
Abstract
The monoterpene indole alkaloids (MIAs) from Madagascar periwinkle (Catharanthus roseus) are secondary metabolites of high interest due to their therapeutical values. Secologanin, the monoterpenoid moiety incorporated into MIAs, is derived from the plastidial methyl-d-erythritol 4-phosphate (MEP) pathway. Here, we have cloned a cDNA encoding hydroxymethylbutenyl diphosphate synthase (HDS), a MEP pathway enzyme, and generated antibodies to investigate the distribution of transcripts and protein in MIA-producing aerial tissues. Consistent with our earlier work, transcripts for the genes encoding the so-called early steps in monoterpenoid biosynthesis (ESMB) enzymes (HDS, others MEP pathway enzymes and geraniol 10-hydroxylase) were preferentially co-localized to internal phloem associated parenchyma (IPAP) cells. By contrast, transcripts for the enzyme catalysing the last biosynthetic step to secologanin, secologanin synthase, were found in the epidermis. A coordinated response of ESMB genes was also observed in cell cultures stimulated to synthesise MIAs by hormone treatment, whereas no changes in SLS expression were detected under the same experimental conditions. Immunocytolabelling studies with the HDS-specific serum demonstrated the localisation of HDS to the plastid stroma and revealed that HDS proteins were most abundant in IPAP cells but could also be found in other cell types, including epidermal and mesophyll cells. Besides showing the existence of post-transcriptional mechanisms regulating the levels of HDS in C. roseus cells, our results support that intercellular translocation likely plays an important role during monoterpene-secoiridoid assembly.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
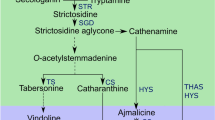
The anticancer Monoterpene Indole Alkaloids (MIAs) produced in Catharanthus roseus are derived from the condensation of the indole moiety tryptamine with the monoterpene-secoiridoid moiety secologanin. The biosynthesis of secologanin requires both the methyl-d-erythritol 4-phosphate (MEP) pathway and the monoterpene-secoiridoid pathway (Contin et al. 1998; Chahed et al. 2000; Veau et al. 2000; Fig. 1). The plastidial MEP pathway synthesizes IPP and DMAPP for the production of isoprenoids such as monoterpenes, carotenoids, gibberellins, abscissic acid, some cytokinins, and the side chain of chlorophyll, tocopherol, phylloquinone, and plastoquinone (Lichtenthaler et al. 1997; Lichtenthaler 1999; Rodríguez-Concepción and Boronat 2002; Kasahara et al. 2002; Fig. 1). On the other hand, the cytosolic mevalonate pathway synthesizes IPP and DMAPP for sesquiterpenes, prenylated proteins and triterpenes such as phytosterols (Adam et al. 1999; Lichtenthaler 1999; Eisenreich et al. 2001). So far, three genes from the MEP pathway encoding deoxyxylulose 5-phosphate synthase (DXS), deoxyxylulose 5-phosphate reductoisomerase (DXR), and methylerythritol 2,4-diphosphate synthase (MECS), and two genes from the monoterpene-secoiridoid pathway encoding two cytochrome P450 monooxygenases (CYP450), geraniol 10-hydroxylase (CYP76B6; G10H) and secologanin synthase (CYP72A1; SLS) have been cloned in C. roseus (for a review see Oudin et al. 2007).

Methyl erythritol phosphate (MEP) pathway and monoterpene-secoiridoid pathway featuring comparison of cell-specific gene-expression and enzyme-localisation patterns using three techniques in C. roseus aerial organs. Solid lines represent a single enzymatic conversion whereas dashed lines indicate multiple enzymatic steps. Abbreviations: DXS, 1-Deoxy-D-xylulose 5-phosphate (DXP) synthase; DXR, DXP reductoisomerase; HDS, Hydroxymethylbutenyl 4-diphosphate synthase; G10H, Geraniol 10-hydroxylase; SLS, Secologanin synthase. The genes encoding enzymatic steps from DXS to G10H represent the so-called early steps in monoterpene biosynthesis (ESMB) genes as compared to SLS catalyzing the last step of monoterpene-secoiridoid pathway. The cell-specific gene-expression/enzyme localisation patterns come from the following references: DXS, DXR and G10H (Burlat et al. 2004; Murata and De Luca 2005); SLS (Irmler et al. 2000; Murata and De Luca 2005); HDS (this study)
Two major model systems are currently used for studying MIA biosynthesis in C. roseus: undifferentiated cell suspensions that accumulate intermediate MIA or side branch products such as the hypotensive MIA ajmalicine, and whole plants, with particular interest in the aerial body which is so far the only system able to produce the highly valuable heterodimeric MIA vinblastine and vincristine used in anti-cancer treatments (Levêque et al. 1996; van der Heijden et al. 2004). Different approaches have been developed to improve the MIA production of undifferentiated C. roseus cell suspensions, including feeding experiments, chemical treatments, and genetic transformation of cell lines (see for reviews van der Heijden et al. 2004; Giglioli-Guivarc’h et al. 2006). Effects of phytohormones have also been widely studied. For instance in C. roseus cell cultures, auxins such as 2,4-dichloro-phenoxy-acetic acid (2,4D) negatively influence MIA accumulation, whereas addition of cytokinins in an auxin-free medium improves alkaloid production (Décendit et al. 1992, 1993; Yahia et al. 1998). Jasmonic acid and its volatile derivative methyljasmonate are also known to induce MIA accumulation in C. roseus cells and in seedlings (Aerts et al. 1994; Gantet et al. 1998). So far, despite the use of different strategies, sustainable improvement of the production of the valuable MIAs has failed in C. roseus cells. Consequently, many efforts are currently directed toward elucidation of the molecular basis of MIA biosynthesis and its regulation. Important advances have been recently made to understand the control at the cellular level of MIA biosynthesis in C. roseus aerial organs (see for reviews Kutchan 2005; Mahroug et al. 2006a). Transcripts for enzymes catalyzing early steps of the monoterpenoid biosynthesis (ESMB), including DXS, DXR, MECS and G10H (Fig. 1), were localized to the internal phloem associated parenchyma (IPAP) by in situ hybridisation (Burlat et al. 2004). By contrast, downstream enzymes of the MIA pathway such as SLS were restricted to epidermal cells both at the transcript and protein levels, whereas the last-two steps in vindoline biosynthesis, a precursor of the anticancer dimeric alkaloids, were found to occur in laticifers and idioblasts of the same organs (St-Pierre et al. 1999; Irmler et al. 2000). These studies revealed that multiple cell types are involved in the biosynthesis of MIAs, suggesting multicellular translocation processes of intermediates during MIA biosynthesis. More recently, the localisation of transcripts encoding G10H, SLS and tabersonine 16-hydroxylase (CYP71D12; T16H), another CYP450 involved in the late steps of MIA biosynthesis, by laser-capture microdissection combined with RT-PCR, led to an alternative model in which epidermal cells would be a major site of MIA production (Murata and De Luca 2005).
