
Abstract
Diterpenoid phytoalexins such as momilactones and phytocassanes are produced via geranylgeranyl diphosphate in suspension-cultured rice cells after treatment with a chitin elicitor. We have previously shown that the production of diterpene hydrocarbons leading to phytoalexins and the expression of related biosynthetic genes are activated in suspension-cultured rice cells upon elicitor treatment. To better understand the elicitor-induced activation of phytoalexin biosynthesis, we conducted microarray analysis using suspension-cultured rice cells collected at various times after treatment with chitin elicitor. Hierarchical cluster analysis revealed two types of early-induced expression (EIE-1, EIE-2) nodes and a late-induced expression (LIE) node that includes genes involved in phytoalexins biosynthesis. The LIE node contains genes that may be responsible for the methylerythritol phosphate (MEP) pathway, a plastidic biosynthetic pathway for isopentenyl diphosphate, an early precursor of phytoalexins. The elicitor-induced expression of these putative MEP pathway genes was confirmed by quantitative reverse-transcription PCR. 1-Deoxy-d-xylulose 5-phosphate synthase (DXS), 1-deoxy-d-xylulose 5-phosphate reductoisomerase (DXR), and 4-(cytidine 5′-diphospho)-2-C-methyl-d-erythritol synthase (CMS), which catalyze the first three committed steps in the MEP pathway, were further shown to have enzymatic activities that complement the growth of E. coli mutants disrupted in the corresponding genes. Application of ketoclomazone and fosmidomycin, inhibitors of DXS and DXR, respectively, repressed the accumulation of diterpene-type phytoalexins in suspension cells treated with chitin elicitor. These results suggest that activation of the MEP pathway is required to supply sufficient terpenoid precursors for the production of phytoalexins in infected rice plants.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Upon pathogen attack, plants activate a complex series of responses leading to the local and systemic induction of broad-spectrum antimicrobial defenses. The activation of defense responses is associated with the increased expression of a large number of genes involved in rapid and localized cell death, the production of pathogenesis-related proteins, and the accumulation of antimicrobial compounds called phytoalexins (Stoessl 1980; Subba and Strange 1994; VanEtten et al. 1994). Of the 15 phytoalexins that have been isolated from rice, 14 are polycyclic diterpenoid phytoalexins and the other is a flavonoid-type phytoalexin named sakuranetin. The diterpenoid phytoalexins have been classified into four groups according to their carbon skeletons: phytocassanes A–E (Koga et al. 1995, 1997; Yajima and Mori 2000), oryzalexins A–F (Akatsuka et al. 1985; Kato et al. 1993, 1994), momilactones A and B (Kato et al. 1973; Cartwright et al. 1981), and oryzalexin S (Tamogami et al. 1993). The antimicrobial activity of phytoalexins has been evaluated in a spore germination experiment using the blast fungus Magnaporthe grisea (Peters 2006).
Proposed biosynthetic pathways of the above phytoalexins are shown in Fig. 1. The biosynthetic pathways for diterpenoid phytoalexins have been well established from the cyclization steps from geranylgeranyl diphosphate (GGDP) to the diterpene hydrocarbons ent-cassa-12,15-diene, ent-sandaracopimaradiene, 9βH-pimara-7,15-diene, and stemar-13-ene, precursors of the four types of diterpenoid phytoalexins. Six diterpene cyclase genes (OsCyc1, OsCyc2, OsDTC1, OsDTC2, OsKS4, and OsKS10) have been identified and shown to cause the two successive cyclizations of GGDP into the above diterpene hydrocarbons (Cho et al. 2004; Nemoto et al. 2004; Otomo et al. 2004a, b). However, the genes responsible for the biosynthetic steps after the diterpene hydrocarbons have not been identified, although the participation of cytochrome P450 monooxygenases and a dehydrogenase in these steps has been suggested (Kato et al. 1995; Atawong et al. 2002; Peters 2006) (Fig. 1).

Diterpenoid biosynthesis pathways in rice. The MEP pathway leading to geranylgeranyl diphosphate biosynthesis and the proposed diterpenoid phytoalexin pathway. G3P, glyceraldehyde-3-phosphate; DXP, 1-deoxy-d-xylulose 5-phosphate; MEP, 2-C-methyl-d-erythritol 4-phosphate; CDP-ME, 4-(cytidine 5′-diphospho)-2-C-methyl-d-erythritol; CDP-ME2P, 2-phospho-4-(cytidine 5′-diphospho)-2-C-methyl-d-erythritol; MECDP, 2-C-methyl-d-erythritol 2,4-cyclodiphosphate; HMBDP, 1-hydroxy-2-methyl-2-(E)-butenyl 4-diphosphate; IPP, isopentenyl diphosphate; DMAPP, dimethylallyl diphosphate; GGDP, geranylgeranyl diphosphate; and CDP, copalyl diphosphate. Enzymes in the MEP pathway and six diterpene cyclases are indicated in bold: DXS, DXP synthase; DXR, DXP reductoisomerase; CMS, CDP-ME synthase; CMK, CDP-ME kinase; MCS, MECDP synthase; HDS, HMBDP synthase; HDR, HMBDP reductase; IPI, IPP isomerase; GGPS, GGDP synthase; OsCyc1, syn-CDP synthase; OsCyc2, ent-CDP synthase; OsDTC1, ent-cassa-12,15-diene synthase; OsDTC2, stemar-13-ene synthase; OsKS4, 9βH-pimara-7,15-diene synthase; and OsKS10, ent-sandaracopimaradiene synthase. The steps specifically inhibited by 5-ketoclomazone (KCZ) and fosmidomycin (FOS) are indicated
In contrast, the biosynthetic steps before GGDP are understood as early terpenoid biosynthesis in plants. This process includes the steps for supplying the basic C5 precursors isopentenyl diphosphate (IPP) and dimethylallyl diphosphate (DMAPP), as well as the step that produces GGDP by the action of GGDP synthase on IPP and DMAPP (Scolnik and Bartley 1994; Okada et al. 2000). In plants, two distinct pathways produce IPP and DMAPP: the mevalonate (MVA) pathway and the methylerythritol phosphate (MEP) pathway (Lichtenthaler et al. 1997). Various isoprenoids, including sesquiterpenes, triterpenes, sterols, brassinosteroids, and prenylquinones, are synthesized in the cytoplasm from IPP derived from the MVA pathway (Heintz et al. 1972; Nes and Venkatramesh 1999; Lange and Ghassemian 2003). Many diterpenoids, including gibberellins, abscisic acids, carotenoids, and phytol chains of chlorophylls, are separately synthesized in plastids from IPP and DMAPP, both derived from the MEP pathway (Lichtenthaler 1999; Rohmer 1999). Like the enzymes responsible for the MEP pathway, the diterpene cyclases involved in phytoalexins biosynthesis are thought to be localized in plastids (Cho et al. 2004; Nemoto et al. 2004). Thus, diterpenoid phytoalexins are predicted to be produced through the MEP pathway in plastids. However, there has been no report of the identification of the biological roles of genes that are thought to function in the MEP pathway in rice.
