
Abstract
The ChillPeach database was developed to facilitate identification of genes controlling chilling injury (CI), a global-scale post-harvest physiological disorder in peach. It contained 7,862 high-quality ESTs (comprising 4,468 unigenes) obtained from mesocarp tissues of two full-sib progeny contrasting for CI, about 48 and 13% of which are unique to Prunus and Arabidopsis, respectively. All ESTs are in the Gateway® vector to facilitate functional assessment of the genes. The data set contained several putative SNPs and 184 unigenes with high quality SSRs, of which 42% were novel to Prunus. Microarray slides containing 4,261 ChillPeach unigenes were printed and used in a pilot experiment to identify differentially expressed genes in cold-treated compared to control mesocarp tissues, and in vegetative compared to mesocarp tissues. Quantitative RT-PCR (qRT-PCR) confirmed microarray results for all 13 genes tested. The microarray and qRT-PCR analyses indicated that ChillPeach is rich in putative fruit-specific and novel cold-induced genes. A website (http://bioinfo.ibmcp.upv.es/genomics/ChillPeachDB) was created holding detailed information on the ChillPeach database.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
World production of peaches and nectarines is about 11 million tons (Lurie and Crisosto 2005). The major producing countries are China, Italy, and the United States in the northern hemisphere, and Chile, South Africa and Australia in the southern hemisphere. Fresh-market peaches are produced in the northern hemisphere from April through September, and in the south, from November to March. Americans eat approximately 4.41 lbs of peaches per capita per year. This consumption level is low when compared to other fresh fruit such as apple (35.28 lbs) and banana (19.85 lbs) (Crisosto 2006). A major reason for low consumption of fresh peach and nectarine is a physiological disorder called chilling injury (CI) or internal breakdown. CI development is the consequence of storing fruit in cold temperatures to extend fruit market life.
Marketing and shipment of peach fruit, either within a large country such as the United States or China, or export to other countries can require a lengthy storage period. Also, the global industry can take advantage of the on and off seasons between the two hemispheres to exchange fruit for marketing. This requires long periods (at least 3–5 weeks of market life—shipment, handling at production and receiving) between the time fruit is harvested to when it finally reaches consumers. CI is encountered in all these cases, and it is responsible for consumer dissatisfaction and reduced consumption.
CI symptoms include lack of juiciness (mealiness or woolliness), flesh browning, black pit cavity, flesh translucency (gel breakdown), red pigment accumulation (bleeding), loss of flavor, development of off flavors, and failure to ripen (Lurie and Crisosto 2005). Because most of these symptoms develop during fruit ripening after cold storage, the CI problem is usually not noticed until fruit reaches consumers (Bruhn et al. 1991; Crisosto 2006), thereby reducing consumer satisfaction.
Physiology of CI has received considerable attention. Several postharvest treatments have been proposed to mitigate the problem of CI in peach and nectarine fruit. These include warming interruptions during cold storage, treatment with plant growth regulators, controlled atmosphere, and intermittent warming and controlled delayed cooling (for review see Lurie and Crisosto 2005). Pre-harvest factors influencing CI were also reviewed by Lurie and Crisosto (2005) including fertilization, irrigation regimes, canopy position of fruit, and crop load and size. Manipulation of these factors has provided only short-term solution to CI problem. A long-term solution is highly desirable and relies on understanding the genetic basis of its inheritance. Elucidation of the inheritance mechanism will enable the breeding of new CI-resistant cultivars, and provide a better understanding of the problem within existing cultivars.
Progress is currently being made in unraveling the genetics of CI. A peach linkage map (Pop-DG) developed by crossing a processing peach cultivar ‘Dr. Davis’ with a fresh market cultivar ‘Georgia Belle’ is proving very useful for identifying genes and molecular markers associated with CI control (Peace et al. 2005a; Ogundiwin et al. 2007). One major quantitative trait locus (QTL) and a few minor QTLs have been localized for mealiness, browning and bleeding using the Pop-DG map (Peace et al. 2005a, 2006; Ogundiwin et al. 2007). A gene encoding a cell wall modifying enzyme, endopolygalacturonase (endoPG) co-localized with the major QTL affecting both mealiness and bleeding (Peace et al. 2005a, b). Another gene in the anthocyanin biosynthesis pathway, leucoanthocyanidin dioxygenase (PpLDOX), mapped to the same genomic region where the major browning QTL was identified (Ogundiwin et al. 2008).
Genomic information is accumulating for fruit crops in the Rosaceae family such as apple (Newcomb et al. 2006), apricot (Grimplet et al. 2005), peach (Horn et al. 2005; ESTree Consortium 2005), and strawberry (Folta et al. 2005), and there are current whole genome sequencing efforts for apple and peach. Extensive Rosaceae expressed sequence tag (EST) data is accessible from the Genome Database for Rosaceae (GDR: www.bioinfo.wsu.edu/gdr) for functional genomic study of fruit quality. The first publicly available peach microarray was μPeach 1.0, constructed by the Italian Consortium for Genomics in Prunus (ESTree Consortium 2005). This microarray contains 4,806 unigenes expressed in peach fruit, 70% of which were expressed during fruit ripening (ESTree Consortium 2005). Given the significance of CI to the stone fruit industry, a dedicated genomics toolkit is indispensable to expedite CI-resistance gene discovery. Such toolkit should be enriched with sequences of genes that are directly involved with CI development to improve the precision of candidate gene mapping. We report here the development of a specialized database (ChillPeach) to target genes expressed during CI development, and the construction of a cDNA microarray containing probes for them. Fruit tissue obtained from two full-sib progeny individuals of Pop-DG with contrasting susceptibility to CI and subjected to various cold storage durations and ripening regimes were used to develop the ChillPeach EST collection. The features of this EST collection, the results of a preliminary test of the microarray developed from it, and validation by real-time quantitative RT-PCR (qRT-PCR) of a set of cold-responsive genes identified in the microarray analysis are also reported as proof of the applicability of the ChillPeach dataset to CI functional genomics study.
Materials and methods
Plant material and CI measurements
Fruit tissue from the mesocarp of two progeny trees of the Pop-DG mapping population were used for developing EST libraries. These two genotypes represent extremes of susceptibility (one resistant, one susceptible) to mealiness and internal browning in Pop-DG. The CI phenotype of the two genotypes was confirmed over 3 years of observation prior to this study. For both, fruit tissue was obtained at several stages of cold storage and ripening as follows: M = mature at 12–14 lb fruit firmness, R = mature with 2–4 days ripening at 20°C to 2–3 lb firmness, TR = tree-ripe—picked at 2–3 lb firmness, CS-1 = M + 1 week cold storage at 5°C, CSR-1 = M + CS-1 + R, CS-2 = M + 2 weeks cold storage at 5°C, CSR-2 = M + CS-2 + R. For CI induction, fruit were forced-air cooled at 0–2°C within 6 h of harvest and then stored at 5°C with 90% relative humidity. At 1 and 2 weeks after cold storage, observations were made on the mesocarp for mealiness and browning after fruit were cut in halves through the suture plane. Mesocarp tissue was collected from three fruit for each treatment listed above, bulked and immediately frozen in liquid nitrogen before storing at −80°C until it was used for RNA isolation. For microarray experiments, tissues from leaves and roots of the two full-sib progeny were also used.