We report here the cloning of a new C. roseus cDNA encoding a MEP pathway enzyme, hydroxymethylbutenyl diphosphate synthase (HDS, also named GCPE). A comparative gene expression analysis showed a coordinated regulation of ESMB genes (including HDS) at the transcript level in hormone-treated cultured cells and a co-localisation of such transcripts to IPAP cells. By contrast, SLS displayed a distinct pattern of spatial and hormone-responsive expression. Additional immunocytolabelling studies were conducted for HDS, showing very high levels of this MEP pathway enzyme in the stroma of plastids from IPAP cells but also lower levels in epidermal and mesophyll cells. Our results support that multicellular translocation processes are required to supply enough substrates for the differentially-localized enzymes of the monoterpene-secoiridoid pathway, despite the presence of low levels of HDS (and likely other biosynthetic enzymes) in most cell types. The involvement of different cell types in the biosynthesis of isoprenoid-derived primary and/or secondary metabolites is also discussed.
Materials and methods
Plant material, cell growth and culture conditions
Two month-old periwinkle (Catharanthus roseus [L.] G. Don, Apocynaceae) plants were grown under greenhouse conditions. Periwinkle suspension cells (line C20D) were maintained on a 7-day-growth cycle in the B5 medium of Gamborg et al. (1968) supplemented with 58 mM sucrose and 4.5 μM 2,4D (maintenance medium, MM). For experimental purposes, 7-day-old cells were subcultured in one of the following 3 media: the maintenance medium (MM); an “inductive” medium (IM) ie MM without 2,4D; a “production” medium (PM), ie IM to which 5 μM zeatin was added on the third day of culture. MeJa (Sigma, L’Isle d’Abeau, France) was added at a concentration of 100 μM to C20D cells grown on IM at the fourth day of culture.
Ajmalicine quantification
For all experiments and as control of hormonal effects, content of ajmalicine (chosen as a marker of MIA accumulation) was measured on 7-day old cells. Aliquots of 50 mg freeze-dried cells were used for ajmalicine determination according to Yahia et al. (1998).
Isolation and cloning of C. roseus HDS cDNA
The databases available on the NCBI web page were searched using the sequence of Arabidopsis thaliana gene product as a query and the TBLASTN algorithm. Degenerate primers were derived from conserved regions in plant ESTs and A. thaliana translated gene sequences. Forward 5′-ATGCG(T/C)ATTGG(A/C)ACAAA(C/T)CA-3′ primer and reverse 5′-TC(T/C)CCTGG(A/T)CCATT(A/T/C)AC(G/A/T)AT-3′ primer (Operon, Cologne, Germany) were used to amplify a 1367 bp cDNA internal fragment by RT-PCR from 1 μg of total RNA extracted from alkaloid-producing cells of C. roseus. Reverse transcription was carried out using Thermoscript reverse transcriptase (Fisher Bioblock Scientific, Illkirch, France) at 60°C followed by a RNase H digest and further standard PCR amplification according to the program: 94°C for 45 s, 56°C for 45 s and 72°C for 1 min. Purified PCR product was cloned into pGEMT-easy vector (Promega, Charbonnières-les-bains, France) and sequenced. Analysis of sequence allowed to design additional specific primers to isolate respectively, 5′-end by asymmetric PCR on an oriented periwinkle cDNA library as previously described (Oudin et al. 1999) and 3′-end by 3′-RACE-PCR according to the manufacturer’s instructions (Fisher Bioblock Scientific). After sequencing, 5′-ATGGCGACCGGAACAGTT-3′ (forward primer) and 5′-TTTTTAGTCTTCTGCAGGAGG-3′ (reverse primer) specific oligonucleotides (Operon) were used to amplify the entire open reading frame by RT-PCR as described above. A 2223 bp cDNA was obtained, further purified, cloned into pGEM-T easy vector and sequenced. The full-length cDNA sequence was designed as HDS (GenBank accession number AY184810).
Sequence analyses
DNA sequencing was carried out by Genome Express SA (Meylan, France). Sequence analyses were performed using the available NCBI web programs and the ClustalW software (Thompson et al. 1994). Plastid targeting signals were predicted with ChloroP (www.cbs.dtu.dk/services/ChloroP/).
Production of His-tHDS fusion protein and antibody preparation
The forward 5′-GCGGATCCGTTTCTGTGATAAAGAACTCC-3′ and reverse 5′-TAGGTACCTTAGTCTTCTGCAGGAGGATC-3′ oligonucleotides (Operon) were used to amplify a truncated HDS cDNA (named tHDS) corresponding to the predicted coding sequence of the mature protein fused to a 6 × His tag. tHDS was isolated by RT-PCR as described above except the use of Platinum Pfx high fidelity DNA polymerase (Fisher Bioblock Scientific). The program was as followed: 94°C for 30 s, 50°C for 45 s and 68°C for 2 min 30 s. After purification, the PCR product was cloned into the BamHI–KpnI sites of pQE-30 (Qiagen, Courtaboeuf, France). The recombinant vector was designed as pQE-tHDS. The recombinant protein was expressed in E. coli BL21 by co-transformation of pQE-tHDS with the plasmid pUBS520 allowing expression of the rare AGG, AGA tRNA Arg codon (Brinkmann et al. 1989). Transformed E. coli cells were grown at 37°C in Luria–Bertani medium supplemented with ampicillin (100 μg ml−1) under vigourous agitation (250 rpm) until absorbance at 600 nm reached 0.8. After addition of 3 mM isopropyl-β-d-thiogalactoside (IPTG), the cells were allowed to grow for 4 h under the same conditions. His-tHDS protein was then purified with Ni2+ columns following the protocol supplied by the manufacturer (Qiagen). His-tHDS fusion protein, purified as previously described, was used for immunization of two New Zealand rabbits. The immunization protocol and the characterization of antibody sera were carried out by Agro-Bio SA (La Ferté St-Aubin, France).
RNA extraction and northern blot analysis
Frozen cells or plant organs were ground to a fine powder in liquid nitrogen. Total RNAs were extracted with RNAeasy Plant mini kit (Qiagen). Total RNAs (10 μg) were fractionated on a 2.2 M formaldehyde/1.2% (w/v) agarose gel according to Sambrook et al. (1989) and blotted onto a nylon membrane (Hybond N+, Amersham-Biosciences, Orsay, France). Hybridisations and washes were performed as described in Courdavault et al. (2005a). For probe preparations, full-length (DXR, HDS, G10H, SLS and RPS9) or partial (DXS +1 to + 1769) cDNAs were [P32]-labelled with the Prime-a-gene labelling kit (Promega).
