
Abstract
Promoters play key roles in conferring temporal, spatial, chemical, developmental, or environmental regulation of gene expression. Promoters that are subject to specific regulations are useful for manipulating foreign gene expression in plant cells, tissues, or organs with desirable patterns and under controlled conditions, and have been important for both basic research and applications in agriculture biotechnology. Recent advances in genomics technologies have greatly facilitated identification and study of promoters in a genome scale with high efficiency. Previously we have generated a large T-DNA tagged rice mutant library (TRIM), in which the T-DNA was designed with a gene/promoter trap system, by placing a promoter-less GUS gene next to the right border of T-DNA. GUS activity screens of this library offer in situ and in planta identifications and analyses of promoter activities in their native configurations in the rice genome. In the present study, we systematically performed GUS activity screens of the rice mutant library for genes/promoters constitutively, differentially, or specifically active in vegetative and reproductive tissues. More than 8,200 lines have been screened, and 11% and 22% of them displayed GUS staining in vegetative tissues and in flowers, respectively. Among the vegetative tissue active promoters, the ratio of leaf active versus root active is about 1.6. Interestingly, all the flower active promoters are anther active, but with varied activities in different flower tissues. To identify tissue specific ABA/stress up-regulated promoters, we compared microarray data of ABA/stress induced genes with those of tissue-specific expression determined by promoter trap GUS staining. Following this approach, we showed that the peroxidase 1 gene promoter was ABA up-regulated by 4 fold within 1 day of exposure to ABA and its expression is lateral root specific. We suggest that this be an easy bioinformatics approach in identifying tissue/cell type specific promoters that are up-regulated by hormones or other factors.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Promoters play key roles in conferring temporal, spatial, chemical, developmental, or environmental regulation of gene expression. They usually contain essential cis-acting elements that interact with transcription factors for regulation of transcription upon perception of upstream signals. Promoters could be constitutively active or become active only upon induction, and may have differential strength under different conditions. Promoters that are subject to specific regulations are useful for manipulating foreign gene expression in plant cells, tissues, or organs with desirable patterns and under controlled conditions, and have been important for both basic research as well as applications in agriculture biotechnology. Recent advances in genomics technologies have greatly facilitated identification and study of promoters in a genome scale with high efficiency. In this review, by using rice as a working model, we will discuss how promoters could be identified with high throughput genomics approaches based on T-DNA insertional mutagenesis of the rice genome and DNA microarray analyses of the rice gene expression profile.
The T-DNA tagged rice mutant library (TRIM) is designed for gene/promoter trapping
Rice (Oryza sativa L.) is one of the most important crops in the world and is the principle food of nearly 50% of the world’s population. Rice shares a high degree of synteny with other cereals and has served as an excellent model cereal crop for genomics research due to its relatively small genome size and ease of transformation. Recent release of the completed rice (japonica cv. Nipponbare) genome sequence unraveled a wealth of approximately 37,500 non-transposable element related genes, of which majority have no assigned functions and close to 30% have no known homologues in Arabidopsis (IRGSP 2005). In this context, the establishment of high-throughput methods for investigating gene functions is of prime importance. T-DNA insertional mutagenesis across the rice genome is one of the most straightforward approaches for assigning a function to a particular sequence and to identify the gene that causes a particular phenotype (Jeong et al. 2002, 2006; Sallaud et al. 2004). Recently, by using T-DNA with functions of gene/promoter trap, gene knockout and gene activation tagging, we have generated a rice mutant library containing 55,000 lines (Hsing et al. 2007). Close to 20,000 flanking sequence tags (FSTs) have been resolved and assigned to the rice genome, and a searchable FST database is available at the Taiwan Rice Insertional Mutants (TRIM) website (http://www.trim.sinica.edu.tw). These resources offer highly valuable tools for high throughput rice functional analyses on a genome scale using both forward and reverse genetic approaches.
Conventional approaches with attempts to identify functions of promoters rely mainly on isolation of individual promoters, fusion of promoters to reporter genes, and analyses of promoter activities through transient or transgenic expression assays. These approaches are time-consuming and labor-intensive, particularly the transgenic approach imposes difficulty for high-throughput promoter analysis. Additionally, the copy numbers (dose effect) and insertion sites (positional effect) of transgenes in the genome of transgenic plants make the precise characterization of promoters complicated. The development of T-DNA insertional mutagenesis offers an in situ and in planta approach for identification and analysis of promoter activity in their native configuration in the genome.