Synthesis of cDNA and library construction
Two libraries were constructed: one with full-length non-normalized ESTs (PP1), and the other with full-length normalized ESTs (PPN). Total RNA was isolated from 4 g of pooled mesocarp tissue of three fruit in each treatment using the method described by Meisel et al. (2005). For both libraries, 10 μg RNA of M, TR and R, and 30 μg RNA of CS1, CSR-1, CS-2 and CSR-2 were pooled and used for cDNA synthesis. The PPN library was slightly different from PP1 because the former contained additional 60 μg RNA of mealy fruit from the CSR-2 treatment. Purification of poly(A)+ RNA from pools was conducted with Oligotex® mRNA Purification System (Qiagen) following manufacturer instructions. The Clontech’s SMART PCR cDNA Synthesis kit was used to construct the libraries, and primers SMART IV oligonucleotide and CDS III/3′ (SMARTTM cDNA Library Construction Kit Clontech) containing SfiI restriction site were used.
Normalization of the PPN library was carried out using the properties of the enzyme DSN (Duplex-specific nuclease) (Evrogen) as described in Zhulidov et al. (2004) but with modification (M.C. Marques and M.A. Perez-Amador, personal communication) in the primers as indicated below. Briefly, in the DSN normalization method, double stranded cDNA is denatured and subsequently allowed to re-hybridize. Following re-association, the double stranded DNA fraction (formed by most abundant transcripts) is degraded by DSN and the equalized single stranded fraction is amplified by PCR. The primers used for the first and second rounds of amplification of normalized cDNA were M1-5′ (5′-AAGCAGTGGTATCAACGCAGAGT-3′) and M1-3′ (5′-ATTCTAGAGGCCGAGGCGG-3′), and M2-5′ (5′-AAGCAGTGGTATCAACGCAG-3′) and M2-3′ (5′-ATTCTAGAGGCCGAGGCG-3′), respectively.
In both libraries, cDNA was size-selected by agarose gel electrophoresis, and only fragments that were 0.5 to 4 kb long were used. cDNAs were digested with SfiI and cloned into Gateway vector pENTR1A (Invitrogen) modified to contain a SfiI restriction sites in the polylinker (kindly provided by M.C. Marques and M.A. Amador, unpublished).
Ligated products were used to transform One Shot MAX Efficiency DH5α-T1 Competent Cells (Invitrogen) following manufacturer instructions. The selection of transformed colonies was carried out on LB agar plates supplemented with kanamycin. Randomly-selected clones were grown overnight in standard selective bacterial growth media, and plasmids were isolated by alkaline lysis using Perfect-Prep kit (Eppendorf). Sequencing reactions were carried out on plasmids from the 5′-end of the cDNA inserts, using an ABI 3100 capillary automatic sequencer (Applied Biosystems) with fluorescent dye terminator technology.
Sequence analysis and functional annotation
ESTs processing, assembly and annotation was performed by using EST2uni (http://bioinf.comav.upv.es/est2uni), an open parallel software package for automated EST pre-processing, assembly and unigene annotation (Forment et al. 2008). EST2uni analysis pipeline featured the following standard EST analysis tools: Raw sequences and base confidence scores were obtained from raw chromatogram files using the program PHRED (Ewing and Green 1998; Ewing et al. 1998). Low-quality and cloning vector regions were removed from the sequences with LUCY (Chou and Holmes 2001), and ESTs that were left with <100 non-vector good-quality bases after trimming were discarded from further analyses. Repetitive elements and low-complexity regions were masked with RepeatMasker (http://www.repeatmasker.org) and SeqClean (http://compbio.dfci.harvard.edu/tgi/software), respectively. For repeat masking in this study, the eucotyledons-specific repeats database was used. Vector sequence contaminants were also removed with SeqClean, using NCBI’s UniVec database (http://www.ncbi.nlm.nih.gov/VecScreen/UniVec.html). Assembly of reads in contigs and singletons to estimate the redundancy of the ESTs, determine the consensus sequences of the redundant ones, and obtain the unigene set, was made with TGICL (Pertea et al. 2003), using the following default parameters: 30 bases minimum overlap length, 94% minimum percent identity for overlaps, and 30 bases maximum length of unmatched overhangs. Poly(A/T) tails and open reading frames (ORFs) were predicted using ESTScan (Iseli et al. 1999). ESTScan was also used to obtain reverse complimentary sequences of the unigenes when necessary.
Di-, tri-, and tetra-nucleotide simple sequence repeats (SSR) were detected with Sputnik (http://espressosoftware.com/pages/sputnik.jsp). Putative single nucleotide polymorphisms (SNPs) were found by EST2uni using a locally developed algorithm. As ESTs have frequent sequencing errors, only positions with a quality score above 39 were considered, and sequence discrepancies between ESTs in the same contig were marked as putative SNPs only if the polymorphism was confirmed by more than one EST in the contig. Because cDNA libraries were constructed using oligo-dT primer for the reverse transcriptase reaction and ESTs were obtained by 5′-end sequencing, unigenes were aligned with the Arabidopsis complete proteins database to determine if they included the translation initiation codon and predict the presence of full-length cDNA clones for each unigene.
For the functional annotation of ChillPeach unigenes, BLASTx was carried out in EST2uni against: (1) the UniRef90 non-redundant protein clusters database (http://www.ebi.ac.uk/uniref; Suzek et al. 2007; downloaded October 2006: UniProtKB release 8.9 of October 2006), (2) the predicted full set of Arabidopsis thaliana proteins provided by TAIR (http://www.arabidopsis.org; downloaded September 2006: TAIR6 of November 2005), and (3) the predicted full set of Oryza sativa proteins provided by TIGR (http://www.tigr.org; downloaded October 2006: TIGR rice pseudomolecule release 4.0 of January 2006). BLASTn searches were also made in EST2uni against the following sets of public peach DNA sequences: (1) the Prunus persica sequences at GenBank (downloaded from NCBI October 2006), (2) the peach EST collection at ESTreeDB (http://www.itb.cnr.it/estree; downloaded December 2006: Release 5 of September 2006), (3) the Prunus EST collection at GDR (http://www.bioinfo.wsu.edu/gdr/genus/prunus; downloaded December 2006: EST Prunus Unigene version 3 of June 2006), (4) the TIGR transcript assembly database for all public plant ESTs, or PlantTA (Childs et al. 2007; http://plantta.tigr.org; downloaded December 2006), and (5) the TIGR transcript assembly database for all public Prunus persica ESTs (downloaded December 2006: Release 2 from June 2006). All these analyses were performed using BLAST default parameters and an arbitrary non-stringent threshold of 10−5 E-value.