Protein preparation and gel blot analysis
Total protein samples were obtained from 500 mg cells and various plant organs ground in liquid nitrogen and thawed in 1 ml cold lysis buffer containing 100 mM Tris pH 7.5, 1 mM EDTA, 1 mM DTT, 1 mM PMSF, 1 mM MgCl2 and protease inhibitors cocktail (Complete EDTA-free, Roche, Meylan, France). To eliminate cell debris, the lysates were centrifuged at 25,000 × g for 25 min at 4°C. Protein concentration was determined with Bradford reagent (Bio-Rad, Marnes La Coquette, France) using BSA as a standard and then electrophoretically separated on a 10% SDS gel according to the method of Laemmli (1970). To have comparable conditions in our protein analysis, 10 μg of total protein were used in all the analyses performed. To verify equal protein loading, a parallel gel was run and stained with Coomassie Blue. After gel equilibration in transfer buffer (48 mM Tris, 39 mM glycine, 20% (v/v) methanol), proteins were transferred to nitrocellulose membranes at 15 V for 15 min, using a semi-dry transfer system (Bio-Rad). The membranes were then incubated in blocking buffer containing 5% (w/v) non-fat dry milk in TTBS (20 mM Tris-HCl pH 7.5, 0.5 M NaCl, 0.05% (w/v) Tween 20) for 30 min at room temperature. The HDS polyclonal antiserum or nonimmune rabbit serum were diluted at a range of 1:50,000–1:250,000 in blocking buffer and incubated for 1 h at room temperature and overnight at 4°C. Blots were then washed 3 times for 10 min in TTBS at room temperature. An anti-rabbit immunoglobulin alkaline phosphatase conjugate (Sigma) was used as a secondary antibody at a 1:5000 dilution in blocking buffer. After 1 h incubation at room temperature and several washes in TTBS, the membranes were revealed in NBT/BCIP chromogenic substrate (Roche) [nitro blue tetrazolium chloride (NBT) 350 μg ml−1; 5-bromo 4-chloro-3-indolyl phosphate (BCIP) 175 μg ml−1, in TBS100-MgCl2 (100 mM Tris–HCl pH9, 100 mM NaCl, 10 mM MgCl2)].
E. coli complementation
The complementation assay was carried out using the Escherichia coli strain EcAB3–3 carrying a synthetic operon that allows to rescue an otherwise lethal deletion of the HDS-encoding gene gcpE by mevalonate supplementation (Campos et al. 2001a). After electroporation of competent cells with pQE-tHDS and plasmid pUBS520, transformants were selected as previously described (Querol et al. 2002). The ability of periwinkle protein to rescue the lethal loss of HDS activity in bacteria was tested by streaking independent colonies on plates without mevalonate.
Tissue fixation, embedding in paraffin, and sectioning
RNase-free conditions were strictly observed for all steps. Glassware was baked for 8 h at 180°C and non-disposable plasticware was incubated 10 min in an aqueous 3% H2O2 solution before washing in DEPC-treated water. Mature C. roseus grown in green house were harvested in late spring-early summer and rapidly fixed in FAA and embedded in Paraplast as previously described (St-Pierre et al. 1999; Burlat et al. 2004; Mahroug et al. 2006b). Serial sections (10 μm) were spread on silane-coated slides overnight at 40°C, and paraffin was removed using xylene (two times 15 min) before rehydration in an ethanol gradient series up to DEPC-treated water.
In situ hybridisation
The protocol used was previously described (St-Pierre et al. 1999; Burlat et al. 2004; Mahroug et al. 2006b). Full-length HDS cDNA cloned in pGEM-T (Promega) was used for the synthesis of sense and anti-sense digoxigenin-labelled RNA probes. For DXS, DXR, G10H and SLS, previously described plasmids were used for the transcription of riboprobes (Irmler et al. 2000; Burlat et al. 2004). Following prehybridisation, hybridisation of the digoxigenin-labelled probes and washing, the riboprobes were immunodetected using a sheep anti-digoxigenin Fab fragments–alkaline phosphatase conjugate (Roche), and the conjugates were visualized using an overnight incubation in the NBT/BCIP chromogenic substrate (Roche).
Cytochemistry
The callose contained within the sieve plates of the conducting-phloem sieve-elements was visualized using aniline blue staining in a 1% aqueous solution.
Immunocytochemistry
Serial sections of paraplast-embedded C. roseus aerial organs were prepared in a similar manner as for in situ hybridisation. All steps were performed at room temperature. Blocking of non-specific putative aldehyde functions remaining after the fixation was performed with 30-min incubation in TTBS (10 mM Tris–HCl, pH 7.8, 500 mM NaCl, 0.3% (w/v) Triton X-100) containing 0.15 M glycine. Sections were washed in TTBS and incubated for 30 min in TTBS-milk containing 5% (w/v) non-fat dry milk to block non specific protein-protein interactions. HDS immune- and pre-immune sera were used at a 1:200 dilution in TTBS-milk for a 2-h-incubation. Following 3 rinses of 10 min in TTBS, the sections were incubated for 1 h in an anti-rabbit immunoglobulin alkaline phosphatase conjugate (Sigma) diluted 1:500 in TTBS-milk. Unbound sera were washed three times for 10 min in TTBS and rapidly in water before color development either for 10 min or overnight in the NBT/BCIP solution described for western blot analysis. The slides were finally washed, dried and mounted with immersion oil under cover slips.
Light microscopy
In situ hybridisation and immunocytochemistry experiments were observed under bright field conditions with an epifluorescence microscope (Olympus BX51) equipped with a digital camera (Olympus DP50) and the corresponding software (Olympus analySIS). Aniline blue-induced fluorescence of callose was imaged with an UV excitation filter set (330–385 nm excitation filter, 420 nm cutting filter). Merging of in situ hybridisation and aniline blue staining views captured on the same area was realized with the analySIS software.
Transmission electron microscopy immunogold labelling
The sample fixation and embedding protocol was essentially as previously described (Burlat et al. 2001) with a few modifications. C. roseus young developing leaves were successively fixed for 2 h in freshly prepared 1.25% glutaraldehyde, 2% paraformaldehyde in 0.05 M sodium phosphate buffer pH 7.5, rinsed 3 times for 10 min in the same buffer, progressively dehydrated in an ethanol series (30, 50, 70, 80, 95, 100, 100, 100% ethanol 10 min each) and infiltrated in a mixture of LRWhite acrylic resin (medium grade, Sigma) and 100% ethanol (1:2, 1:1, 3:1, v/v for 1 h each at room temperature) with subsequent pure LRWhite changes (2–3) within one week at 4°C. Sample embedding was performed in 0.2 ml gelatin capsules with a 24-h polymerization at 50°C. Ultrathin (100 nm) sections were performed on a Reichert-Jung ultramicrotome using a Drukker diamond knife. Sections were collected on a droplet of water contained within a plastic ring. The immunogold labelling protocol was essentially realised as previously described (Burlat et al. 1997) with a few modifications. The plastic rings containing the ultrathin sections were successively carried on 50 μl droplets of the different solutions deposited either on parafilm spread on the bench for rinsing and blocking steps, or on 96-well silicon mold (Euromedex, Mundolsheim, France) placed within a humid chamber for antibodies incubation. Blocking of remaining aldehyde sites was achieved with a 30-min incubation in TBS (10 mM Tris–HCl, pH 7.8, 500 mM NaCl) containing 0.15 M Glycine, followed by 5 rinses in TBS for 2 min each. Following a 30-min incubation in TBS-milk (5% w/v non fat dry milk), the sections were incubated in 1:10–1:160 two-fold serial dilutions of immune- or pre-immune sera in TBS-milk for 1 h at room temperature followed by an overnight period at 4°C (1:10–1:20 being the optimal dilution range). The sections were washed 7 times for 2 min in TBS and were further incubated in proteinA-10 nm gold particle conjugates (Amersham-Biosciences) diluted 1:25 in TBS-milk. Finally, after washing 5 times for 2 min in TBS and 3 times for 2 min in distilled water, the sections were deposited on formvar-coated 200 mesh nickel grids (Sigma). Post-staining employed 2% (w/v) uranyl acetate aqueous solution for 2 min followed by 2 quick rinses in distilled water. The observations were made using a Jeol JEM 1010 transmission electron microscope (TEM) operating at 80 kV equipped with a Gatan Bioscan camera allowing to capture digital pictures. For the semi-quantitative analysis of immunogold labelling densities, low magnification pictures were captured and printed on paper. The same area was then observed with high magnification and the number of gold particles present on each plastid was posted on the paper prints. To measure the surface of each plastid, the digital files were calibrated with the scale bar present on the prints using the AnalySIS software (Olympus), and number of plastid per cells, plastid surface figures and gold particle numbering for each plastid in each cell types were further utilised to calculate the labelling densities expressed in number of gold particle per μm2 of plastids sections and number of gold particle per cell sections.