In the T-DNA insertional mutagenesis approach, the T-DNA used for tagging the rice genome was designed with a gene/promoter trap system, by placing a promoter-less reporter gene (GUS) next to the right border of T-DNA. Expression of GUS occurs only when the T-DNA is inserted within a transcriptional unit of the rice genome and in correct orientation. Multiple splice donor and acceptor sequences are placed in front of the GUS gene, with splicing aligned in all reading frames, which allows fusion of the upstream exon to the GUS gene regardless of insertion position is at exon or intron of the tagged gene. GUS expression with this gene/promoter trap system provides a direct information on the expression pattern of the tagged gene. This approach has been successfully applied for the identification of various promoters from several T-DNA tagged rice mutant libraries (Jeon et al. 2000; Jeong et al. 2002; Jung et al. 2003; Lee et al. 2004; Hsing et al. 2007). However, it should be noted that GUS is a very stable enzyme, thus down regulation of gene expression is unlikely detected with this approach. For the same reason, gene regulation at the level of protein stability, such as those mediated by ubiquitin-related processes, may not be reflected by GUS staining. Also, since some T-DNA insertion mutants contain more than one inserted T-DNA (Hsing et al. 2007), positive GUS staining could be attributed to any of these T-DNAs, thus genetic segregation analysis is needed to ascertain the precise rice flanking sequence involved. After potential rice promoter sequences are revealed, recapitulation experiments should be carried out by transforming rice with these sequences linked to GUS (or other reporter gene coding region) to determine whether these sequences are indeed sufficient for promoter functions.
Two binary vectors, pTag4 and pTag8, both contain the promoter-less GUS gene next to the right borders of T-DNA, were used for generating the TRIM library. pTag8 is different form pTag4 by containing eight copies of the CaMV35S enhancers next to the left border that could function in either orientation and at a considerable distance for transcriptional activation of a nearby gene (Hsing et al. 2007). One advantage of the gene/promoter trap system through the GUS activity screen is that genes or promoters could be identified in heterozygotes due to its dominant gain-of-function nature. Another advantage of this system is that the GUS staining could be easily performed at high throughput in test tubes for samples collected in the laboratory and field. Based on this approach, many promoters active in various organs and tissues, e.g., flowers, seeds, leaves, and roots, of transgenic rice have been identified (some examples shown in Fig. 1).

Diagrams highlighting tissue specific GUS staining in T-DNA insertion transgenic rice plants. Panels on the left half indicate the basic rationale and vegetative tissues used in these experiments. Panels on the right half show results from flowers, seeds, leaves, and roots. Cross sections of leaves and roots are also presented
Systematic GUS activity screens of the TRIM library for genes/promoters active in vegetative tissues
Previously, GUS activity screens of the TRIM library identified many putative stress-responsive and spatially and temporally regulated genes at high frequency. In a GUS activity screen of 1,500 transgenic lines, from a TRIM sub-population tagged by pTag4, for genes/promoters responsive to stresses, 4.1% of lines were GUS positive, with 3.1% responsive (up- or down-regulated) to osmotic (sorbitol), 2.4% to salt (NaCl), 1.3% to low (4°C) and 1.1% to high (45°C) temperature stresses in leaves and roots (Hsing et al. 2007). In that study, T-DNA appeared to tag more frequently on genes responsive to osmotic and salt stresses than to temperature stresses. It is not clear whether the higher numbers of putatively tagged genes reflect the numbers of genes involved in osmotic and salt stresses in comparison to temperature stresses, or whether the tissue culture and transformation process facilitated preferential integration of T-DNA into osmotic and salt responsive genes.