Unigenes were annotated with the description of the most similar UniRef90 cluster of proteins. When no significantly similar UniRef90 cluster was found, unigenes were annotated with the first informative (i.e., not containing words such as “unknown”, “anonymous”, or “hypothetical”) description of the BLAST hits, if any, against the rest of databases analyzed, in the following order: TIGR Plant transcripts, TIGR Peach transcripts, Arabidopsis proteins, Oryza sativa proteins, GenBank Prunus persica DNA sequences, GDR Prunus ESTs, and ESTreeDB. Unigenes were annotated as highly similar to the first BLAST hit when the E-value was lower than 10−15. BLASTX hits with an E-value higher than 10−10 were not considered for annotation. Gene Ontology (GO) annotation of the Arabidopsis most similar protein was used for GO annotation of the peach unigenes.
A bi-directional BLAST comparison was also performed with Arabidopsis and rice sequence databases to obtain a set of putative orthologs. In these analyses, two sequences were considered orthologs when each one was the first hit in a BLAST search with the other.
Microarray slide printing
A microarray was constructed to represent all the unigenes in the ChillPeach database. To reduce spot cross-hybridization within gene families in the microarray experiments (Everstz et al. 2001), a set of unigene clusters or “superunigenes”, grouping different unigenes with extensive sequence overlap (more than 300 bp with more than 90% identity, and covering more than 50% of the length of one of the unigenes), was obtained from the cDNA clone collection using BLAST. One cDNA clone that was the best representative for each superunigene was selected to be printed on the glass slides. The two criteria used in choosing representative cDNA clone were: (1) EST sequence length greater than 300 bp and covering at least 90% of the unigene consensus sequence, and (2) GC content not greater than 80% in a 70 base-long sliding window. Where more than one clone in a superunigene satisfied all the criteria, the one with longest 5′ sequence was selected to ensure that full-length clones were used for slide printing. Where no clone in a superunigene satisfied all the criteria, the criteria were progressively relaxed until a representative clone was selected.
DNA probes for microarrays were obtained by PCR from each of the selected cDNA clones in the cDNA plasmid collection. PCR reactions were carried out in a final volume of 100 μl using 4 ng plasmid DNA, 400 nM of each primer, and 200 μM dNTPs. Primers used were pENTR-forward (5′-GGCTTTAAAGGAACCAATTCAG-3′) and pENTR-reverse (5′-GCAATGCTTTCTTATAATGCCAAC-3′). Amplifications were performed in a 96-well format and PCR conditions consisted of an initial denaturation at 94°C for 5 min, followed by 30 cycles of 94°C for 30 s, 57°C for 30°s and 72ºC for 2 min 30 s, and a final 7 min extension at 72°C. The reaction products were analyzed on agarose gels and PCR products were purified using ExcelaPure 96-Well UF Plate (EdgeBioSystems). Finally, only representative clones with a single product in the PCR reaction were printed in the microarray.
Before printing, purified PCR products corresponding to 4,261 selected cDNA clones were transferred to 384-well plates at a final concentration of 100–200 ng/μl in 5 μl PRONTO™ (Universal spotting buffer, Corning) and reorganized using a robotic liquid dispenser (MultiPROBRE® Iiex Robotic Liquid Handling System, Packard BioScience Company). Lucidea Universal ScoreCard (Amersham) spike controls were diluted in 100 ng/μl PRONTO™ and spotted on the array for quality evaluation. Each of the calibration and negative controls from the Lucidea kit were spotted several times across the whole area of the array. Every selected ChillPeach clone was spotted once. All samples were spotted on Corning UltraGAPS glass slides, using a MicroGrid II spotting device from Biorobotics. After printing, slides were cross-linked at 150 mJ and stored.
Microarray hybridization, scanning and data analyses
For microarray experiments, equal amounts of RNA samples from CS-1, CSR-1, CS-2, and CSR-2 were pooled to form the cold-treated sample (CT), and equal amounts of RNA samples from M, R, and TR were pooled to form the non-cold treated sample (NC). RNA was also isolated from leaves and roots of the two full-sib progeny, pooled and treated as vegetative sample (Vg). A pool of equal amounts of RNA from all mesocarp tissue and vegetative samples was made and treated as a pool reference (PR). To obtain differential gene expression values, three technical replicates from each sample pool—CT, NC, and Vg—were hybridized and CT versus NC and Vg versus NC obtained after each of the log2 ratios of fluorescence values were normalized against those of the PR.
RNA samples for microarray hybridization were amplified using the method of Van Gelder et al. (1990). Briefly, 1 μg of total RNA of each sample (CT, NC, and Vg) and PR was amplified and aminoallyl-labelled using MessageAmp® II aRNA kit (Ambion, http://www.ambion.com) and 5-(3-aminoallyl)-2′-deoxyuridine-5′-triphosphate (aa-dUTP, Ambion), following manufacturer’s instructions. Approximately 40–50 μg of amplified RNA (aRNA) was obtained. For each sample and the PR, 7.5 μg of aminoallyl-labelled aRNA was re-suspended in 0.1 M Na2CO3 (pH 9.0) and labelled with either Cy3 or Cy5 Mono NHS Ester (CyTM Dye Postlabelling Reactive Dye Pack, Amersham). The samples were purified with MegaclearTM (Ambion) following manufacturer instructions. Incorporation of Cy3 and Cy5 was measured using 1 μl of the probe in a Nanodrop spectrophotometer (Nanodrop Technologies Inc.; http://www.nanodrop.com/).
Microarray hybridization of samples and PR to the ChillPeach microarray slides was performed manually using Telechem Hybridization Chambers (Corning), following manufacturer instructions. Briefly, slides were re-hydrated and UV-cross-linked, and then pre-hybridized 45 min at 42°C in 5× SSC, 0.1%SDS, 0.1 mg/ml BSA, 10 mM EDTA pH 8, washed twice for 30 s in milliQ water (Millipore) and in isopropanol for 30 s. Arrays were drained by centrifugation at 528g for 2 min. For each hybridization, 100 pmol of each Cy5-labelled sample was mixed with 100 pmol of Cy3-labelled PR. Fluorescence values were normalized against those of the reference PR by dividing the log2 values obtained at 635/532 nm. Three technical replicates were made, one of them dye-swapped. Mixture was dried in a speed-vac, and re-suspended in 34 μl water, 4 μl EDTA 0.5 M pH 8 and 2 μl polyA (10 μg/μl). This mix was heated for 3 min at 95°C and 40 μl of 2× hybridization buffer (50 μl formamide 50 μl, 25 μl 20× SSC, 2 μl 10%SDS) was added. Hybridization was done overnight at 42°C.