Large amounts of diterpenoid phytoalexins accumulate in the culture medium of elicitor-treated rice cells. This phytoalexins accumulation occurs because an adequate supply of substrate is present, resulting from the activation of an unknown gene together with those of known diterpene cyclases. To better understand inducible phytoalexins biosynthesis in rice, we used oligomicroarrays to survey the genes whose transcription levels change following elicitor treatment of suspension-cultured rice cells. The microarray analysis revealed that putative MEP-pathway genes exhibit elicitor-induced expression with profiles similar to those of diterpene cyclase genes. Molecular and chemical approaches showed that OsDXS, OsDXR, and OsCMS are functional enzymes that catalyze the first three steps in the MEP pathway, and that the application of ketoclomazone and fosmidomycin, inhibitors of DXS and DXR, respectively, repress the accumulation of diterpene-type phytoalexins in suspension-cultured cells induced by elicitor. These results suggest that activation of the MEP pathway is required for the production of sufficient amounts of phytoalexins to supply terpenoid precursors in plastids.
Materials and methods
Plant materials and chemical treatment
Suspension-cultured rice cells (Oryza sativa L. cv. Nipponbare) were maintained as described by Cho et al. (2004). Six days after subculturing, the cells were subjected to treatment with the elicitor N-acetylchitooctaose at 1 ppm, and the DXS inhibitor 5-ketoclomazone at 50 μM, the DXR inhibitor fosmidomycin at 100 μM, or the HMG-CoA reductase inhibitor mevastatin at 5 μM (Sauret-Gueto et al. 2006). A small amount of callus was continually harvested at various time points after elicitor treatment for the extraction of total RNA. For phytoalexins measurements, 0.5-ml samples of culture medium were collected from each sample. Leaf discs (6 mm diameter) were punched from latest leaf of 6 weeks after seedling. Discs were treated with JA (500 μM), UV-irradiation (254 nm, 20-cm distance for 20 min), or CuCl2 (500 μM) and incubated for 72 h under light and humid condition at 25°C. H2O treatment was used as a negative control. After incubation, phytoalexins or total RNA were extracted.
RNA preparation and fluorescent labeling of probes
Total RNA was extracted from elicitor-treated rice cells using Sepasol-RNA I Super (Nacalai Tesque) and purified using an RNeasy Plant Mini Kit (Qiagen K.K., Tokyo, Japan) following the manufacturer’s instructions. The total RNA was used as a template to synthesize Cy-3- or Cy-5-conjugated deoxy CTP-labeled cRNA probes using the Low Input Linear Amplification and Labeling kit (Agilent), following the manufacturer’s instructions. Cy-3- and Cy-5-labeled cRNAs were purified using an RNeasy Mini Kit.
Microarray data acquisition and cluster analysis
Agilent Technologies custom microarrays (G4138A) were hybridized according to the manufacturer’s instructions. cRNA synthesized from an untreated sample (0 h) was used as a reference, and two time-course replicate analyses were performed using RNA prepared from independent cell cultures. Microarray slides were scanned using a microarray scanner (G2565B Agilent), and the resulting output files (tiff images) were imported into the Feature Extraction software (Version 9.1 Agilent). Spot and background intensities from scanned slides were quantified and data files were normalized using the software algorithm. The results were filtered to identify candidate clones at all time points with expression more than 2.5-fold greater than that of the references in both experiments. Data from the selected clones were imported into Multi Experiment Viewer (TMEV4, The Institute of Genomic Research, http://www.TIGR.org) for cluster analysis. A hierarchical clustering analysis based upon the average linkage and cosine correlation was then used to cluster genes on the y-axis using TMEV4.
RT-PCR and quantitative real-time PCR
For each sample, 1 μg of total RNAs was treated with 10 units of DNase (Roche Diagnostics) to remove contaminating genomic DNA, and then used for reverse transcription (RT) to synthesize first-strand cDNA using an oligo-dT primer and a Superscript III Reverse Transcriptase kit (Invitrogen, Carlsbad, CA). cDNA samples diluted 10-fold were subjected to polymerase chain reaction (PCR) (OsCyc1, OsCyc2, OsDTC1, OsDTC2, OsKS4, OsKS10, and UBQ) or quantitative RT-PCR (OsDXS3, OsDXR, OsCMS, OsCMK, OsMCS, OsHDS, OsHDR, and UBQ) using SYBR Green technology on an ABI PRISM 7300 Real-Time PCR System (Applied Biosystems, Foster City, CA). Raw data of qRT-PCR were analyzed using the ΔCT (difference in threshold cycles) method, and the results were expressed as relative mRNA values normalized to the expression level of UBQ (ubiquitin fused to ribosomal protein L40, D12629) as an internal control. For each sample, the mean value from triplicate amplifications was adapted to calculate the transcript abundance. Primers for amplification were designed with the aid of Primer Express software (Applied Biosystems), and sequences of the amplified products were confirmed using an ABI310 Genetic Analyzer (Applied Biosystems). The primers used were as follows: UBQ-F (5′-GGACTGGTTAAATCAATCGTCA-3′) and UBQ-R (5′-CCATATACCACGACCGTCAAAA-3′); OsCyc1-F (5′-TGACGAGGCTGGGCATATC-3′) and OsCyc1-R (5′-TCTGGAGTCCAGTTCCTGAAA-3′); OsCyc2-F (5′-TTAGGAAAATGGTTGACTAC-3′) and OsCyc2-R (5′-ATCGACTAAATTCATCTCAC-3′); OsDTC1-F (5′-TTCATCTCTGTCACTTTTTCTTTTT-3′) and OsDTC1-R (5′-ATCCCACGAAGTCATCCAC-3′); OsDTC2-F (5′-CCAGCGTCTTCTACCAGGAG-3′) and OsDTC2-R (5′-AAAATGTTCAGCGAGGGAGA-3′); OsKS4-F (5′-CGCCTTTGTAACTCTAAGGTA-3′) and OsKS4-R (5′-ACGTAAAAGGCTTGTATATC-3′); OsKS10-F (5′-TGATCGTCCATTTCAGCTCT-3′) and OsKS10-R (5′-TTCTCAATCAGCGCGATGTA-3′); OsDXS3-F (5′-GGGGGAGGTTCCAGTAAGAA-3′) and OsDXS3-R (5′-TCATTTTGCATTTGGAAGCA-3′); OsDXR-F (5′-GCTCCATGCATAGTCAGCAG-3′) and OsDXR-R (5′-GCACGGACGAACGATTTATT-3′); OsCMS-F (5′-ACGGGATGGACTTGAGGTCA-3′) and OsCMS-R (5′-TTATTTCTCATTCATCAGGCG-3′); OsCMK-F (5′-CCCAAAATTAGCCCATTCAA-3′) and OsCMK-R (5′-GTCGACTTTCTCGTGCGTCT-3′); OsMCS-F (5′-TCGACCTTTATGTTGGCAAG-3′) and OsMCS-R (5′-CATGCTACGGCCTACTCCTC-3′); OsHDS-F (5′-GGAAAAGAACCATTCCACCTT-3′) and OsHDS-R (5′-TGTCACTTGGTATGCCCGTA-3′); and OsHDR-F (5′-CTGATGGCTTGGTGAAGGTT-3′) and OsHDR-R (5′-CAGCACATGCCGTAGTATGC-3′).