Results
Isolation of a C. roseus cDNA encoding HDS and generation of specific antibodies
The entire coding region of Catharanthus roseus HDS was amplified using a homology-based PCR approach. The full-length HDS cDNA sequence contained an open-reading frame of 2223 bp encoding a protein of 740 amino acids with an estimated molecular mass of 81.9 kD. Multiple alignment analysis of the amino acid sequence deduced from the C. roseus HDS nucleotide sequence with E. coli protein and two plant protein sequences revealed 84.6% and 89.7% sequence identity with A. thaliana and L. esculentum proteins, respectively (Fig. 2). The sequence analysis also showed the presence of a N-terminal extension defining a putative plastid-targeting sequence of 38 aa, as previously described for all MEP pathway enzymes from plants. The multiple alignment also revealed the presence of a large additional internal domain of 269 aa, characteristic of plant HDS proteins but absent in the E. coli sequence (Fig. 2; Querol et al. 2002; Rodríguez-Concepción et al. 2003), and the presence of three conserved cysteines which are the ligands of the [4Fe–4S]2+ centre in the HDS protein family (Fig. 2; Seeman et al. 2005).

Multiple alignment of HDS sequences. C. roseus (C.r, AA02474), L. esculentum (L.e, AA015447), A. thaliana (A.t, AF434673) and E. coli (E.c, P27433) HDS sequences are deposited in the Genbank and Swiss-Prot databases. Identical residues are highlighted in black boxes and conservative changes in gray boxes. A grey striped bar shows the sequence of the predicted plastid-targeting peptide from C. roseus protein using the ChloroP algorithm. Arrowhead indicates the amino terminus of the truncated HDS (tHDS) from C. roseus used to complement an HDS-deficient E. coli mutant. Asterisks (*) mark the position of the three conserved cysteine residues
In order to confirm the biochemical activity of C. roseus protein, a complementation assay was carried out using the E. coli EcAB3-3 strain, in which a disruption of the gcpE gene encoding HDS results in a lethal phenotype (Campos et al. 2001b). For this assay, the pQE-tHDS plasmid was constructed by subcloning a truncated HDS cDNA (tHDS) lacking the predicted plastid transit peptide into the pQE30 expression vector (Fig. 2). The pQE-tHDS plasmid was used to transform EcAB3–3 cells, engineered with a synthetic operon to synthesize IPP and DMAPP from exogenously supplied mevalonate (Campos et al. 2001a). Our results showed that the cloned C. roseus HDS cDNA encodes a protein able to complement the lethal HDS deficiency of the E. coli mutant (Fig. 3). Thus, functional analysis in E. coli demonstrated that we isolated the C. roseus HDS functional orthologue.

Complementation of HDS-deficient E. coli with the C. roseus mature HDS protein. Cells of the HDS-deficient EcAB3-3 strain were transformed with a construct to express the predicted C. roseus mature HDS protein (tHDS), or with the original pQE30 vector as a control. After recovering the generated EcAB3–3 strains (pQE-tHDS and pQE30) on plates supplemented with 1 mM MVA to rescue the lethal deletion of the chromosomal HDS gene, colonies were streaked on new plates with (+) or without (−) MVA and incubated at 37°C
To our knowledge, there are no reports concerning the regulation at the protein level of any MEP pathway enzyme in C. roseus. To generate the specific antibodies required for such studies, we first expressed the recombinant tHDS protein in E. coli cells (Fig. 4A) and purified this His-tagged protein by affinity chromatography on Ni2+-columns (Fig. 4B). Polyclonal antibodies raised against the His–tHDS fusion protein were then generated and tested against purified His–tHDS, and crude extracts from C. roseus cell suspensions and plant organs. In all the samples, a unique band was immunodetected by the polyclonal antibodies with a molecular mass around 78 kD corresponding to the mature protein without signal peptide (Fig. 4C). We also noticed a slight shift of the band obtained with tHDS as compared to the three other samples which can be attributed to the presence of the His-tag in the recombinant tHDS (Fig. 4C).

Expression, purification and immunodetection of HDS enzyme. (A) Protein crude extract of E. coli cells harbouring the empty pQE-30 vector (pQE) or pQE-tHDS (p-tHDS). (B) Purification profile of the recombinant tHDS used to raise antibodies. (C) Control of the antiserum specificity against the purified recombinant tHDS, a crude extract from C. roseus cells (CE) and plant aerial organs (YL: young leaves; FB: flower buds). M, protein standard
In situ hybridisation and immunocytolabelling
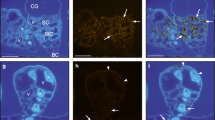
The first goal of this study was to investigate the distribution of HDS-encoding transcripts in aerial organs of C. roseus and to compare it with that of other ESMB genes (DXS, DXR and G10H) and SLS. In situ hybridisation experiments were performed on serial sections from several aerial organs of C. roseus. As depicted on Fig. 5, labelling with HDS anti-sense probes appeared to be restricted to adaxial parts of the vasculature regions in young developing cotyledons and leaves (Fig. 5A, G), whereas no significant background was observed with the corresponding sense probes used on serial sections (Fig. 5B, H). This labelling pattern was essentially identical to those obtained for the other ESMB genes (Fig. 5C–E, I, J, K), but distinct from that of SLS, whose transcripts were mostly found in the epidermis (Fig. 5F, L). ESMB genes were previously described to be expressed in internal phloem parenchyma cells (Burlat et al. 2004). Confusion on the nature of these specialised cells appeared in a recent study using laser capture and RT-PCR analyses, since they were assimilated as so-called “vascular cells” (Murata and De Luca 2005). Better characterization of the MEP pathway-expressing specialised cells and of the surrounding cells was achieved in young developing stems, which contain more developed vascular bundles (Fig. 6A). The conducting region of external and internal phloem was revealed by aniline blue staining of callose in sieve plates (Fig. 6B). Cells expressing HDS were located on the same exact section in an internal (adaxial) zone next to the blue-spotted internal phloem conducting region (Fig. 6A, C). To avoid confusion with parenchymatous cells contained within the phloem, we renamed these cells internal phloem associated parenchyma (IPAP) cells. The same predominant localisation in IPAP cells was obtained on cotyledons and petals cross-sections with the HDS antisense probes, with no background observed with the corresponding sense probes on serial sections (Fig. 7A–J). Interestingly, when the distribution of the HDS protein was investigated on serial sections by immunocytolabelling with the HDS-specific antibody, labelling was not only observed in IPAP cells but also in epidermal cells and, to a lower extent, in mesophyll cells (Fig. 7D–L). Longer incubation times with the chromogenic substrates resulted in a strong labelling of all cell types (Fig. 7F, G, M, N). Similar results were obtained in young developing leaves (Supplementary Fig. 1).