After hybridization, slides were washed in 2× SSC, 0.1% SDS for 5 min at 42°C, 0.1× SSC, 0.1% SDS for 10 min at room temperature, 0.1× SSC for 5 min at room temperature four times, and 0.01× SSC for 5 min at room temperature four times. Arrays were drained by centrifugation at 528g for 2 min. Slides were scanned with a GenePix 4000B scanner (Axon Instruments) at 10 μm resolution, 100% laser power, and different PMT values to adjust the ratio to 1.0. Microarray images were analyzed and globally normalized using GenePix 4.1 (Axon Instruments) software. Only spots with background-subtracted intensity greater than two-fold the mean background intensity in at least one channel were selected for analysis. Data files were imported into Acuity 4.0 (Axon Instruments) and normalized by using the Lowess normalization method. Finally, only spots with valid values in at least five of the six analyzed hybridizations were considered for further analyses. Mean and standard deviation of values were calculated from each sample as log2 values, and later normalized to the median of the expression in NC sample.
To detect differentially expressed genes in cold-treated fruits or in vegetative tissues compared to non-cold treated fruits, data were analyzed with the SAM package (Significance Analysis of Microarray, Tusher et al. 2001). Statistical significance was assessed using two class (unpaired) SAM analysis, with a false discovery rate of 5%, q-value ≤0.05, and two-fold change cut-off. ChillPeach genes up-regulated in cold-treated tissues were compared with ColdArrayDB (http://cold.stanford.edu/cgi-bin/data.cgi)—a database that contains global expression profiles of Arabidopsis genes in response to cold. The search conditions used were: chip type—23 k, experiment—cold treatment, growth medium—plate and soil, expression level >50, and fold change >2.
Real-time qRT-PCR analysis
Thirteen genes were selected from the microarray results for qRT-PCR analysis. Total RNA was isolated and purified from treatments M, CS-1, and CSR-1 of each of the two full sib progeny as described above in two biological replicates.
One microgram of total RNA was used to synthesize first-strand cDNA using the SuperScript first-strand synthesis system for RT-PCR (Invitrogen). cDNA samples were diluted in a total volume of 100 ml. Two microliter of cDNA was used for qRT-PCR using SYBR Green PCR master mix (Applied Biosystems) following manufacturer’s recommendations and an ABI Prism 7000 sequence detection system (Applied Biosystems). Each biological replicate was assayed in triplicate. Gene-specific oligonucleotide primers were designed using Primer Express® version 2.0 software (Applied Biosystems). Primer information is shown in Supplemental Table 1. Expression levels for target genes were calculated relative to Initiation Factor elF-4-Gamma using the DDthreshold cycle (Ct) method (Applied Biosystems). Pairwise comparisons were made on the \( 2^{{ - \Updelta \Updelta C_{T} }} \) values between the two full sib progeny and among the three treatments (M, CS-1 and CSR-1) using Student’s t-test.
Results
ChillPeach EST assembly
Two dedicated cDNA libraries PP1 (non-normalized) and PPN (normalized) were generated from mesocarp of two full-sib peach progeny that contrast for susceptibility to mealiness and browning (major symptoms of CI). All clones are in the Gateway® vector. Sequence information of 8,432 clones randomly selected from both (768 ESTs from PP1 and 7,664 from PPN) is available in the ChillPeach database (Table 1). The nucleotide sequence data have also been submitted to GenBank and assigned the accession numbers FC860626–FC868487. After vector and low quality sequence trimming, 7,868 high quality ESTs were obtained with average sequence length of 640 bases, 7,058 (90%) of which were longer than 500 bases. Following sequence assembly of this EST dataset based on 30 bases minimum overlap length, 94% minimum percent identity for overlaps, and 30 bases maximum length of into 1,480 contigs (4,468 unigenes total), indicating a redundancy of 43% for both unmatched overhangs, 2,988 singletons were identified and the remaining ESTs clustered libraries. The number of ESTs per contig ranged from 2 to 34, while most contigs (83%) contained four or less ESTs.
Using EST2uni, a structured MySQL database was created and populated with information about all the data including cDNA libraries, isolated clones, raw sequences, and results of all bioinformatics analyses performed. The website to access this ChillPeach database is http://bioinfo.ibmcp.upv.es/genomics/ChillPeachDB. The website provides a powerful data mining tool with an advanced querying interface and high integration among all kinds of data.
ChillPeach novelty
When compared with other public databases the ChillPeach EST database showed some uniqueness both at nucleotide (sequence novelty obtained from BLASTn searches) and protein (functional novelty obtained from BLASTx searches) levels (Table 2). BLASTn and BLASTx matches of the ChillPeach unigenes to the NCBI (National Center for Biotechnology Information) Prunus EST collection showed that 2,331 (52%) ChillPeach unigenes did not share significant nucleotide sequence similarity, and 28% were functionally unique. Of the remaining 2,137 with similar sequences in the NCBI Prunus EST collection, at least 31% had either full length or were longer than their NCBI counterparts at the 5′ UTR. Similar searches of Prunus ESTs in the TIGR PlantTA (http://www.plantta.tigr.org), ESTree (http://www.itb.cnr.it/estree), and GDR (http://www.bioinfo.wsu.edu/gdr) databases revealed that 52–72% of ChillPeach unigene sequences were new to these databases. Functional uniqueness to these databases ranged between 26 and 55%. Out of the total 4,468 ChillPeach unigenes, 563 sequences (13%) have no Arabidopsis ortholog and 179 (4%) have no ortholog in any plant database based on results of BLASTx search of all plant transcripts in the PlantTA of TIGR. Analysis of those sequences corresponding to Arabidopsis orthologs indicated that more than 45% of them were likely to be full length.
SSRs and SNPs
Sequence information of the ChillPeach unigene set revealed the existence of 184 perfect di-, tri-, and tetranucleotide microsatellites or SSRs representing 4% of all ChillPeach unigenes (Table 3). Out of these 184 EST SSRs, 77 (42%) are novel to Prunus. The number of observed dinucleotide SSRs was more than twice that of trinucleotides and ten times more than tetranucleotides. All but three ChillPeach dinucleotides were AG repeats, representing 97% of the dinucleotide repeats and 58% of all SSRs. Only two AC repeats and one AT repeat were observed, and no CG repeat was present in the dataset. Among the trinucleotide repeats, AAG repeats were the most abundant (34%) followed by ATC repeats (19%). Analysis of the positions of the SSRs relative to putative initiation (Met) and stop codons in the ChillPeach dataset showed that 109 (59%) fell in the putative 5’UTR region, and 14 (7.6%) were found in the 3’UTR region, while 56 (30%) were located within the coding sequence and the positions of the remaining five repeats could not be determined.