Plasmid construction
First-strand cDNA was used as a PCR template to amplify the OsDXS3d58 cDNA using the primers OsDXS3d58-F (5′-CACCGGATCCATCGACTACTCCGGCGAG-3′) and OsDXS3d58-R (5′-AAGCTTTCAGCTGAGCTGAAGTGCCT-3′). The 1,984-bp amplified fragment was digested with BamHI and HindIII and then fused between the same sites in the pQE30 vector to yield the plasmid pQE-OsDXS3, which expresses a hexahistidine-tagged protein. An OsDXR cDNA fragment was prepared from the plasmid pFLC-1-OsDXR (AK099702) obtained from the Rice Genome Resource Center (Rice Genome Project of the National Institute of Agrobiological Sciences, Japan). The 1,490-bp SmaI-XbaI fragment of OsDXR was fused between the same sites in the pUC18 vector to yield the plasmid pUC-OsDXR, which expresses OsDXR fused with the LacZ alpha peptide. To isolate the OsCMS cDNA, the Arabidopsis AtCMS (At2g26930) gene product sequence was used as a query in a BLASTX search of rice, because the predicted amino acid sequence of OsCMS in the Rice Genome Automated Annotation System (http://www.ricegaas.dna.affrc.go.jp/) (Sakata et al. 2002) did not exhibit high similarity with known CMS proteins. The longest OsCMS cDNA that encodes a highly homologous region was amplified by PCR using first-strand cDNA as a template with the primers CMSorf-F (5′-CAGAATTCTATGCCAAAACAATATCTACC-3′) and CMSorf-R (5′-CACTGCAGCTATTTCTCATTCATCAGGCG-3′). The 621-bp amplified fragment was digested with EcoRI and PstI and then fused between these sites in the pSTV28 vector to yield the plasmid pST-OsCMSt. The sequence of the truncated OsCMS cDNA was deposited in GenBank under the accession number AB296384.
Complementation analysis
The E. coli dxs disruptant DXM3 (Kim et al. 2006) and the dxr disruptant DYM1 (Takahashi et al. 1998; Kuzuyama et al. 1999) were grown on Luria-Bertani (LB) medium supplemented with 0.01% 2-C-methyl-d-erythritol (ME) and 50 μg/ml kanamycin (Km), and were transformed with pQE-OsDXS3 and pUC-OsDXR, respectively. Transformants of each were selected on LB + ME solid medium containing 50 μg/ml ampicillin (Amp) and then transferred to LB + Amp medium lacking ME. The E. coli ispD mutant NMW33 (Kuzuyama et al. 2000), harboring the pTMV20km plasmid, which carries the MVA pathway operon consisting of the mevalonate kinase, phosphomevalonate kinase, and diphosphomevalonate decarboxylase genes of Streptomyces sp. CL190, was grown on LB + Km supplemented with 0.01% mevalonolactone (MVL) and transformed with pST-OsCMS. The transformant was first selected on LB containing 35 μg/ml chloramphenicol (Cm) and MVL, and then transferred onto LB + Cm medium lacking MVL. The plasmids pQE-DXS, pMEW73, and pMNMW33-1 were used as positive controls harboring the native E. coli genes DXS, DXR, and CMS, respectively (Takahashi et al. 1998; Kuzuyama et al. 1998b, 2000).
Phytoalexins measurements
A total of 0.5-ml samples of media from rice cell cultures treated with elicitor and/or inhibitor were extracted three times with 0.5 ml of ethyl acetate at an approximate pH of 5.8. The combined ethyl acetate extracts were evaporated to dryness. The residues were dissolved in 5 ml of 70% aqueous methanol. Leaf discs were immersed in 200 μl of 70% aqueous methanol directly and heated at 95°C twice. About 5-μl samples of the resulting solutions were subjected to HPLC-ESI-MS/MS. An Agilent 1100 separation module (Agilent Technologies, Palo Alto, CA, USA) equipped with a Pegasil C18 column (150 × 2.1 mm in diameter; Senshu Scientific, Tokyo, Japan) was used with 70% aqueous acetonitrile containing 0.1% acetic acid as a solvent at a flow rate of 0.2 ml/min. All of the diterpenoid phytoalexins tested by HPLC-ESI-MS/MS were analyzed with a quadrupole tandem mass spectrometer (API-3000, Applied Biosystems Instruments, Foster City, CA, USA) outfitted with an electrospray ion source in positive-ion mode. Nitrogen was used as the collision gas. The ES capillary was set at 3.0 kV and the source temperature was 400°C. Other parameters were optimized by the spectrometer software (Applied Biosystems Instruments). The diterpenoid phytoalexins was determined with combinations of precursor/product ions; m/z 315/271 for momilactone A, m/z 331/269 for momilactone B, m/z 317/299 for phytocassanes A, D, and E, m/z 335/317 for phytocassane B and m/z 319/301 for phytocassane C in the MRM mode.