HDS is co-expressed with the other Early Steps in Monoterpenoid Biosynthesis (ESMB) genes (DXS, DXR, G10H) in internal phloem associated parenchyma (IPAP) cells whereas SLS is specifically expressed in epidermis. Paraffin-embedded serial sections of young aerial organs (developing cotyledons and leaves) of C. roseus were hybridised with digoxigenin-labelled transcripts. Hybridised transcripts were localized with anti-digoxigenin-alkaline phosphatase conjugates followed by BCIP-nitroblue tetrazolium color development. (A–F) serial cross-sections of cotyledons 3-days after imbibition; (G–L) serial longitudinal sections of the revoluted leaf base. The anti-sense probes used for mRNA labelling (A, C–F, G, H–L) and control hybridisation with sense RNA probes (B, H) are mentioned on the figure. Bars: 100 μm

Detailed characterization of the specialised IPAP cells expressing ESMB genes. The internal (adaxial) phloem associated parenchyma cells (IPAP) that express the ESMB genes represent a subset of the mesophyll cells located just above -and not within- the internal (adaxial) conducting phloem (ICP). A young developing C. roseus stem cross-section was hybridised with HDS anti-sense probes and photographed under bright-field conditions to show the typical labelling of IPAP (A). After in situ hybridisation, the section was further incubated in aniline blue aqueous solution and imaged using an UV excitation filter set to show the typical white-blue fluorescent spots of callose within the sieve plates allowing to locate the conducting elements of both phloems (external conducting phloem, ECP and internal conducting phloem, ICP) (B). Both pictures A and B were merged using the red and blue channel, respectively (C). Note that callose labelling within ECP and ICP was not overlapping with the HDS transcript labelling. X, xylem tracheids. Bars: 100 μm

Comparison of HDS transcript and protein cytolocalisation in cotyledons and petals. In addition to IPAP cells, constituting the most active site in HDS gene expression, immunocytolabelling using two substrate-incubation times reveal low level of HDS protein in the epidermis and mesophyll cells, where HDS transcripts were not detected by in situ hybridisation. Serial cross-section from young developing cotyledon (A–G) and flower buds taken at the petal level (H–N) were hybridised with HDS anti-sense probes (A, B, H, I), HDS sense probes (C, J), anti-HDS immune serum (D, F, K, M) and HDS pre-immune serum (E, G, L, N). Detection of the alkaline phosphatase secondary conjugates was carried out with a colorimetric reaction in BCIP/NBT solution for 16 h (A-C, F-G, H-J, M-N) or for 10 min (D, E, K, L). Note the typical specific labelling of internal phloem associated parenchyma (IPAP) both in cotyledons (A, B) and petals (H, I) with long incubation time with substrate to reveal the transcripts by in situ hybridisation. Also, note that a short incubation time with substrate following the immunolabelling allowed detection of labelling within the epidermis in addition to the IPAP (D, K). Finally, note that immunolabelling using a long development time permitted to detect additional specific signals in the epidermis and mesophyll cells (F, M). The corresponding negative controls did not show significant background. Bars: 100 μm except for (A) bar: 200 μm and (H) bar: 500 μm
In order to study the sub-cellular localisation of HDS epitopes and to conduct a semi-quantitative analysis of labelling density between IPAP, epidermis and mesophyll cells, the HDS-specific immune serum was further used to carry out immunogold labelling and transmission electron microscopy (TEM) observation (Fig. 8). The immunogold labelling was specifically observed in the stroma of the plastids in all cell types (Fig. 8). This sub-cellular localisation is fully consistent with the plastidic location of the MEP pathway enzymes in other plant species (Lichtenthaler et al. 1997; Lichtenthaler 1999; Araki et al. 2000; Bouvier et al. 2000; Carretero-Paulet et al. 2002). In good agreement with the results obtained on paraffin-embedded sections, epidermis and mesophyll cells displayed weak immunogold labelling compared to that observed in IPAP cells (Fig. 8). A semi-quantitative study was further carried out to check the specificity and the reproducibility of the immunogold labelling (Table 1). A total of 763 plastids were analysed in five cell types, each for their size (plastid surface) and density (number of plastids and plastid surface per cell section), and for the number of gold particles, in order to determine labelling density figures expressed either in gold particle/μm2 of plastid section and gold particle/cell section (Table 1). Interestingly, each cell type had a characteristic plastid size. The normalised labelling density (gold particle/μm2) observed in IPAP plastids was circa 50 times higher than that observed in mesophyll plastids and circa 10 times higher than the labelling density observed in epidermis plastids (Table 1). When integrating the size and number of plastids per cell section, the labelling density of one IPAP cell section was circa 100–250 and 15–30 times higher than the labelling density observed in one epidermis and one mesophyll cell section, respectively (Table 1; Supplementary Fig. 2).

Immunogold labelling of HDS in the stroma of plastids from C. roseus leaf cells confirms the occurrence of HDS-rich cells (internal (adaxial) phloem associated parenchyma, IPAP) and HDS-poor cells (epidermis and mesophyll cells). Ultrathin cross-sections of C. roseus leaves where used for immunogold labelling at the transmission electron microscopy level. (A) Very low abundant -but specific- labelling (one gold particle) was occasionally observed in the stroma of epidermis plastids (P). (B) Typical dense labelling that was specifically observed in the stroma of two IPAP plastids (P). (C) Typical dense labelling that was specifically observed in the stroma of IPAP plastids (P, low right) and of the very low abundant—but specific—labelling (one gold particle) in the stroma of a plastid in a neighbouring palissadic parenchyma cell (P, top left). In all cases, note that no background was observed in other sub-cellular compartments such as mitochondrions (M), vacuoles (V), neither in cytoplasm (C) nor in cell wall (CW). See Table 1 and Sup. Fig. 2 for semi-quantitative analysis of these immunolabellings
Expression profiles of ESMB genes and SLS in undifferentiated cell suspensions
To further investigate the regulation of the expression of HDS and other ESMB genes involved in MIA biosynthesis, we used the C20D cell line as a model system. These undifferentiated C. roseus cell suspensions can modify their MIA production level capacity in response to exogenous treatments (Giglioli-Guivarc’h et al. 2006). Thus, MIAs do not accumulate in a medium containing auxin, whereas MIA production is induced at day 5 of subculture in an auxin-free medium. MIA production can be further enhanced by addition of the cytokinin zeatin (Décendit et al. 1992; Chahed et al. 2000; Veau et al. 2000; Papon et al. 2005). Methyljasmonate (MeJa) also induces the biosynthesis of MIAs in C. roseus cell suspension cultures (Gantet et al. 1998) and seedlings (Aerts et al. 1994) by coordinately switching on the expression of multiple genes of the MIA biosynthetic pathway, including G10H (Menke et al. 1999a; Geerlings et al. 2000; van der Fits and Memelink 2000; Collu et al. 2001). The ability of MeJa to induce MIA accumulation in C20D cells was previously described (Gantet et al. 1998). However, its effect on the expression of genes of the MIA biosynthetic pathway has not been reported in this cell line, except for strictosidine synthase (Chatel et al. 2003).