Sequence analysis of the ESTs within each of the 396 contigs containing at least four ESTs showed occurrence of 70 (18%) single nucleotide polymorphisms (SNPs). These putative SNPs were confirmed by at least two EST pairs within the respective contigs. When more than one SNP was found in a unigene, their distribution in the ESTs was found consistent with the existence of different alleles.
Functional annotation
Of the 4,468 ChillPeach unigenes, only 163 could not be annotated with an informative description other than “expressed protein”, “hypothetical protein”, etc., based on their similarity to sequences in external databases (see “Material and methods”). However, a total of 2,851 ChillPeach unigenes (64%) that shared significant similarity with Arabidopsis proteins fell into various Gene Ontology (GO) functional classes other than ‘unknown’, as determined by the GO term assigned to the most similar Arabidopsis protein. Another 1,054 unigenes were similar to Arabidopsis proteins annotated as “biological process unknown”, “molecular function unknown” and/or “cellular component unknown”. Only 13% of the unigenes (563 sequences) did not have a match in Arabidopsis and could not be annotated with GO functional terms. Of the annotated unigenes, 2,165 (76%) were annotated in the Molecular Function ontology, 1,699 (60%) in the Biological Process ontology, and 1,926 (68%) in the Cellular Component ontology (Fig. 1).

Functional distribution of ChillPeach unigenes based on GO functional categorization
Of the 2,148 ChillPeach unigenes with Prunus sequence novelty, 1,855 were assigned to the Molecular Function ontology, 844 to the Biological Processes ontology, and 1,386 to the Cellular Component ontology. Of the 1,103 ChillPeach unigenes with Prunus functional novelty, 868 were annotated in the Molecular Function ontology, 367 in the Biological Process ontology, and 668 in the Cellular Component ontology. Unigenes containing SSRs were also classified, and of the 184 total, 131 fell into the Molecular Function ontology, 68 in the Biological Process ontology, and 109 in the Cellular Component ontology. Novel unigenes and those with SSRs shared similar distributions within the ontology classes with those conducted for the entire ChillPeach unigene set.
Microarray analysis
We demonstrated the usefulness of ChillPeach unigenes in microarray studies by conducting a targeted microarray study comparing cold-treated peach fruit mesocarp tissue to non-cold treated, and by comparing vegetative tissue to mesocarp tissue.
Cold-responsive ChillPeach ESTs
In our study of differential gene expressions in cold-treated peach fruit, 399 genes were identified as differentially expressed (287 up-regulated and 112 down-regulated) in cold-treated mesocarp tissue sample using two criteria: a False Discovery Rate (FDR) and q-values <0.05. Of the 287 up-regulated genes, 48 (16.7%) were novel Prunus genes, 40 (14%) had no Arabidopsis orthologs, and 14 were novel to both Prunus and Arabidopsis. In contrast, only 8 of the 112 down-regulated genes (7%) were novel in Prunus and 7 genes (6%) had no Arabidopsis orthologs.
Of the 287 up-regulated cold-responsive genes, there were 74 with ≥2.0-fold increase, and 27 of these had ≥2.8-fold increase (Table 4). The gene α-L-arabinofuranosidase topped the list of genes with significant expression in cold-treated mesocarp tissue samples with 10.3-fold increase, closely followed by a dehydrin 2 gene (8.5-fold increase), a chitinase gene (6.5-fold increase), a little protein 1 gene (5.9-fold increase) and a protein kinase gene (4-fold increase). Of the 74 genes with ≥2.0-fold increase 28 were novel to Prunus. For genes down-regulated in cold-treated mesocarp tissue, there were nine genes with <0.5-fold expression, two of which were novel to Prunus (Table 4). Five of these were heat shock proteins. Others were CBF1, pectinesterase PPE8B precursor, granule-bound starch synthase 1 (chloroplast precursor), and T17B22.3 protein genes. Of the 74 genes with >2.0 fold change, 59 had Arabidopsis orthologs. When these 59 ChillPeach genes were compared to the expression profile of the ColdArrayDB, 23 of these were also found to be differentially expressed in cold-treated Arabidopsis vegetative tissue (Table 5); 11 genes were up-regulated and 12 were down-regulated. Of the remaining 36 genes, 30 were not responsive to cold in Arabidopsis vegetative tissue (fold-change <2), and six were absent in the expression profile of the ColdArrayDB (Table 6).
Fruit versus vegetative tissue microarray
A total of 950 genes (434 up-regulated and 516 down-regulated) were identified as significantly differentially expressed (FDR and q-values <0.05) in vegetative tissue sample compared with non-cold treated mesocarp tissue. Of the 434 genes that were preferentially expressed in vegetative tissue, 69 (14.9%) were novel Prunus genes, 66 (15.2%) had no Arabidopsis orthologs, and 21 (5%) were novel to both. Of the 515 down-regulated genes, 66 (12.8%) were novel Prunus genes, 52 (10%) had no Arabidopsis orthologs, and 11 were new to both. Among those with higher values in vegetative tissue, 16 had ≥210-fold increase in expression compared to mesocarp tissue (Table 7). Sixty-three genes had 25- to 210-fold, and 260 genes had 4- to 32-fold increase in expression, respectively. Most of these genes were easily identifiable markers of photosynthetic tissues.
A total of 515 genes were significantly over-expressed in mesocarp tissue as compared to the vegetative tissue. Of these, expression of 45 genes was 25 to 210 fold higher in mesocarp than in vegetative tissues and therefore could be good candidates for mesocarp-specific genes. Eleven of the genes with >27 fold increase in mesocarp are shown in Table 7. Of the differentially expressed genes, 322 showed expression ratios of 22 to 25 fold higher in mesocarp than in vegetative tissues.
qRT-PCR analysis
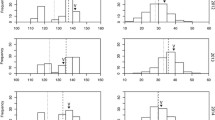
The results of single gene qRT-PCR analysis of all selected 13 genes corroborated the results of microarray analysis (Fig. 2, Table 8). The 13 genes comprised of six genes that were cold-induced in both peach and Arabidopsis (ColdArrayDB) (Table 5), five genes that were up-regulated in peach but not reported in Arabidopsis (Table 6), and two genes that were up-regulated in peach but down regulated in Arabidopsis (Table 5). Both microarray and qRT-PCR analyses showed that all the 13 genes were up-regulated in cold treated mesocarp tissues of both progeny (Fig. 2). The most notable difference in the qRT-PCR analysis was observed for Thau (Thaumatin-like precursor) and Chit (Chitinase) genes where their relative expression in some of the treatments was about 10 times the relative expression of other genes (Fig. 2). Expression of Thau and Chit increased by 1,107-fold and 185-fold, respectively, in CS-1 compared to M of the resistant progeny; and by 167-fold and 60-fold, respectively, in the susceptible progeny (Table 8). In addition, ST1 (sulfate transporter) also had 167-fold change in the resistant progeny. Cold-induced genes with greater fold change in the resistant progeny compared to the susceptible progeny were Thau, Chit, ST1, Ara (α-L-arabinofuranosidase), B-Gal (beta-galactosidase precursor), AGAT (Alanine-glyoxylate aminotransferase 2), ARG2 (Indole-3-acetic acid-induced protein ARG2), CCR (cinnamoyl-CoA reductase) PGDH (D-3-phosphoglycerate dehydrogenase chloroplast precursor) and ZEP (zeaxanthin epoxidase). Two genes had higher expression in the susceptible progeny than the resistant progeny: Ank (ring zink finger ankyrin) and Anth (anthocyanidin-3-O-glucosyltransferase). The fold change of PGIP (polygalacturonase inhibiting protein) was similar for both CI-resistant and susceptible progeny.