Results
Synchronous expression of diterpene cyclase genes triggered by chitin elicitor treatment
We previously reported that OsDTC1 and OsDTC2 are inducibly expressed by elicitor treatment (Cho et al. 2004; Nemoto et al. 2004). To further investigate the expression profiles of diterpene cyclase genes, we performed reverse-transcriptase PCR (RT-PCR) using total RNA prepared from suspension-cultured rice cells treated with chitin elicitor for 0, 2, 4, 6, 8, 10, 12, or 24 h. All six diterpene cyclase genes shown in Fig. 1 exhibited transient expression patterns that peaked at 4–8 h after elicitor treatment (Fig. 2). Given that the accumulation of phytoalexins was previously detected from 8 h after elicitor treatment, the expression profiles of these genes are consistent with roles in phytoalexins biosynthesis. This result prompted us to search for novel genes that are elicitor-inducible and involved in the biosynthesis of phytoalexins.

Expression profiles of diterpene cyclase genes analyzed by RT-PCR. The total RNAs used in RT-PCR were isolated from rice cells exposed to 1 ppm chitin elicitor for the indicated times and a pair of the gene-specific primers. As an internal standard, the rice ubiquitin fused to ribosomal protein gene UBQ was amplified by RT-PCR using the gene-specific primers
Identification of genes involved in phytoalexins biosynthesis using a 22-k rice oligomicroarray based on full-length rice cDNAs
We screened for elicitor-responsive genes that may be involved in phytoalexins biosynthesis using a highly efficient comprehensive analysis with a rice oligo microarray. cRNA probes for the microarray analysis were synthesized from the total RNA used in the RT-PCR analysis of diterpene cyclase genes described above. The Cy-3 labeled cRNA probe from the 0-h time point sample was used as a reference, and the Cy-5 labeled cRNA probes from seven other time points (2, 4, 6, 8, 10, 12, and 24 h) were compared against the 0-h reference. As described in the “Materials and Methods,” the genes that were upregulated greater than 2.5-fold at each of the time points were identified. Of 21,495 genes on the microarray, 1,000 genes were upregulated by the elicitor treatment, and the diterpene cyclase genes OsCyc1, OsCyc2, and OsDTC1 were among the 1,000 genes. The OsDTC2 gene was excluded during the sorting because one of the microarray analyses exhibited less than 2.5-fold upregulation of the gene, and the OsKS4 and OsKS10 genes are not included in probes on rice 22-k microarray. The upregulated genes were subjected to hierarchical clustering using the cosine correlation and average linkage methods to be sorted into groups of genes with similar expression patterns following elicitor treatment. As shown in Fig. 3A, the genes were categorized into three expression pattern groups: a group of 544 genes whose expression quickly increased and then decreased rapidly after elicitor treatment, named the early-induced expression-1 (EIE-1) node; a group of 262 genes whose expression quickly increased and then decreased slowly after the treatment, named the early-induced expression-2 (EIE-2) node; and a group of 194 relatively late-induced genes, named the late-induced expression (LIE) node. The complete lists of these genes are available in the supplemental data (Tables S1–S3). Because the three diterpene cyclase genes (OsCyc1, OsCyc2, and OsDTC1) were among the 194 genes in the LIE node, other candidate genes responsible for phytoalexins biosynthesis were also expected to be in the LIE node. Therefore, focusing on the remaining 191 genes in the LIE node, we found that five genes annotated to encode the MEP pathway enzymes 1-deoxy-d-xylulose 5-phosphate synthase (DXS), 1-deoxy-d-xylulose 5-phosphate reductoisomerase (DXR), 4-(cytidine 5′-diphospho)-2-C-methyl-d-erythritol kinase (CMK), 1-hydroxy-2-methyl-2-(E)-butenyl 4-diphosphate synthase (HDS), and 1-hydroxy-2-methyl-2-(E)-butenyl 4-diphosphate reductase (HDR) were present in the LIE node (Fig. 3B). The genes of two related proteins involved in the MEP pathway, 4-(cytidine 5′-diphospho)-2-C-methyl-d-erythritol synthase gene (CMS) and 2-C-methyl-d-erythritol 2,4-cyclodiphosphate synthase gene (MCS), have also been annotated in the rice genome, but were not in the LIE node because MCS was excluded during the sorting as a gene activated less than 2.5-fold, and CMS was not spotted on the microarray. We tentatively named these seven genes OsDXS, OsDXR, OsCMS, OsCMK, OsMCS, OsHDS, and OsHDR. The genes for HMG-CoA synthase, HMG-CoA reductase, and mevalonate kinase, all of which are involved in the mevalonate pathway, an alternative IPP biosynthesis pathway, were not induced by elicitor treatment in our microarray analysis (Table S4).

Hierarchical clustering of expression profiles of genes induced by the elicitor. (A) Hierarchical clustering sort of the 1,000 elicitor-induced genes, such that similar genes appear near each other. Shades of red indicate increased relative expression; shades of green indicate decreased relative expression. The EIE-1 and -2 nodes (blue and green bars) and the LIE node (red bar) are indicated. (B) Hierarchical clustering sort of the LIE node. Elicitor-induced genes possibly involved in the MEP pathway are indicated by arrows with accession numbers. (C) Number of clones involved in different metabolic pathways
Figure 3C shows the expected functions of the 194 genes in the LIE node. The LIE node includes 71 genes (about 36% of the total) that encode enzymes of several biosynthesis pathways: genes that encode enzymes involved in terpenoid synthesis, amino acid synthesis and metabolism (shikimate and phenylpropanoid pathways), flavonoid synthesis, and lipid catabolism, and genes for P450s and dehydrogenases. Since P450s and dehydrogenases have been suggested to be involved in the downstream oxidation of diterpene hydrocarbons leading to phytoalexins, the P450s and dehydrogenases in the LIE node could be promising candidates for phytoalexins biosynthetic genes (Kato et al. 1995; Atawong et al. 2002; Peters 2006). The remaining 123 genes in the LIE node appear to be unrelated to biosynthetic pathways. Approximately 18% of the genes in the LIE node have known functions or had previously been identified as pathogenesis-related proteins. Only five genes encoding transcription factors are included in the LIE node.