The expression of ESMB genes and SLS was studied by RNA blot hybridisation in C20D cells following several hormonal treatments inducing MIA production (Figs 9A, 10A). Depletion of 2,4D increased the level of transcripts of all the ESMB genes analyzed (HDS, DXS, DXR, and G10H) from day 4 onward. The induction of ESMB gene expression correlated with MIA accumulation, which started at day 5 (Chahed et al. 2000; Veau et al. 2000; data not shown). Moreover, addition of zeatin further increased MIA accumulation and transcript levels of all ESMB genes (Fig. 9A). When cells were grown on an auxin-free medium treated with 100 μM of MeJa at day 4 of culture and then harvested at different time points, DXS, DXR, HDS and G10H transcript levels were strongly increased after 24 h (Fig. 10A). In contrast, none of the hormonal treatments did significantly affect the expression of SLS or RPS9 (Menke et al. 1999b), a gene used as a control for equal loading of RNA (Figs. 9A, 10A; Courdavault et al. 2005a). The accumulation pattern of HDS protein was also investigated in C20D cell suspensions by western blot analysis. The level of the HDS protein was analysed in total protein extracts (10 μg) from the same samples used for RNA analysis. In agreement with the transcript analysis (Fig. 9A), HDS protein levels were enhanced in auxin-depleted medium from day 4 onward (Fig. 9B) and 24 h after treatment with MeJa (Fig. 10B). In spite of significant changes in the HDS transcript abundance, however, addition of zeatin only slightly affected the level of the corresponding protein. Taken together, our results suggest a co-regulated expression of the ESMB genes (but not of SLS) and, in the case of HDS, a post-transcriptional regulation of protein levels in C. roseus cell cultures.

Expression analyses of Early Step in Monoterpenoid Biosynthesis (ESMB) genes and SLS in C20D cells. (A) RNA gel blot analysis of DXS, DXR, HDS, G10H (ESMB genes), and SLS transcript accumulation in C20D cells grown on MM, IM or PM media. Total RNA was extracted from C. roseus cells harvested from day 3–7 of subculture. Equal loading of RNA (ten μg) was checked by ethidium bromide (EtBr) staining. (B) Expression analysis of the HDS enzyme by western blot. Total protein extracts were isolated from the same batch of C. roseus cells used for RNA blots. Each lane contained 10 μg of total protein

Jasmonate-responsive expression of the ESMB genes and SLS in C20D cells. (A) Accumulation of DXS, DXR, HDS, G10H (ESMB genes), SLS and RPS9 transcripts in C20D cells grown either in IM medium (control) or in IM medium supplemented with 100 μM of methyljasmonate (MeJa). Total RNA was extracted from cells harvested half an hour, eight or twenty-four hours after MeJa addition. Northern blot analysis was done using 10 μg of total RNA. Equal loading was confirmed by ethidium bromide (EtBr) staining. (B) Protein gel blot analysis of HDS in C20D cells. Total protein extracts were isolated from cells cultured as described above for northern blot analysis
Discussion
In situ hybridisation studies had previously shown that specialised internal phloem parenchyma cells from C. roseus aerial organs specifically express at least three of the MEP pathway genes (DXS, DXR, MECS) and G10H (Burlat et al. 2004; Fig. 1), all of them belonging to the so-called ESMB group (Courdavault et al. 2005a). On the other hand, the last step in secologanin biosynthesis catalysed by SLS appeared to occur in the epidermis, as demonstrated by in situ hybridisation and immunolocalisation studies (Irmler et al. 2000; Fig. 1). This suggested the occurrence of an intercellular translocation event, from specialised internal phloem parenchyma cells to epidermal cells, of an unknown metabolite included within the 10-hydroxygeraniol-to-loganin portion of the monoterpene-secoiridoid pathway (Burlat et al. 2004; Fig. 1). This model added more complexity to the previously suggested intercellular translocation of downstream intermediates in the MIA pathway from the epidermis to laticifer-idioblasts, which was deduced from the localisation of transcripts and proteins of tryptophan decarboxylase (TDC) and strictosidine synthase (STR) in the epidermis and desacetoxyvindoline 4-hydroxylase (D4H) and deacetylvindoline 4-O-acetyl transferase (DAT) in laticifers and idioblasts (St-Pierre et al. 1999).
In a recent elegant study using a combination of laser capture microdissection/RNA isolation and amplification/RT-PCR, G10H and SLS were shown to be expressed in a similar manner in “vascular cells” and epidermis, as well as in laticifers and, more weakly, in idioblasts and mesophyll (Murata and De Luca 2005). The authors suggested that most of the MIA pathway genes up to T16H, including the ESMB genes and SLS, were co-expressed in the epidermis, which was hypothesised as a major site of the alkaloid factory. The so-called “vascular cells” were presented as a terpene factory expressing only the genes ranging from DXS to SLS. Their conclusion somehow challenged our results and raised a few questions for which we aimed to provide an answer.
First, we note that the laser captured “vascular cells” presented in Murata and De Luca work excluded the specialised internal phloem parenchyma cells. We previously described these cells as chlorophyll containing parenchymatous cells located above the internal conducting phloem (Burlat et al. 2004; Courdavault et al. 2005b). Here, we provide a more detailed characterization to emphasize the distinction between the internal phloem conducting elements and the HDS-expressing specialised cells, and we rename these cells internal phloem associated parenchyma (IPAP) cells to avoid confusion with parenchymatous cells in direct contact with the sieve tubes/companion cells complex (Fig. 6). As a consequence of the exclusion of IPAP cells from the laser-captured “vascular cells”, Murata and De Luca (2005) probably underestimated the abundance of the transcripts highly expressed in this tissue. Next, Murata and De Luca (2005) proposed a common expression profile for G10H and SLS. Unfortunately, the relative transcript level between various cell types was not semi-quantitatively estimated in their study. Even if, in agreement with Murata and De Luca (2005), our data show that the localisation of HDS protein occurs not only in IPAP cells but also in the mesophyll and the epidermis, our results clearly indicate that semi-quantitative analysis of transcript and protein abundance is important to assess the relative role of IPAP cells and epidermis in terpenoid metabolism. Indeed, our immunolocalisation studies show that one IPAP cell section displays 100–250 times more HDS epitopes than one epidermal cell section, and 15–30 times more HDS epitopes than one mesophyll cell section. We can not totally exclude that the low level of HDS protein in epidermal cells would be sufficient to supply the demand of terpenoid precursors for MIA biosynthesis, and that the high level of HDS protein in IPAP cells could be dedicated to isoprenoid molecules that remain to be identified. However, our semi-quantitative HDS immunological data combined with the common expression profile of all ESMB genes within the IPAP cells (Burlat et al. 2004; this study), and the distinct pattern of SLS expression (Irmler et al. 2000; this study) lead us to contradict Murata and De Luca’s model and to reinforce our model of an intercellular translocation event of a monoterpene secoiridoid metabolite from IPAP cells to epidermis.