In another GUS activity screen of 2,023 transgenic lines, from a TRIM sub-population tagged by pTag8, for genes/promoters expressed in vegetative tissues under normal growth conditions, 15.4% of lines were GUS positive, with 8.5% in leaves, 0.7% in roots, and 6.2% in both leaves and roots, and a 2-fold higher frequency of T-DNA insertion into leaf active genes than into root active genes (Hsing et al. 2007). Recently, we extended the GUS activity screens of the TRIM mutant library tagged by pTag8 for genes/promoters expressed in leaves and roots. Among 7,338 lines screened, 814 lines (11.2%) showed GUS positive, with 4.4% in leaves, 0.4% in roots, and 6.3% in both leaves and roots, and a 1.6-fold higher frequency of T-DNA insertion into leaf active genes than into root active genes (Table 1). The higher frequency of T-DNA insertion into leaf active genes than into root active genes could be due to higher number of genes expressed in leaves than in roots. However, DNA microarray analyses using the same age of rice seedlings (2-week-old) indicated that there are only 20% more genes expressed in leaves than in roots (C-Y Hong and T-H D Ho unpublished results), which would not account for the 1.6–2-fold increase in the frequency of T-DNA insertion into leaf active genes. Another explanation could be that genes expressed in leaves are more accessible to T-DNA insertion during the transformation process.
GUS activity screens of transgenic rice seedlings, for genes/promoters expressed in leaves and roots, indicated that gene trap with pTag8 resulted in a 3-fold higher frequency than with pTag4, very likely due to the presence of the multimerized CaMV35S enhancers in pTag8 (Hsing et al. 2007). A 2-fold higher frequency of gene trap by GUS activity screening of transgenic rice seedlings with a T-DNA containing four copies of the CaMV35S enhancer has also been reported (Jeong et al. 2002). One important observation is that the CaMV35S enhancers enhance endogenous gene expression without altering expression patterns in most T-DNA tagged transgenic rice (Jeong et al. 2002, 2006).
Systematic GUS activity screens of the TRIM library for promoters active in reproductive tissues
The flower initiates the sexual reproductive cycle in all flowering plants. Essential steps of sexual reproduction, meiosis, and fertilization, take place in the flower, leading to fruit and seed production that are of great importance in the life cycle of plant and for food production. Rice breeding programs that combine modern biotechnology and conventional breeding practices for manipulation of flower reproduction could play a pivotal role for maximal food production. Such advanced rice breeding programs could be applied in improving important agronomic traits, for example, those control flowering time, floral organogenesis, male or female sterility, and fertilization that are important in hybrid seed production and yield of rice. Additionally, flowers are one of the most sensitive organs in plants to environmental stresses, therefore, adaptations of flowers to environmental stresses could significantly increase crop yield. In rice, low temperature (<20°C) causes tapetal cells to swell, which leads to nutrient starvation and subsequent death of pollen mother cells (Imin et al. 2004). Dehydration also exhibits an adverse effect on the development of anther and pollen grains, which causes the greatest reduction in grain yield (Cruz and O′Toole 1984; Boojung and Fukai 1996).
Identification of genes specifically expressed at different developmental stages and in specific tissues of flowers is one of the most important steps to understand functions of these genes in the sexual reproduction of rice. Isolation of promoters of these genes not only provides essential tools for studying how rice sexual reproduction is regulated, but also is useful for controlling expression of transgenes at the right time in the right place during sexual reproduction of rice. Despite of the importance of these subjects, studies of genes/promoters involved in the sexual reproduction in rice and other cereals have been scarce.
Recently, we performed GUS activity screens of the TRIM mutant library for genes/promoters expressed during the sexual reproductive stage of rice. In this effort, rice leaves and flowers (spikelets) on the day of anthesis were collected from field and immediately stained for GUS activity. As shown in Table 2, GUS activity screens of two T-DNA tagged populations, each containing 666 and 646 transgenic lines, revealed that 19.7% and 24.6% of lines were GUS positive in both leaves and flowers, indicating a high efficiency of the gene/promoter trap system. The frequencies of GUS positive in flowers (18.8% and 24.1%) were significantly higher than in leaves (6.6% and 9.1%). Surprisingly, all lines showed GUS positive in flowers were also GUS positive in anthers. The frequencies of anther specific GUS expression in two populations were 10.5% and 10.8%. Frequency of GUS positive in other tissues of flowers was much lower, ranging from 0.8% to 7.4% in two populations. However, GUS activity in roots of these two populations has not been performed yet. Considering that there is a 2-fold higher frequency of T-DNA tagging in leaf-active genes than root-active genes (Hsing et al. 2007), the frequency of flower specific GUS expression could be slightly lower than those shown in Table 2.