Expression profiles of putative MEP-pathway genes
The changes in the transcript levels of the MEP-pathway-gene homologs that exhibited late-induced expression in the microarray analysis were confirmed by qRT-PCR analysis. Gene-specific primers were designed for all seven genes of the MEP pathway using sequences in the 3′ untranslated regions. Including the OsCMS gene, whose cDNA was not present in the database, all of the genes were clearly induced by elicitor treatment (Fig. 4) and showed expression profiles similar to those of diterpene cyclase genes (Fig. 2). Kim et al. reported that the rice genome contains three OsDXS-related sequences (OsDXS1-3), and the expression of OsDXS3 is induced in rice plants by UV irradiation (Kim et al. 2005). This result is consistent with our result that expression of the OsDXS gene (AK100909), which is identical to OsDXS3, is specifically induced by the chitin elicitor. In contrast, the expression of two other homologs OsDXS1 (AK104851) and OsDXS2 (AK064944) remains at a basal level or less after elicitor treatment in our microarray analysis (Table S5). Based on the designation of Kim et al. we named the elicitor-induced OsDXS gene OsDXS3. We also found that, in addition to OsDXS3, all of the possible genes in the MEP pathway in rice are upregulated upon elicitor treatment. This result also suggests that the MEP pathway is coordinately activated along with the genes responsible for diterpenoid phytoalexins production.

Expression profiles of possible genes in the MEP pathway in cultured-cells. The total RNAs used for expression profiling were isolated from rice cells exposed to 1 ppm chitin elicitor for the indicated times. Values indicate relative mRNA levels normalized to the expression of the UBQ gene, and the maximal value in each experiment with different primers was arbitrarily set to 1.0. A, OsDXS3; B, OsDXR; C, OsCMS; D, OsCMK; E, OsMCS; F, OsHDS; G, OsHDR. The results are the average of at least three independent experiments; bars indicate the standard deviation of the mean
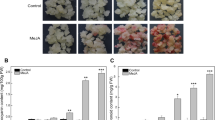
Diterpenoid phytoalexins production is also induced by jasmonic acid (JA), UV-irradiation, or CuCl2 treatment in rice leaves (Tamogami et al. 1997; Kodama et al. 1988a, b). To investigate whether the coordinate expression of the MEP-pathway-gene observed in suspension cultured rice cells is seen in rice plants as well, we analyzed mRNA levels of these genes in rice leaves treated with JA, UV-irradiation, and CuCl2 by qRT-PCR. As a result, expressions of all MEP-pathway-genes were found to be induced by any elicitation similar to that of OsKS4 in rice plants. Phytoalexins production was also confirmed in leaves treated by these elicitations (Fig. 5). These results suggest that the involvement of the MEP pathway in the phytoalexins production in planta was inferred from the synchronous expressions of genes in the MEP pathway in rice plants.

Expression analysis of possible genes in the MEP pathway in rice leaves. The total RNAs used for expression profiling were isolated from rice leaf discs exposed with H2O (N), JA (J), UV (U), or CuCl2 (C) for 72 h. Expression of the OsKS4 gene was shown as a positive control for these treatments. Values indicate relative mRNA levels normalized to the expression of the UBQ gene. Inset shows levels of phytoalexins production in the treated leaf discs
Functional analysis of OsDXS3, OsDXR, and OsCMS
The qRT-PCR analysis revealed the synchronous upregulation of all of the putative MEP-pathway genes by elicitor treatment. However, evidence of the functions of these genes in rice was lacking. To examine the functions of these genes, we performed complementation analysis using E. coli mutants defective in the MEP-pathway genes. The E. coli DXM3 and DYM1 strains, which are disrupted in the DXS and DXR genes, can only survive in culture medium supplemented with methylerythritol (ME) because the MEP pathway is essential for the growth of E. coli. The E. coli CMS-disrupted strain NMW33, harboring the plasmid pTMV20Km, which carries the mevalonate pathway gene cluster, can only grow in the presence of supplementary mevalonolactone (MVL) (Fig. 6). These mutants were transformed with the plasmids pQE-OsDXS3, pUC-OsDXR, or pST-OsCMS, which harbor the OsDXS3, OsDXR, and OsCMS cDNAs, to generate the DXM3/OsDXS3, DYM1/OsDXR, and NMW33/OsCMS strains, respectively. As shown in Fig. 6, when these transformants were spread on LB medium without supplemental ME or MVL, all transformants carrying the rice homologs of the MEP-pathway genes showed normal growth, similar to that of the control strains carrying the native genes for the E. coli MEP pathway. These results indicate that OsDXS3, OsDXR, and OsCMS encode functional enzymes that are involved in the MEP pathway, and that this complementation system is a powerful tool for confirming that genes from various organisms function in the MEP pathway. Since these genes, thought to encode the rice MEP pathway enzymes, exist as single copies in the rice genome, except for OsDXS, the remaining genes (OsCMK, OsMCS, OsHDS, and OsHDR) probably also encode functional enzymes. However, these functions also need to be experimentally confirmed.

Complementation of the E. coli DXM3, DYM1, and NMW33 mutants. The DXM3, DYM1, and NMW33 mutants cannot grow on LB without supplemented 2-C-methyl-d-erythritol (ME), which is used by E. coli in place of MEP (Duvold et al. 1997), or mevalonolactone (MVL). The DXM3, DYM1, and NMW33 mutants transformed with plasmids harboring OsDXS3 (A), OsDXR (B), or OsCMS (C), respectively, were grown for three days at 37°C on LB plates containing (below) or lacking (top) 0.01% (V/V) ME or MVL. Plasmids harboring the EcDXS, EcDXR, or EcCMS genes were used as positive controls and empty vectors (pQE30, pUC18, or pSTV28) were used as negative controls, respectively
Decreased accumulation of phytoalexins in rice cells treated with inhibitors of the DXS and DXR proteins
The synthesis of diterpenoids in plants is thought to begin in plastids from the common precursor GGDP. Diterpenoid-type phytoalexins such as momilactones and phytocassanes are also thought to be synthesized in plastids from GGDP derived from the MEP pathway. To investigate the contribution of the MEP pathway to the production of diterpenoid-type phytoalexins in rice, the chemical inhibitors KCZ and FOS, which suppress the metabolic steps catalyzed by OsDXS and OsDXR, respectively (Sakakibara et al. 2005, Kuzuyama et al. 1998a), were added to suspension-cell culture media, and then phytoalexins levels were measured after elicitor treatment. When either 100 μM FOS or 50 μM KCZ were added to the culture media, the production of momilactones and phytocassanes that normally occurs in untreated cells was strongly suppressed in inhibitor-treated cells. In contrast, treatment of 5 μM mevastatin (MEV), an inhibitor for HMG-CoA reductase in the MVA pathway, was shown to be less effective than FOS and KCZ treatments to the phyntoalexins production; the phytoalexins levels in the FOS-treated and the MEV-treated cells at 48 h after elicitation were about 30 and 90% of those in rice cells treated with the elicitor alone, respectively (Fig. 7). These results strongly suggest that in plastids, the MEP pathway is responsible for the biosynthesis of diterpene-type phytoalexins in rice.

