA expression. ATL18 was used to monitor equal loading (TIFF 3522 kb)
Supplementary Fig. 4
qRT-PCR analysis of ATHB18 in Col-0 seedlings grown in high R/FR (0) and then exposed to low R/FR for the indicated times. The histogram shows the relative expression levels of ATHB18 in high and low R/FR light. Each value is the mean of three separate quantitative PCR reactions normalized to actin 2. Relative transcript abundance of ATHB18 at each time point is normalized to its relative level in Col-0 seedlings in high R/FR (0) (TIFF 1690 kb)
Supplementary Fig. 5
Kinetic of induction of HAT2, IAA19 and IAA29 by low R/FR light. qRT-PCR analysis of HAT2, IAA19 and IAA29 in Col-0 seedlings grown in high R/FR (0) and then exposed to low R/FR for the indicated times. The histograms show the relative expression levels of HAT2, IAA19 and IAA29 in high and low R/FR light. Each value is the mean of three separate quantitative PCR reactions normalized to actin 2. Relative transcript abundance of HAT2, IAA19 and IAA29 at each time point is normalized to their relative levels in Col-0 seedlings in high R/FR (0) (TIFF 1963 kb)
Supplementary Fig. 6
HAT2, but not other light-regulated HD-Zip II genes, is induced by auxin. qRT-PCR analysis of HD-Zip II γ and δ genes in Col-0 seedlings untreated (DMSO) or treated for the indicated times with IAA (1 μM). The graphs show the relative expression levels of ATHB2, HAT1, HAT2, HAT3 and ATHB4 in untreated and IAA-treated seedlings. Each value is the mean of three separate quantitative PCR reactions normalized to actin 2. Relative transcript abundance of ATHB2, HAT1, HAT2, HAT3 and ATHB4 at each time point is normalized to their relative levels in DMSO-treated seedlings at the same time points. The standard deviation for all values is ≤0.2 (TIFF 1731 kb)
Supplementary Fig. 7
Root digital in situ analyses of the HD-Zip II genes. Expression data from Birnbaum et al. (2003) were deduced from and visualized by the Arabidopsis eFP Browser (Winter et al. 2007; http://bar.utoronto.ca/efp/cgi-bin/efpWeb.cgi) (TIFF 6819 kb)
Supplementary Fig. 8
Shoot digital in situ analyses of the HD-Zip II genes. Expression data from Schmid et al. (2005) were deduced from Arabidopsis eFP Browser (Winter et al. 2007; http://bar.utoronto.ca/efp/cgi-bin/efpWeb.cgi) and used to plot histograms. (a) Chart shows absolute expression levels of HD-Zip II genes in rosette leaves from 17 day-old seedlings. Expression levels in leaf #2, #4, #6, #8, #10 and #12 were depicted with differently green-colored bars; (b) Chart shows absolute expression levels of HD-Zip II genes in the shoot apices from 7 day-old (vegetative), 14 day-old (transition) and 21 day-old (inflorescence) seedlings (TIFF 1418 kb)
Supplementary Fig. 9
Flower digital in situ analyses of the HD-Zip II genes. Expression data in flower stages 12 and 15 from Schmid et al. (2005) were deduced from and visualized by the Arabidopsis eFP Browser (Winter et al. 2007; http://bar.utoronto.ca/efp/cgi-bin/efpWeb.cgi). Sep, sepals; pet, petals; stam, stamens; carpels (TIFF 3071 kb)
Supplementary Fig. 10
HD-Zip II genes’ expression in 35S::HAT1, 35S::HAT2 and 35S::ATHB2 plants. Total RNAs were extracted and subjected to qRT-PCR using UPL probes and primers specific to the genes indicated in the left of the histograms (TIFF 1998 kb)
Rights and permissions
About this article
Cite this article
Ciarbelli, A.R., Ciolfi, A., Salvucci, S. et al. The Arabidopsis Homeodomain-leucine Zipper II gene family: diversity and redundancy. Plant Mol Biol 68, 465–478 (2008). https://doi.org/10.1007/s11103-008-9383-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11103-008-9383-8