For a total of 186 transgenic lines showing flower specific GUS expression, the frequencies of GUS expression in various tissues were also determined. As shown in Table 3, GUS expression was detected in anthers of all lines. The frequency was highest for GUS expression in both anther and stigma and next in anther, stigma, ovary, and lodicule.
For two populations of transgenic lines showing anther specific GUS expression (Table 2), apparent differential promoter strength was observed based on the intensity of GUS staining. As shown in Table 4, frequency was highest for lines showing moderately GUS staining, and lowest for lines showing high GUS staining.
Combining microarray analysis with GUS promoter trap system for identifications of tissue-specific ABA/stress up-regulated promoters
We have been interested in searching for tissue specific ABA/stress up-regulated promoters for the obvious reason that they could be used to drive the expression of transgenes in appropriate tissues only when plants are under stress. Microarray analysis has now been routinely used to study gene expression profile for the whole genome. Microarray databases of various stress and hormone (including ABA) regulated gene expression in Arabidopsis and rice are available (Kikuchi et al. 2003). With the completion of the rice genomic sequencing project and availability of extensive full-length cDNA databases, sequences on rice microarray chips can be used to cover the activity of most genes in the rice genome. However, it is not easy to reveal tissue specific gene expression profiles using microarrays unless RNA samples from dissected tissues are used, which could not easily be carried out due to difficulty of collecting sufficient quantities of certain tissues/cell types (Lee et al. 2005; Nawy et al. 2005). In order to identify tissue specific ABA/stress up-regulated genes in rice, we have dissected ABA-treated rice plants into shoots and roots before RNA samples for microarray analysis were isolated. There are at least 1,500 genes substantially induced by ABA in rice roots with 47 of them up-regulated by more than 10 fold (Fig. 2). Interestingly, the ABA regulated gene expression profile in shoots was substantially different from that in roots, with roots having three times ABA up-regulated genes as in shoots (Table 5). On the other hands, shoots have more ABA down-regulated genes than in roots (Table 5). It should be noted that this is in contrast to the GUS promoter-trap data that show almost twice as many leaf promoters as root promoters (Table 1). Two potential mechanisms could account for these differences. First, the tissue specificity of ABA regulated genes (Table 5) is different from those of constitutively expressed genes (Table 1). Second, the CaMV35S enhancers used in the pTag8 construct may have a stronger effect on the constitutive expression of leaf specific genes.

Distribution of ABA up-regulated genes in rice roots as determined by microarray analysis. About 3-week-old TNG67 rice plants were treated with or without 20 μM ABA for 1 day before shoot and root tissues were harvested. Poly(A)-containing RNA was isolated from these samples, and cDNA was prepared using the poly(A)-RNA as template. The cDNA sample from plants treated without ABA were labeled with Cy3 and that from ABA treated sample labeled with Cy5. After mixing, the cDNAs were allowed to hybridize with Agilent 22K rice chips synthesized based on the full-length cDNA database compiled by Kikuchi et al (2003)
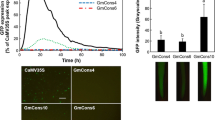