Effects of inhibitors of the MEP pathway on the production of diterpenoid phytoalexins. Diterpenoid phytoalexins (momilactones A and B and phytocassanes A–E) were extracted from media collected at various time points (0, 24, 48, and 120 h) after treatment with elicitor and inhibitors, and the phytoalexins levels were determined by HPLC-ESI-MS/MS. Solid circles indicate treatment with 1 ppm N-acetylchitooctaose (elicitor), solid squares indicate treatment with elicitor and 100 μM fosmidomycin (FOS), solid triangles indicate treatment with elicitor and 50 μM 5-ketoclomazone (KCZ), and open circles indicate treatment with elicitor and 5 μM mevastatin (MEV). The results are the averages of at least three independent experiments; bars indicate the standard deviation of the mean
Discussion
By analyzing the gene expression in elicitor-treated suspension-cultured rice cells using a 22-k rice oligomicroarray, we narrowed down the genes responsible for the early stage of phytoalexins biosynthesis, the MEP pathway, which supplies GGDP. Hierarchical clustering sorted the genes that were differentially expressed in response to elicitor treatment into three groups with similar expression patterns. Almost 80% of the elicitor-induced genes were clustered into the early-induced expression-1 (EIE-1) and EIE-2 nodes. The remaining genes were clustered into the late-induced expression (LIE) node, which contains many genes involved in terpenoid biosynthesis, including the phytoalexins biosynthetic genes. The LIE node contained the genes OsCyc1, OsCyc2, and OsDTC1, all of which are maximally expressed at around 8 h after elicitor treatment, suggesting that narrowing down phytoalexins biosynthetic genes using microarrays is a reliable approach. In fact, in addition to the MEP-pathway genes, genes for cytochrome P450 monooxygenases and dehydrogenases, both of which have been suggested as being involved in phytoalexin biosynthesis (Atawong et al. 2002; Peters 2006), are also present in the LIE node. Since these genes are strong candidates for involvement in phytoalexins biosynthesis, their biochemical functions are currently being analyzed by our group.
Kim et al. have reported that OsDXS3, one of the three DXS homologs in rice, is inducibly expressed by the UV irradiation of rice leaves (Kim et al. 2005). Expression of the MtDXS2 gene, a DXS homolog of the legume Medicago truncatula, has also been reported to be strongly stimulated in roots upon their colonization by mycorrhizal fungi, correlated with the accumulation of carotenoids (Walter et al. 2002). This evidence suggests that the activation of the first committed step in the MEP pathway catalyzed by DXS is a common response in plants under both biotic and abiotic stresses. Our results further indicate that, in addition to the DXS gene, six more genes possibly involved in the MEP pathway are synchronously expressed upon treatment with chitin elicitor. Moreover, these seven genes of the MEP pathway exhibit expression patterns very similar to those of diterpene cyclase genes responsible for phytoalexins biosynthesis. This suggests that the activation of the MEP pathway and downstream phytoalexins biosynthetic pathways is coordinately controlled by an unidentified transcriptional regulation mechanism. Guevara-Garcia et al. have reported that coordinated regulation at the transcript level of all the genes in the MEP pathway in Arabidopsis is observed during development (Guevara-Garcia et al. 2005). However, the synchronous regulation of MEP-pathway gene expression in rice we demonstrate here must be different from the already reported regulation in Arabidopsis, because the elicitor-induced expression of the MEP pathway genes in rice is a transient event that recovers from an activated state to a basal state within 12 h. On the other hand, the composition of the MEP-pathway genes appears to be similar among plant species. For example, the genomes of both rice and Arabidopsis contain three highly homologous DXS genes and single copies of the six other MEP-pathway genes. Furthermore, in these plants, the basal expression levels of the CMS genes, which encode 4-(cytidine 5′-diphospho)-2-C-methyl-d-erythritol synthase, are very low, because neither the ESTs nor the full-length cDNA sequences for the rice OsCMS genes have been deposited in the database, and the Arabidopsis CMS gene (AtMECT) has not been able to be detected by northern blot analysis (Rohdich et al. 2000; Okada et al. 2002).
There have been some reports that the MEP pathway is post-transcriptionally modulated in Arabidopsis; Guevara-Garcia et al. have reported that the MEP pathway enzyme levels are regulated at translational or post-translational levels in response to developmental cues and changes in the MEP pathway flux (Guevara-Garcia et al. 2005), and Sauret-Güeto et al. have reported that a loss of function of the chloroplast-targeted exoribonuclease polyribonucleotide phosphorylase (PNPase) resulted in the post-transcriptional up-regulation of DXR and three other enzymes of the MEP pathway (DXS, HDS, and HDR) (Sauret-Güeto et al. 2006). On the other hand, we have demonstrated in this study that synchronous and transient expression at mRNA level of all the genes in the MEP pathway in rice upon elicitor treatment. Therefore, although it might be expected that post-transcriptional modulation also occurs during development in rice plant as a common regulatory system similar to that in Arabidopsis, we consider that the MEP pathway is likely to be regulated at transcription level upon elicitor-induced phytoalexins biosynthesis in rice. Further experiments will be needed to prove regulation of the MEP pathway in rice at protein level.
GGDP is a common precursor in the branch point of the diterpenoid metabolic pathway. In addition to phytoalexins, many important diterpene compounds are synthesized using the same GGDP pool in plastids, including the plant hormone gibberellin and photosynthetic pigments. Therefore, it is highly possible that the MEP pathway is an important origin of the GGDP used in the synthesis of all plastidial diterpenoids, including phytoalexins. Our phytoalexins measurement data indicated that, following elicitor treatment, more than 700 μg/g FW of phytoalexins accumulated in the culture medium (Fig. 7). This productivity is much greater than that of the plant hormone gibberellin, which is produced in an organ- and time-specific manner at a level of around several ng/g FW using the same GGDP pool as diterpene phytoalexins synthesis. Therefore, it appears that the activation of the MEP pathway in infected rice is essential for the production of sufficient secondary metabolites, including phytoalexins.
In this study, we demonstrated that the inducible production of phytoalexins in rice occurs as a consequence of the coordinate expression of genes responsible for both the early and downstream stages of phytoalexins biosynthesis. This coordinate expression also suggests that the same or similar mechanisms elicit the transcription of these genes. However, the mechanism of the synchronous regulation of the expression of this series of genes is still unknown. One potential approach would be a search for a factor that regulates the coordinate expression of elicitor-inducible genes in the rice oligomicroarray analysis.