Genes chosen for data validation by real time qRT-PCR. Shown are relative levels of differential gene expression among treatments. Genes in group B were charted on a different scale because their expression in some of the treatments was about 10 times the expression of genes in group A. The data represented the mean of two biological and three technical replicates. Gene expression levels were normalized against peach Initiation Factor elF-4-Gamma. The level of each analyzed gene transcript in mature (M) susceptible sample was set to one and the level of the remaining sample was calculated relative to this reference. Error bars show the standard error of the mean for each treatment
The qRT-PCR analysis also showed that the expression of all genes dropped drastically during ripening (CSR-1), although the expression of some genes such as Chit and Thau (in both progeny), Ara and B-Gal (in susceptible progeny only), and PGIP, ST1 and ZEP (in resistant progeny only) was still significantly higher in CSR-1 than in normal fruit (M) (Table 8).
Discussion
ChillPeach database (http://bioinfo.ibmcp.upv.es/genomics/ChillPeachDB) has been created containing a set of functional genomic tools to facilitate speedy detection of genetic factors responsible for CI in peach and nectarine and, possibly, other stone fruits. The ChillPeach EST libraries were developed using two peach genotypes that contrast for CI but have a common genetic background as they were progeny of a single cross (Pop-DG). ‘Dr. Davis’ and ‘Georgia Belle’, parents of the two genotypes, are canning and fresh market peach cultivars, respectively. ‘Dr. Davis’ is resistant to CI while ‘Georgia Belle’ is susceptible. The two progeny were observed for 3 consecutive years for expression of CI and one was consistently resistant and the other consistently susceptible.
A peach genetic linkage map was earlier constructed using the Pop-DG segregating population from which the two genotypes used in this study were chosen (Peace et al. 2005a, 2006; Ogundiwin et al. 2007). Quantitative genetic analyses of CI symptoms have also been conducted and several QTLs have been identified using the Pop-DG map (Peace et al 2005a, 2006; Ogundiwin et al. 2007). This linkage map is syntenic with the Prunus reference T × E linkage map (Aranzana et al. 2003; Dirlewanger et al. 2004). This direct correspondence between the two maps allows studies in Pop-DG to benefit from the bin mapping strategy developed from the T × E population (Howad et al. 2005). The genomic positions of putative CI genes that are identified through microarray analyses involving ChillPeach unigenes can therefore be determined using the Pop-DG map directly or indirectly through T × E bin mapping, and correspondence with positions of CI QTLs will be a strong indication of the involvement of the respective genes in CI.
About half of the EST sequences in the ChillPeach database have not been reported for Prunus in available databases including NCBI, TAIR, PlantTA, ESTree, and GDR. This high novelty was probably due to the normalization of the PPN library. Usually, 10–20 abundant genes (several thousand mRNA copies per cell) account for at least 20% of the cellular mRNA mass, several hundred genes of medium abundance (several hundred mRNA copies per cell) comprise 40–60% of the mRNA mass, and several thousand rare genes (<10 mRNA copies per cell) may account for 20–40% of the mRNA mass (Carninci et al. 2000). Hence, straightforward random sequencing of clones from standard cDNA libraries is inefficient for discovering rare transcripts, owing to the repeated occurrence of intermediately and highly abundant cDNA (Zhulidov et al., 2004). This was the case with the PP1 library. In the PPN library, however, the prevalence of clones representing abundant transcripts was decreased by normalization before sequencing. This significantly increased the efficiency of random sequencing and the discovery of novel Prunus genes.
Therefore the large contribution this limited EST dataset is making to already available databases reflects the paucity of tree fruit functional genomic information and justifies its development. The database should aid the discovery of genes controlling CI that would otherwise be missed if analyses were to rely solely on previously available Prunus ESTs. This database should also be of interest to other tree fruit functional genomics researchers who may be involved in other aspects of fruit physiology. All ChillPeach ESTs are cloned in a pENTR1A GATEway® vector (Invitrogen), and this should facilitate rapid sub-cloning in a range of expression and gene silencing vectors to make assays of gene function much easier.
It is common to observe SSRs in cDNA sequence datasets, and the ChillPeach EST database was not different in this regard. A total of 184 perfect di, tri- and tetranucleotide repeats were observed among the 4,468 unigenes, representing 4% frequency. This frequency is rather low compared to 17% SSR frequency in apple non-redundant EST sequences reported by Newcomb et al. (2006). The inbreeding nature of peach compared to outbreeder apple, and the fact that the apple EST dataset was obtained from extensive tissue types of seven cultivars as opposed to only mesocarp tissue of two full-sib peach genotypes, could explain the low SSR frequency reported here. Despite the total SSR frequency disparity there was good correspondence in many structural components of the SSRs between the two datasets. The dinucleotide repeat group in the ChillPeach SSR dataset contained almost exclusively (97%) AG repeats. AC and AT repeats had 1.8 and 0.9% frequency, respectively, and no occurrence of CG repeat was observed. AG repeats was also reported to dominate the dinucleotide repeats in apple with 88% frequency, AC and AT repeats had 4 and 7.6% frequency, and CG repeats was almost non-existent with 0.05% frequency (Newcomb et al. 2006). Among the trinucleotide repeats, AAG repeats were the most frequent for both ChillPeach and apple. A greater number of the SSRs were found in the 5′ UTR compared to the 3′ UTR, as reported for apple (Newcomb et al. 2006). SSRs are valuable molecular markers and those derived from ESTs such as ChillPeach ESTs may prove to be a good addition to Prunus mapping efforts, giving researchers a plethora of functional markers to choose from. A large proportion (42%) of the ChillPeach EST SSRs is new to Prunus and approximately 30% of the SSRs were located within coding regions.