Interestingly, we also notice a striking association between the response of biosynthetic genes to various hormonal signals in undifferentiated C. roseus cell suspensions and the cell-specific expression of the same genes in planta. Indeed, we clearly demonstrate that the IPAP-specific ESMB genes (from DXS to G10H) similarly respond to auxin depletion and cytokinin or methyljasmonate addition in undifferentiated cell suspensions, unlike the epidermis-specific SLS gene, whose expression does not significantly change with the treatments (Table 2). Our data are consistent with a recent report combining a genome-wide transcript profiling by cDNA-amplified fragment-length polymorphism combined with metabolic profiling of MeJa elicited C. roseus cells (Rischer et al. 2006). The authors observed, in response to MeJa, increases in gene tag expression for DXS, HDS and G10H but not for SLS. Moreover, in C20D cells, Courdavault et al. (2005a) also reported that CaaX–prenyltransferases catalysing prenylation of proteins, are essential for the expression of ESMB genes (including DXS, DXR and G10H) while expression of genes involved in later steps of MIA biosynthesis (such as epidermis-specific SLS, TDC and STR) was independent of protein prenylation. The distinct coordinated regulatory processes of ESMB genes vs SLS occurring in C20D cells is reminiscent of the IPAP-specific versus epidermis-specific expression profiles originally existing in planta. The activation of the MEP pathway genes in parallel or preceding the activation of specific downstream pathways was already reported in tomato (Botella-Pavia et al. 2004) and Arabidopsis (Guevara-Garcia et al. 2005), underlining the importance of a coordinated regulation of the MEP pathway with downstream pathways leading to isoprenoid end products to make sure that the required precursors will be supplied when needed. Finally, besides the control by transcriptional events, the existence of post-transcriptional regulation of HDS accumulation can be deduced from the lack of a good correlation between transcript and protein levels in zeatin-treated cell cultures (Fig. 9). This is in agreement with recent reports showing that HDS and other enzymes of the MEP pathway are regulated by post-transcriptional mechanisms (Guevara-Garcia et al. 2005; Rodríguez-Concepción 2006; Sauret-Güeto et al. 2006). Very little is known about the post-transcriptional regulation of the genes of the MIA biosynthetic pathway, including the MEP pathway, in C. roseus. Its significance will require future analysis, in particular a comparison of accumulation of the whole set of C. roseus MEP pathway proteins.
In addition to the terpenoid-derived complex alkaloids in C. roseus, the MEP pathway fuels numerous primary metabolism pathways such as those involved in the biosynthesis of hormones (some cytokinins, gibberellins and abscissic acid) and photosynthesis actors such as carotenoids and the side chain of chlorophyll, tocopherol, phylloquinone, and plastoquinone (Arigoni et al. 1997; Lichtenthaler et al. 1997; Lichtenthaler 1999; Kasahara et al. 2002). The results we previously obtained using in situ hybridisation led us to postulate that some intercellular transportation processes might occur for isoprenoid-derived primary metabolites in a similar manner as for monoterpene secoiridoids and MIAs (Burlat et al. 2004). Here, the localisation of HDS protein in the plastids of non-IPAP cells (such as those in the epidermis and mesophyll) may indicate the presence of an active MEP pathway in these cells to supply precursors for the biosynthesis of essential, housekeeping isoprenoids, questioning the necessity for a multicellular transportation. MIA production may require much higher levels of isoprenoid precursors than those needed for normal cell functions, therefore explaining the relative low density of HDS labelling observed in epidermis and mesophyll plastids compared to IPAP cell plastids. Indeed, our semi-quantitative analysis revealed that in C. roseus, one mesophyll cell section accumulates 15–30 times less HDS epitopes than one IPAP cell section. Even if the epitope density does not necessarily reflects the corresponding enzyme activity, specialised secondary metabolites (MIAs) may necessitate a high density of enzymes concentrated within a few specialised cells during a relatively short period of time and within peculiar developmental conditions, whereas primary metabolites such as chlorophyll may be supplied by a lower density of the same enzyme distributed within a large number of mesophyll cells and express in a more constitutive manner. However, it is also possible that the MEP pathway-enriched IPAP cells participate in the biosynthesis of isoprenoid moieties that can be further transported to other cell types to build isoprenoid-derived primary metabolites. Interestingly, DXS and DXR promoter–GUS fusions expressed in transgenic Arabidopsis plants have shown a strong expression along vascular tissues of young developing leaves and cotyledons, whereas weaker levels were found in mesophyll cells of the same organs (Estévez et al. 2000; Carretero-Paulet et al. 2002). Several other recent studies focused on the non-cell autonomous processes occurring in Arabidopsis and tobacco during the biosynthesis of chlorophylls (Nagata et al. 2002; Gutiérrez-Nava et al. 2004) and gibberellin (Silverstone et al. 1997; Itoh et al. 1999; Yamaguchi et al. 2001), further suggesting that intercellular transportation of isoprenoid-derived primary or secondary metabolites might be a more widespread phenomenon than previously thought.
Abbreviations
- DXS:
-
1-Deoxy-d-xylulose 5-phosphate synthase
- DXR:
-
1-Deoxy-d-xylulose 5-phosphate reductoisomerase
- ESMB:
-
Early steps in monoterpenoid biosynthesis
- G10H:
-
Geraniol 10-hydroxylase
- HDS:
-
Hydroxymethylbutenyl 4-diphosphate synthase
- HMBPP:
-
Hydroxymethylbutenyl 4-diphosphate
- IM:
-
Inducing medium
- IPAP:
-
Internal phloem associated parenchyma
- MECS:
-
2C-methyl-d-erythritol 2,4-diphosphate synthase
- MeJa:
-
Methyljasmonate
- MEP:
-
Methyl-d-erythritol 4-phosphate
- MIA:
-
Monoterpene indole alkaloid
- MM:
-
Maintenance medium
- PM:
-
Production medium
- SLS:
-
Secologanin synthase
- STR:
-
Strictosidine synthase
- T16H:
-
Tabersonine 16-hydroxylase
- TEM:
-
Transmission electron microscopy
References
Adam KP, Thiel R, Zapp J (1999) Incorporation of 1-[1-C-13]deoxy-d-xylulose in chamomile sesquiterpenes. Arch Biochem Biophys 369:127–132
Aerts RJ, Gisi D, De Carolis E, De Luca V, Baumann TW (1994) Methyl jasmonate vapor increases the developmentally controlled synthesis of alkaloids in Catharanthus roseus and Cinchona seedlings. Plant J 5:635–643
Araki N, Kusumi K, Masamoto K, Niwa Y, Iba K (2000) Temperature-sensitive Arabidopsis mutant defective in 1-deoxy-d-xylulose 5-phosphate synthase within the plastid non-mevalonate pathway of isoprenoid biosynthesis. Physiol Plant 108:19–24