References
Akatsuka T, Takahashi N, Kodama O, Sekido H, Kono Y, Takeuchi S (1985) Novel phytoalexins (Oryzalexins A, B and C) isolated from rice blast leaves infected with Pricularia oryzae. Part 1: isolation, characterization and biological activities of oryzalexins. Agric Biol Chem 49:1689–1694
Atawong A, Hasegawa M, Kodama O (2002) Biosynthesis of rice phytoalexin: enzymatic conversion of 3beta-hydroxy-9beta-pimara-7,15-dien-19,6beta-olide to momilactone A. Biosci Biotechnol Biochem 66:566–570
Cartwright D, Langcake P, Pryce R, Leworthy D, Ride J (1981) Isolation and characterization of two phytoalexins from rice as momilactones A and B. Phytochemistry 20:535–537
Cho EM, Okada A, Kenmoku H, Otomo K, Toyomasu T, Mitsuhashi W, Sassa T, Yajima A, Yabuta G, Mori K, Oikawa H, Toshima H, Shibuya N, Nojiri H, Omori T, Nishiyama M, Yamane H (2004) Molecular cloning and characterization of a cDNA encoding ent-cassa-12,15-diene synthase, a putative diterpenoid phytoalexin biosynthetic enzyme, from suspension-cultured rice cells treated with a chitin elicitor. Plant J 37:1–8
Duvold T, Bravo JM, Pale-Grosdemange C, Rohmer M (1997) Biosynthesis of 2-C-methyl-d-erythritol, a putative C 5 intermediate in the mevalonate independent pathway for isoprenoid biosynthesis. Tetrahedron Lett 38:4769–4772
Guevara-García A, San Román C, Arroyo A, Cortés ME, de la Luz Gutiérrez-Nava M, León P (2005) Characterization of the Arabidopsis clb6 mutant illustrates the importance of posttranscriptional regulation of the methyl-d-erythritol 4-phosphate pathway. Plant Cell 17:628–643
Heintz R, Benveniste P, Robinson WH, Coates RM (1972) Plant sterol metabolism. Demonstration and identification of a biosynthetic intermediate between farnesyl PP and squalene in a higher plant. Biochem Biophys Res Commun 49:1547–1553
Kato T, Kabuto C, Sasaki N, Tsunagawa M, Aizawa H, Fujita K, Kato Y, Kitahara Y, Takahashi N (1973) Momilactones, growth inhibitors from rice, Oryza sativa L. Tetrahedron Lett 14:3861–3864
Kato H, Kodama O, Akatsuka T (1993) Oryzalexin E, a diterpene phytoalexin from UV-irradiated rice leaves. Phytochemistry 33:79–81
Kato H, Kodama O, Akatsuka T (1994) Oryzalexin F, a diterpene phytoalexin from UV-irradiated rice leaves. Phytochemistry 36:299–301
Kato H, Kodama O, Akatsuka T (1995) Characterization of an inducible P450 hydroxylase involved in the rice diterpene phytoalexin biosynthetic pathway. Arch Biochem Biophys 316:707–712
Kim BR, Kim SU, Chang YJ (2005) Differential expression of three 1-deoxy-D: -xylulose-5-phosphate synthase genes in rice. Biotechnol Lett 27:997–1001
Kim SM, Kuzuyama T, Chang YJ, Song KS, Kim SU (2006) Identification of class 2 1-deoxy-d-xylulose 5-phosphate synthase and 1-deoxy-d-xylulose 5-phosphate reductoisomerase genes from Ginkgo biloba and their transcription in embryo culture with respect to ginkgolide biosynthesis. Planta Med 72:234–240
Kodama O, Suzuki T, Miyakawa J, Akatsuka T (1988a) Ultraviolet-induced accumulation of phytoalexins in rice leaves. Agric Biol Chem 52:2469–2473
Kodama O, Yamada A, Yamamoto A, Takemoto T, Akatsuka T (1988b) Induction of phytoalexins with heavy metal ions in rice leaves. Nippon Noyaku Gakkaishi 13:615–617
Koga J, Shimura M, Oshima K, Ogawa N, Yamauchi T, Ogawasawara N (1995) Phytocassanes A, B, C and D, novel diterpene phytoalexins from rice, Oryza sativa L. Tetrahedron 51:7907–7918
Koga J, Ogawa N, Yamauchi T, Kikuchi M, Ogasawara N, Shimura M (1997) Functional moiety for the antifungal activity of phytocassane E, a diterpene phytoalexin from rice. Phytochemistry 44:249–253
Kuzuyama T, Shimizu T, Takahashi S, Seto H (1998a) Fosmidomycin, a specific inhibitor of 1-deoxy-d-xylulose 5-phosphate reductoisomerase in the nonmevalonate pathway for terpenoid biosynthesis. Tetrahedron Lett 39:7913–7916
Kuzuyama T, Takahashi S, Watanabe H, Seto H (1998b) Direct formation of 2-C-methyl-d-erythritol 4-phosphate from 1-deoxy-d-xylulose 5-phosphate by 1-deoxy-d-xylulose 5-phosphate reductoisomerase, a new enzyme in the non-mevalonate pathway to isopentenyl diphosphate. Tetrahedron Lett 39:4509–4512
Kuzuyama T, Takahashi S, Seto H (1999) Construction and characterization of Escherichia coli disruptants defective in the yaeM gene. Biosci Biotechnol Biochem 63:776–778
Kuzuyama T, Takagi M, Kaneda K, Dairi T, Seto H (2000) Formation of 4-(cytidine 5′-diphospho)-2-C-methyl-d-erythritol from 2-C-methyl-d-erythritol 4-phosphate by 2-C-methyl-d-erythritol 4-phosphate cytidylyltransferase, a new enzyme in the nonmevalonate pathway. Tetrahedron Lett 41:703–706
Lange BM, Ghassemian M (2003) Genome organization in Arabidopsis thaliana: a survey for genes involved in isoprenoid and chlorophyll metabolism. Plant Mol Biol 51:925–948
Lichtenthaler HK (1999) The 1-deoxy-d-xylulose-5-phosphate pathway of isoprenoid biosynthesis in plants. Annu Rev Plant Physiol Plant Mol Biol 50:47–65
Lichtenthaler HK, Schwender J, Disch A, Rohmer M (1997) Biosynthesis of isoprenoids in higher plant chloroplasts proceeds via a mevalonate-independent pathway. FEBS Lett 400:271–274
Nemoto T, Cho EM, Okada A, Okada K, Otomo K, Kanno Y, Toyomasu T, Mitsuhashi W, Sassa T, Minami E, Shibuya N, Nishiyama M, Nojiri H, Yamane H (2004) Stemar-13-ene synthase, a diterpene cyclase involved in the biosynthesis of the phytoalexin oryzalexin S in rice. FEBS Lett 571:182–186