Another class of molecular marker obtainable from EST datasets is SNP. Only 70 SNPs, confirmed by more than two ESTs per contig, were observed from the redundant ChillPeach ESTs. This number appeared lower than reported for apple where 18,408 SNPs were detected from 126,209 redundant EST sequences (Newcomb et al. 2006). However, only 396 contigs of our 4,468 ChillPeach unigenes (8.9%) had four or more ESTs and were those analyzed for SNP detection. This lower redundancy in ChillPeach dataset (due in part to lower number of ESTs, but mainly a direct consequence of the normalization of the PPN library from which most ESTs were sequenced), could partly explain the apparent lower SNP frequency compared to the much larger apple dataset. In addition, as explained in the case of SSR above, higher heterozygosity of an outbreeder apple, and the multiple tissue and cultivars utilized for data collection may be other contributing factors.
To demonstrate the utility of ChillPeach and to generate a cDNA microarray for gene expression analyses of CI in Prunus, a preliminary microarray experiment was conducted. The microarray experiments successfully discriminated differentially expressed genes between cold-treated and normal mesocarp tissues, and between vegetative and mesocarp tissues. Some of these genes may be involved with CI because mesocarp tissues with CI were represented in the cold-treated samples. Of the 4,261 genes on the ChillPeach microarray slides, at least 516 were significantly up-regulated in mesocarp tissue.
Arabidopsis orthologs of cold-induced ChillPeach genes varied in their response to cold treatment when compared to their ChillPeach counterparts. Some Arabidopsis ChillPeach orthologs were similarly up-regulated in both species while some had opposite response (up-regulated in peach but down-regulated in Arabidopsis). Several Arabidopsis orthologs of cold-induced ChillPeach genes were non-responsive to cold treatment in Arabidopsis vegetative tissue, and a few were not found in the Arabidopsis expression profile. The genes with similar expression between the two species supported the utility of ChillPeach for microarray studies while those that were non-responsive to cold or absent in the Arabidopsis expression profile may be fruit-specific genes because vegetative tissues were used in the Arabidopsis experiments as opposed to the fruit tissue in ChillPeach. This is particularly significant for the functional genomics analyses of CI for which the database is designed. CI occurs as a post-harvest physiological disorder in fruit; therefore, it is essential for the success of CI genetic analyses that the platform to be used in global gene expression should be transcriptionally active in the fruit as in the case of ChillPeach ESTs. About 14% of the differentially expressed genes in cold-treated tissue were new Prunus genes and 38% of the genes with ≥2.0-fold increases in expression were novel to Prunus. This underscores the suitability of ChillPeach dataset for tackling CI problems.
Among the highly expressed genes in cold-treated mesocarp tissue were common stress-induced genes in plants including dehydrin 2, chitinase, RING zinc finger ankyrin protein, ABA-inducible protein, BZIP protein BZ2, chalcone synthase, protein kinase, and others. Other classes of genes such as ripening-related, and sugar and amino acid transport genes were also up-regulated. Whether or not this list includes genes involved in CI control will become clearer as this microarray platform is used specifically for this purpose and efforts to answer this question are underway. The down-regulated genes included heat shock proteins (HSPs). Cold temperatures are known to induce expression of HSPs. The reason for the down-regulation of these genes in our microarray experiment is not clear. The cold-treated fruit used for the analysis included those that were sampled immediately after cold treatment and those that were ripened at room temperature for about 48 h after cold storage. The exposure to room temperature could possibly explain in part why HSPs were down-regulated in this study.
Real-time qRT-PCR analysis confirmed the qualitative and quantitative results of microarray analysis for all 13 genes tested. All genes showed induced expression in cold-treated mesocarp tissue in both analyses. When gene expression results of microarray and qRT-PCR analyses are compared at the quantitative level, all 13 genes had greater fold change in qRT-PCR analysis of CS-1 versus M. However, all but two genes in both progenny had lower fold change in qRT-PCR analysis of CSR-1 versus M. It should be noted that microarray experiment compared a pool of normal mesocarp tissues (no cold treatment) to a pool of mesocarp tissues that included CS-1 and CSR-1 treatments. The expression values observed in the microarray analysis seemed to be a balance between the qRT-PCR values for CS-1 and CSR-1. Therefore, there appears to be a general agreement in the quantitative fold change of the genes considered in both analyses.
The validation of microarray results is a further proof of the usefulness of the ChillPeach platform for transcriptome studies in peach. Furthermore, the qRT-PCR analysis revealed some differences in the expression of tested genes between CI-resistant and susceptible full-sib progeny and between tissues subjected to cold treatment (CS-1) and those that were allowed to ripen after cold treatment (CSR-1). The high transcript accumulation of all the genes in cold-treated fruit was drastically depleted within 2–4 days of subsequent ripening. However, the transcript level of many genes in the ripened cold-treated mesocarp was still significantly higher than the transcript level in normal tissue. The depletion of transcripts during ripening could indicate the involvement of these genes in the ripening/softening process. It is possible that the transcripts have been converted to their respective proteins important in the ripening/softening and other associated processes.
CI symptoms (mealiness, browning, bleeding and loss of flavor) are observed during ripening/softening after cold treatment of most CI susceptible peach cultivars. The observed pattern of transcript accumulation of the qRT-PCR-tested genes could indicate their involvement in CI development. Detailed transcriptome analysis focusing on each of the CI symptoms will be necessary to associate these genes with any specific symptom.
Of the 13 genes validated by qRT-PCR, 10 had significantly higher expression in cold-treated CI-resistant progeny compared to its cold-treated susceptible counterpart. Two genes had opposite reaction (i.e. higher expression in susceptible progeny), and the expression of one gene was similar to both progeny. The 12 genes with differential expression in the two full-sib progeny may be associated with genotype differences including resistance/susceptibility to CI. Further studies are needed to confirm the involvement of these genes in CI development. Such studies should consider separate pools of several resistant and susceptible full sib progeny for each of the CI-symptoms.