Although it is relatively easy to identify root specific ABA up-regulated genes based on microarray analysis, further dissection of roots into various parts/tissue types, such as main root, lateral roots, elongation zone, root meristem, quiescent center, root caps, etc., requires a considerable amount of delicate manipulations (Lee et al. 2005; Nawy et al. 2005). As described earlier in this work, the promoter-trap system based on random insertions of T-DNA carrying a promoter-less GUS reporter gene is a useful approach in identifying tissue/cell type specific promoters. More than 7,000 rice lines, among the 60,000 T-DNA insertion lines available in the TRIM library, have been screened for GUS expression. The microarray database and the GUS/promoter trap/GUS expression database can be cross-referenced by performing a Blast search of the ABA up-regulated sequences from the microarray analysis against the TRIM database to match up with T-DNA lines positive with GUS staining. Among the 491 lines showing GUS staining in roots (Table 1), we have identified two genes whose expression is also up-regulated by ABA in roots as revealed by the microarray analysis. One of them encodes a hypothetical protein and the other encodes peroxidase I, and the latter were chosen for further studies. This peroxidase 1 gene (AK060007 on the Agilent 22k rice chip) is up-regulated by ABA in roots by more than 2 fold within 0.5 day of treatment (Fig. 3a and b). The ABA up-regulation reached almost 4 fold after 1 day, and persisted to at least 3 days of ABA treatment (Fig. 3b). However, ABA had no effect on the expression of this gene in shoots (Fig. 3b), suggesting that this is a root specific ABA up-regulated gene. GUS staining of T-DNA insertion line, M27568, which has a promoter-less GUS inserted in the 5’UTR of the peroxidase 1 gene, revealed that only lateral roots, but not the primary root, were stained with GUS activity, indicating that peroxidase 1 gene promoter is highly lateral root specific. Taken together, we were able to deduce that the expression of rice peroxidase 1 gene is lateral root specific and ABA up-regulated. This finding would not have been possible if one had relied on either microarray analysis or promoter trap system alone. Tissue specific gene expression profiling using microarray is limited by the isolation of minute amounts of RNA from specific tissues, which is technically challenging. Furthermore, a 2–3 fold up-regulation by ABA would not have been easily quantified by GUS staining. Therefore, we suggest that cross-referencing between microarray data and promoter-trap/GUS staining observations as demonstrated in this work is a simple approach in the identification of tissue-specific ABA/stress up-regulated promoters. Since we picked up two tissue specific ABA/stress up-regulated genes/promoters from a GUS staining population of only 491 lines (from a total of 7,338 lines so far screened for vegetative tissue expression), this indicates that about 20 lateral root specific ABA up-regulated promoters could be obtained once the whole population of T-DNA mutant population in TRIM (more than 55,000 lines) has been screened for tissue specific GUS expression. It is conceivable that this approach could be adopted for the isolation of tissue/cell type-specific promoters up-regulated by other factors. This approach represents a relatively easy link between two databases, one generated by the semi-quantitative microarray gene expression profiling analysis and the other by the tissue/cell type specific promoter trap system, in revealing additional information regarding special properties of promoters without going through the tedious micro-dissection of specific tissues/cell types.

Identification of an ABA up-regulated lateral root specific rice gene promoter by combinatorial analysis of microarray gene expression profiling and T-DNA insertion promoter-trapping. The list of rice root specific ABA up-regulated genes (∼350 entries was compared with the list of promoter-trapping GUS positive lines (∼450 entries), and two genes were found identical, i.e., both up-regulated by ABA and their promoters capable of support the expression of promoter-less GUS in the T-DNA. One of them was chosen for more detailed analysis shown in this figures. (A) Part of microarray chip with AK060007, encoding peroxidase 1, highlighted with circle. (B) Time course of root-specific ABA up-regulation of peroxidase 1 gene (AK060007). Experimental conditions were similar to that described in Fig 2, except rice plants were treated with or without ABA. (C) GUS staining of T-DNA insertion line M27568