Nes WD, Venkatramesh M (1999) Enzymology of phytosterol transformations. Crit Rev Biochem Mol Biol 34:81–93
Okada K, Saito T, Nakagawa T, Kawamukai M, Kamiya Y (2000) Five geranylgeranyl diphosphate synthases expressed in different organs are localized into three subcellular compartments in Arabidopsis. Plant Physiol 122:1045–1056
Okada K, Kawaide H, Kuzuyama T, Seto H, Curtis IS, Kamiya Y (2002) Antisense and chemical suppression of the nonmevalonate pathway affects ent-kaurene biosynthesis in Arabidopsis. Planta 215:339–344
Otomo K, Kanno Y, Motegi A, Kenmoku H, Yamane H, Mitsuhashi W, Oikawa H, Toshima H, Itoh H, Matsuoka M, Sassa T, Toyomasu T (2004a) Diterpene cyclases responsible for the biosynthesis of phytoalexins, momilactones A, B, and oryzalexins A-F in rice. Biosci Biotechnol Biochem 68:2001–2006
Otomo K, Kenmoku H, Oikawa H, Konig WA, Toshima H, Mitsuhashi W, Yamane H, Sassa T, Toyomasu T (2004b) Biological functions of ent- and syn-copalyl diphosphate synthases in rice: key enzymes for the branch point of gibberellin and phytoalexin biosynthesis. Plant J 39:886–893
Peters RJ (2006) Uncovering the complex metabolic network underlying diterpenoid phytoalexin biosynthesis in rice and other cereal crop plants. Phytochemistry 67:2307–2317
Rohdich F, Wungsintaweekul J, Eisenreich W, Richter G, Schuhr CA, Hecht S, Zenk MH, Bacher A (2000) Biosynthesis of terpenoids: 4-diphosphocytidyl-2C-methyl-d-erythritol synthase of Arabidopsis thaliana. Proc Natl Acad Sci USA 97:6451–6456
Rohmer M (1999) The discovery of a mevalonate-independent pathway for isoprenoid biosynthesis in bacteria, algae and higher plants. Nat Prod Rep 16:565–574
Sakakibara H, Kasahara H, Ueda N, Kojima M, Takei K, Hishiyama S, Asami T, Okada K, Kamiya Y, Yamaya T, Yamaguchi S (2005) Agrobacterium tumefaciens increases cytokinin production in plastids by modifying the biosynthetic pathway in the host plant. Proc Natl Acad Sci USA 102:9972–9977
Sakata K, Nagamura Y, Numa H, Antonio BA, Nagasaki H, Idonuma A, Watanabe W, Shimizu Y, Horiuchi I, Matsumoto T, Sasaki T, Higo K (2002) RiceGAAS: an automated annotation system and database for rice genome sequence. Nucleic Acids Res 30:98–102
Sauret-Güeto S, Botella-Pavía P, Flores-Pérez U, Martínez-García JF, San Román C, León P, Boronat A, Rodríguez-Concepción M (2006) Plastid cues posttranscriptionally regulate the accumulation of key enzymes of the methylerythritol phosphate pathway in Arabidopsis. Plant Physiol 141:75–84
Scolnik PA, Bartley GE (1994) Nucleotide sequence of an Arabidopsis cDNA for geranylgeranyl pyrophosphate synthase. Plant Physiol 104:1469–1470
Stoessl A (1980) Phytoalexins: a biogenetic perspective. Phytopathol Z 99:251–272
Subba R, Strange RN (1994) Chemistry, biology, and role of groundnut phytoalexins in resistance to fungal attack. In: Daniel M, Purkayastha RP (eds) Handbook of phytoalexin metabolism and action. Marcel Dekker, New York, pp 199–227
Takahashi S, Kuzuyama T, Watanabe H, Seto H (1998) A 1-deoxy-d-xylulose 5-phosphate reductoisomerase catalyzing the formation of 2-C-methyl-d-erythritol 4-phosphate in an alternative nonmevalonate pathway for terpenoid biosynthesis. Proc Natl Acad Sci USA 95:9879–9884
Tamogami S, Mitani M, Kodama O, Akatsuka T (1993) Oryzalexin S structure: a new stemarane-type rice plant phytoalexin and its biogenesis. Tetrahedron 49:2025–2032
Tamogami S, Rakwal R, Kodama O (1997) Phytoalexin production elicited by exogenously applied jasmonic acid in rice leaves (Oryza sativa L.) is under the control of cytokinins and ascorbic acid. FEBS Lett 412:61–64
VanEtten HD, Mansfield JW, Bailey JA, Farmer EE (1994) Two classes of plant antibiotics: phytoalexins versus “phytoanticipins”. Plant Cell 6:1191–1192
Walter MH, Hans J, Strack D (2002) Two distantly related genes encoding 1-deoxy-d-xylulose 5-phosphate synthases: differential regulation in shoots and apocarotenoid-accumulating mycorrhizal roots. Plant J 31:243–254
Yajima A, Mori K (2000) Diterpenoid total synthesis, XXXII synthesis and absolute configuration of (−)-phytocassane D, a diterpene phytoalexin isolated from the rice plant, Oryza sativa. Eur J Org Chem 2000:4079–4091
Acknowledgements
We thank Dr Yoshiaki Nagamura and Ms Ritsuko Motoyama of the Rice Genome Resource Center for technical support with the microarray analysis, and also for providing the rice full-length cDNA clone that was developed in the Rice Genome Project of the National Institute of Agrobiological Sciences, Japan, and Prof Tadao Asami in The University of Tokyo for distribution of 5-ketoclomazone. This work was supported by the Program for Promotion of Basic Research Activities for Innovative Biosciences (PROBRAIN).
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below are the electronic supplementary materials.
Rights and permissions
About this article
Cite this article
Okada, A., Shimizu, T., Okada, K. et al. Elicitor induced activation of the methylerythritol phosphate pathway toward phytoalexins biosynthesis in rice. Plant Mol Biol 65, 177–187 (2007). https://doi.org/10.1007/s11103-007-9207-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11103-007-9207-2