References
Aranzana MJ, Pineda A, Cosson P, Dirlewanger E, Ascasibar J, Cipriani G et al (2003) A set of simple-sequence repeat (SSR) markers covering the Prunus genome. Theor Appl Genet 106:819–825
Bruhn CM, Feldman N, Garlitz C, Hardwood J, Ivan E, Marshall M et al (1991) Consumer perceptions of quality: apricots, cantaloupes, peaches, pears, strawberries, and tomatoes. J Food Qual 14:187–195. doi:10.1111/j.1745-4557.1991.tb00060.x
Carninci P, Shibata Y, Hayatsu N, Sugahara Y, Shibata K, Itoh M et al (2000) Normalization and substraction of cap-trapper-selected cDNAs to prepare full-length cDNA libraries for rapid Discovery of new genes. Genome Res 10:1617–1630. doi:10.1101/gr.145100
Childs KL, Hamilton JP, Zhu W, Ly E, Cheung F, Wu H et al (2007) The TIGR plant transcript assemblies database. Nucleic Acids Res 35:D846–D851. doi:10.1093/nar/gkl785
Chou HH, Holmes MH (2001) DNA sequence quality trimming and vector removal. Bioinformatics 17:1093–1104. doi:10.1093/bioinformatics/17.12.1093
Crisosto CH (2006) Short-term approaches to increase peach fruit consumption. Compact Fruit Tree 39:11–14
Dirlewanger E, Graziano E, Joobeur T, Garriga-Caldere F, Cosson P, Howard W et al (2004) Comparative mapping and marker assisted selection in Rosaceae fruit crops. Proc Natl Acad Sci USA 101:9891–9896. doi:10.1073/pnas.0307937101
Consortium ESTree (2005) Development of an oligo-based microarray (μPEACH 1.0) for genomics studies in peach fruit. Acta Hortic 682:263–268
Everstz EM, Au-Young J, Ruvolo MV, Lim AC, Raynolds MA (2001) Hybridization cross-reactivity within homologous gene families on glass cDNA microarrays. Biotechniques 31:1182–1192
Ewing B, Green P (1998) Base-calling of automated sequencer traces using phred. II. Error probabilities. Genome Res 8:186–194
Ewing B, Hillier L, Wendl MC, Green P (1998) Base-calling of automated sequencer traces using phred. I. Accuracy assessment. Genome Res 8:175–185
Folta KM, Staton M, Stewart PJ, Jung S, Bies DH, Jesdurai C et al (2005) Expressed sequence tags (ESTs) and simple sequence repeat (SSR) markers from octoploid strawberry (Fragaria x ananassa). BMC Plant Biol 5:12. doi:10.1186/1471-2229-5-12
Forment J, Gilabert F, Robles A, Conejero V, Nuez F, Blanca JM (2008) EST2uni: an open, parallel tool for automated EST analysis and database creation, with a data mining web interface and microarray expression data integration. BMC Bioinformatics 9:5. doi:10.1186/1471-2105-9-5
Grimplet J, Romieu C, Audergon J-M, Marty I, Albagnac G, Lambert P et al (2005) Transcriptomic study of apricot fruit (Prunus armeniaca) ripening among 13, 006 expressed sequence tags. Physiol Plant 125:281–292. doi:10.1111/j.1399-3054.2005.00563.x
Horn R, Lecouls AC, Callahan A, Dandekar A, Garay L, McCord P et al (2005) Candidate gene database and a transcript map for peach, a model species for fruit trees. Theor Appl Genet 110:1419–1428. doi:10.1007/s00122-005-1968-x
Howad W, Yamamoto T, Dirlewanger E, Testolin R, Cosson P, Cipriani G et al (2005) Mapping with a few plants: using selective mapping for microsatellite saturation of the Prunus reference map. Genetics 171:1305–1309. doi:10.1534/genetics.105.043661
Iseli C, Jongeneel CV, Bucher P (1999) ESTScan: a program for detecting, evaluating, and reconstructing potential coding regions in EST sequences. In: Proceedings of the 7th international conference on intelligent systems for molecular biology. AAAI Press, Menlo Park, pp 138–148
Lurie S, Crisosto CH (2005) Chilling injury in peach and nectarine. Postharvest Biol Technol 37:195–208. doi:10.1016/j.postharvbio.2005.04.012
Meisel L, Fonseca B, González S, Baeza-Yates R, Cambiazo B, Campos R et al (2005) A rapid and efficient method for purifying high quality total RNA from peaches (Prunus persica) for functional genomics analysis. Biol Res 38:83–88
Newcomb RD, Crowhurst RN, Gleave AP, Rikkerink EHA, Allan AC, Beuning LL et al (2006) Analysis of expressed sequence tags from apple. Plant Physiol 141:147–166. doi:10.1104/pp. 105.076208
Ogundiwin EA, Peace CP, Gradziel TM, Dandekar AM, Bliss FA, Crisosto CH (2007) Molecular genetic dissection of chilling injury in peach fruit. Acta Hortic 738:633–638
Ogundiwin EA, Peace CP, Nicolet CM, Rashbrook VK, Gradziel TM, Bliss FA et al (2008) Leucoanthocyanidin dioxygenase gene (PpLDOX): a potential functional marker for cold storage browning in peach. Tree Genet Genomes 4:543–554. doi:10.1007/s11295-007-0130-0
Peace CP, Ahmad R, Gradziel TM, Dandekar AM, Crisosto CH (2005a) The use of molecular genetics to improve peach and nectarine post-storage quality. Acta Hortic 682:403–409
Peace CP, Crisosto CH, Gradziel TM (2005b) Endopolygalacturonase: a candidate gene for Freestone and Melting flesh in peach. Mol Breed 16:21–31. doi:10.1007/s11032-005-0828-3
Peace CP, Crisosto CH, Garner DT, Dandekar AM, Gradziel TM, Bliss FA (2006) Genetic control of internal breakdown in peach. Acta Hortic 713:489–496
Pertea G, Huang X, Liang F, Antonescu V, Sultana R, Karamycheva S et al (2003) TIGR Gene Indices clustering tools (TGICL): a software system for fast clustering of large EST datasets. Bioinformatics 19:651–652. doi:10.1093/bioinformatics/btg034
Suzek BE, Huang H, McGarvey P, Mazumder R, Wu CH (2007) UniRef: comprehensive and non-redundant UniProt reference clusters. Bioinformatics 23:1282–1288. doi:10.1093/bioinformatics/btm098
Tusher VG, Tisbshirani R, Chu G (2001) Significance analysis of microarrays applied to the ionizing radiation response. Proc Natl Acad Sci USA 98:5116–5121. doi:10.1073/pnas.091062498
Van Gelder RN, von Xastrow ME, Yol A, Dement DC, Barchas JD, Eberwine JH (1990) Amplified RNA synthesized from limited quantities of heterogeneous cDNA. Proc Natl Acad Sci USA 87:1663–1667. doi:10.1073/pnas.87.5.1663
Zhulidov PA, Bogdanova EA, Shcheglov AS, Vagner LL, Khaspekov GL, Kozhemyako VB et al (2004) Simple cDNA normalization using Kamchatka crab duplex-specific nuclease. Nucleic Acids Res 32(3):E37. doi:10.1093/nar/gnh031
Acknowledgements
We thank Dr. Abhaya Dandekar for critical review of the manuscript. This research was funded by UC Discovery Grant (bio05-10527) with the Industry-University Cooperative Research Program.
Author information
Authors and Affiliations
Corresponding author
Additional information
Ebenezer A. Ogundiwin and Cristina Martí contributed equally to this publication.
Electronic Supplementary Material
Rights and permissions
About this article
Cite this article
Ogundiwin, E.A., Martí, C., Forment, J. et al. Development of ChillPeach genomic tools and identification of cold-responsive genes in peach fruit. Plant Mol Biol 68, 379–397 (2008). https://doi.org/10.1007/s11103-008-9378-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11103-008-9378-5