References
Boojung H, Fukai S (1996) Effects of soil water deficit at different growth stage on rice growth and yield under upland conditions. 2. Phenology, biomass production and yield. Field Crop Res 48:47–55
Cruz R, O′Toole J (1984) Dry land rice response to an irrigation gradient at flowering stage. Agron J 76:178–183
Hsing YI, Chern CG, Fan MJ, Lu PC, Chen KT, Lo SF, Sun PK, Ho SL, Lee KW, Wang YC, Huang WL, Ko SS, Chen S, Chen JL, Chung CI, Lin YC, Hour AL, Wang YW, Chang YC, Tsai MW, Lin YS, Chen YC, Yen HM, Li CP, Wey CK, Tseng CS, Lai MH, Huang SC, Chen LJ, Yu SM (2007) A rice gene activation/knockout mutant resource for high throughput functional genomics. Plant Mol Biol 63:351–364
Imin N, Kerim T, Rolfe BG, Weinman JJ (2004) Effect of early cold stress on the maturation of rice anthers. Proteomics 4:1873–1882
IRGSP (2005) The map-based sequence of the rice genome. Nature 436:793–800
Jeon JS, Lee S, Jung KH, Jun SH, Jeong DH, Lee J, Kim C, Jang S, Yang K, Nam J, An K, Han MJ, Sung RJ, Choi HS, Yu JH, Choi JH, Cho SY, Cha SS, Kim SI, An G (2000) T-DNA insertional mutagenesis for functional genomics in rice. Plant J 22:561–570
Jeong DH, An S, Kang HG, Moon S, Han JJ, Park S, Lee HS, An K, An G (2002) T-DNA insertional mutagenesis for activation tagging in rice. Plant Physiol 130:1636–1644
Jeong DH, An S, Park S, Kang HG, Park GG, Kim SR, Sim J, Kim YO, Kim MK, Kim SR, Kim J, Shin M, Jung M, An G (2006) Generation of a flanking sequence-tag database for activation-tagging lines in japonica rice. Plant J 45:123–132
Jung K-H, Hur J, Ryu C-H, Choi Y, Chung Y-Y, Miyao A, Hirochika H, An G (2003) Characterization of a rice chlorophyll-deficient mutant using the T-DNA gene-trap system. Plant Cell Physiol 44:463–472
Kikuchi S, Satoh K, Nagata T, Kawagashira N, Doi K, Kishimoto N, Yazaki J, Ishikawa M, Yamada H, Ooka H, Hotta I, Kojima K, Namiki T, Ohneda E, Yahagi W, Suzuki K, Li CJ, Ohtsuki K, Shishiki T, Otomo Y, Murakami K, Iida Y, Sugano S, Fujimura T, Suzuki Y, Tsunoda Y, Kurosaki T, Kodama T, Masuda H, Kobayashi M, Xie Q, Lu M, Narikawa R, Sugiyama A, Mizuno K, Yokomizo S, Niikura J, Ikeda R, Ishibiki J, Kawamata M, Yoshimura A, Miura J, Kusumegi T, Oka M, Ryu R, Ueda M, Matsubara K, Kawai J, Carninci P, Adachi J, Aizawa K, Arakawa T, Fukuda S, Hara A, Hashizume W, Hayatsu N, Imotani K, Ishii Y, Itoh M, Kagawa I, Kondo S, Konno H, Miyazaki A, Osato N, Ota Y, Saito R, Sasaki D, Sato K, Shibata K, Shinagawa A, Shiraki T, Yoshino M, Hayashizaki Y, Yasunishi A (2003) Collection, mapping, and annotation of over 28,000 cDNA clones from japonica rice. Science 301:376–379
Lee SC, Kim JY, Kim SH, Kim SJ, Lee K, Han SK, Choi HS, Jeong DH, An G, Kim SR (2004) Trapping and characterization of cold-responsive genes from T-DNA tagging lines in rice. Plant Sci 166:69–79
Lee JY, Levesque M, Benfey PN (2005) High-throughput RNA isolation technologies. New tools for high-resolution gene expression profiling in plant systems. Plant Physiol 138:585–590
Nawy T, Lee JY, Colinas J, Wang JY, Thongrod SC, Malamy JE, Birnbaum K, Benfey PN (2005) Transcriptional profile of the Arabidopsis root quiescent center. Plant Cell 17:1908–1925
Sallaud C, Gay C, Larmande P, Bes M, Piffanelli P, Piegu B, Droc G, Regad F, Bourgeois E, Meynard D, Perin C, Sabau X, Ghesquiere A, Glaszmann JC, Delseny M, Guiderdoni E (2004) High throughput T-DNA insertion mutagenesis in rice: a first step towards in silico reverse genetics. Plant J 39:450–464
Author information
Authors and Affiliations
Corresponding author
Additional information
Su-May Yu and Swee-Suak Ko equally contributed to this work.
Rights and permissions
About this article
Cite this article
Yu, SM., Ko, SS., Hong, CY. et al. Global functional analyses of rice promoters by genomics approaches. Plant Mol Biol 65, 417–425 (2007). https://doi.org/10.1007/s11103-007-9232-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11103-007-9232-1