Arigoni D, Sagner S, Latzel C, Eisenreich W, Bacher A, Zenk MH (1997) Terpenoid biosynthesis from 1-deoxy-d-xylulose in higher plants by intramolecular skeletal rearrangement. Proc Natl Acad Sci USA 94:10600–10605
Botella-Pavia P, Besumbes O, Phillips MA, Carretero-Paulet L, Boronat A, Rodríguez-Concepción M (2004) Regulation of carotenoid biosynthesis in plants: evidence for a key role of hydroxymethylbutenyl diphosphate reductase in controlling the supply of plastidial isoprenoid precursors. Plant J 40:188–199
Bouvier F, Suire C, d’Harlingue A, Backhaus RA, Camara B (2000) Molecular cloning of geranyl diphosphate synthase and compartmentation of monoterpenes synthesis in plant cells. Plant J 24:241–252
Brinkmann U, Mattes RE, Buckel P (1989) High-level expression of recombinant genes in Escherichia coli is dependent on the availability of the dnaY gene product. Gene 85:109–114
Burlat V, Ambert K, Ruel K, Joseleau JP (1997) Relationship between the nature of lignin and the morphology of degradation performed by white-rot fungi. Plant Physiol Biochem 35:645–654
Burlat V, Kwon M, Davin LB, Lewis NG (2001) Dirigent proteins and dirigent sites in lignifying tissues. Phytochemistry 57:883–897
Burlat V, Oudin A, Courtois M, Rideau M, St-Pierre B (2004) Co-expression of three MEP pathway genes and geraniol 10-hydroxylase in internal phloem parenchyma of Catharanthus roseus implicates multicellular translocation of intermediates during the biosynthesis of monoterpene indole alkaloids and isoprenoid-derived primary metabolites. Plant J 38:131–141
Campos N, Rodríguez-Concepción M, Sauret-Güeto S, Gallego F, Lois LM, Boronat A (2001a) Escherichia coli engineered to synthesize isopentenyl diphosphate and dimethylallyl diphosphate from mevalonate: a novel system for the genetic analysis of the 2-C-methyl-d-erythritol 4-phosphate pathway for isoprenoid biosynthesis. Biochem J 353:59–67
Campos N, Rodríguez-Concepción M, Seeman M, Rohmer M, Boronat A (2001b) Identification of gcpE as a novel gene of the 2-C-methyl-d-erythritol 4-phosphate pathway for isoprenoid biosynthesis in Escherichia coli. FEBS Lett 488:170–173
Carretero-Paulet L, Ahumada I, Cunillera N, Rodríguez-Concepción M, Ferrer A, Boronat A, Campos N (2002) Expression and molecular analysis of the arabidopsis DXR gene encoding 1-deoxy-d-xylulose 5-phosphate reductoisomerase, the first committed enzyme of the 2-C-methyl-d-erythritol 4-phosphate pathway. Plant Physiol 129:1581–1591
Chahed K, Oudin A, Guivarc’h N, Hamdi S, Chénieux JC, Rideau M, Clastre M (2000) 1-Deoxy-d-xylulose 5-phosphate synthase from periwinkle: cDNA identification and induced gene expression in terpenoid indole alkaloid-producing cells. Plant Physiol Biochem 38:559–566
Chatel G, Montiel G, Pré M, Memelink J, Thiersault M, Saint-Pierre B, Doireau P, Gantet P (2003) CrMYC1, a Catharanthus roseus elicitor- and jasmonate-responsive bHLH transcription factor that binds the G-box element of the strictosidine synthase gene promoter. J Exp Bot 54:2587–2588
Collu G, Unver N, Peltenburg-Looman AMG, van der Heijden R, Verpoorte R, Memelink J (2001) Geraniol 10-hydroxylase, a cytochrome P450 enzyme involved in terpenoid indole alkaloid biosynthesis. FEBS Lett 508:215–220
Contin A, van der Heijden R, Lefeber AWM, Verpoorte R (1998) The iridoid glucoside secologanin is derived from the novel triose phosphate/pyruvate pathway in Catharanthus cell culture. FEBS Lett 434:413–416
Courdavault V, Thiersault M, Courtois M, Gantet P, Oudin A, Doireau P, St-Pierre B, Giglioli-Guivarc’h N (2005a) CaaX-prenyltransferases are essential for expression of genes involved in the early stages of monoterpenoid biosynthetic pathway in Catharanthus roseus cells. Plant Mol Biol 57:855–870
Courdavault V, Burlat V, St-Pierre B, Giglioli-Guivarc’h N (2005b) Characterisation of CaaX-prenyltransferases in Catharanthus roseus: relationships with the expression of genes involved in the early stages of monoterpenoid biosynthetic pathway. Plant Sci 168:1097–1107
Décendit A, Liu D, Ouelhazi L, Doireau P, Mérillon JM, Rideau M (1992) Cytokinin-enhanced accumulation of indole alkaloids in Catharanthus roseus cell cultures-The factors affecting the cytokinin response. Plant Cell Rep 11:400–403
Décendit A, Petit G, Andreu F, Doireau P, Mérillon JM, Rideau M (1993). Putative sites of cytokinin action during their enhancing effect on indole alkaloid accumulation in periwinkle cell suspensions. Plant Cell Rep 12:710–712
Eisenreich W, Rohdich F, Bacher A (2001) Deoxyxylulose phosphate pathway to terpenoids. Trends Plant Sci 6:78–84
Estevez JM, Cantero A, Romero C, Kawaide H, Jimenez LF, Kuzuyama T, Seto H, Kamiya Y, Leon P (2000) Analysis of the expression of CLA1, a gene that encodes the 1-deoxyxylulose 5-phosphate synthase of the 2-C-methyl-d-erythritol-4-phosphate pathway in Arabidopsis. Plant Phys 124:95–103
Gamborg OL, Miller RA, Ojima K (1968) Nutrient requirements of suspension cultures of soybean root cells. Exp Cell Res 50:151–158
Gantet P, Imbault N, Thiersault M, Doireau P (1998) Necessity of a functional octadecanoic pathway for indole alkaloid synthesis by Catharanthus roseus cell suspensions cultured in an auxin-starved medium. Plant Cell Physiol 39:220–225
Geerlings A, Ibanez MM, Memelink J, van der Heijden R, Verpoorte R (2000). Molecular cloning and analysis of strictosidine beta-d-glucosidase, an enzyme in terpenoid indole alkaloid biosynthesis in Catharanthus roseus. J Biol Chem 275:3051–3056
Giglioli-Guivarc’h N, Courdavault V, Oudin A, Crèche J, St-Pierre B (2006) Madagascar periwinkle, an attractive model for studying the control of the biosynthesis of terpenoid derivative compounds. In: Teixeira Da Silva JA (ed) Floriculture, Ornamental and Plant Biology, Vol. II. Global Science Books
Guevara-Garcia A, San Roman C, Arroyo A, Cortes ME, Gutiérrez-Nava MdL, Leon P (2005) Characterization of the Arabidopsis clb6 mutant illustrates the importance of posttranslational regulation of the methyl-D-erythritol 4-phosphate pathway. Plant Cell 17:628–643
Gutiérrez-Nava MdL, Gillmor CS, Jiménez LF, Guevara-García A, León P (2004) CHLOROPLAST BIOGENESIS Genes Act Cell and Noncell Autonomously in Early Chloroplast Development. Plant Physiol 135:471–482
Irmler S, Schröder G, St-Pierre B, Crouch NP, Hotze M, Schmidt J, Strack D, Matern U, Schröder J (2000) Indole alkaloid biosynthesis in Catharanthus roseus: new enzyme activities and identification of cytochrome P450 CYP72A1 as secologanin synthase. Plant J 24:797–804
Itoh H, Tanaka-Ueguchi M, Kawaide H, Chen X, Kamiya Y, Matsuoka M (1999) The gene encoding tobacco gibberellin 3β-hydroxylase is expressed at the site of GA action during stem elongation and flower organ development. Plant J 20:15–24
Kasahara H, Hanada A, Kuzuyama T, Takagi M, Kamiya Y, Yamaguchi S (2002) Contribution of the mevalonate and methylerythritol phosphate pathways to the biosynthesis of gibberellins in Arabidopsis. J Biol Chem 277:45188–45194
Kutchan TM (2005) A role for intra- and intercellular translocation in natural product biosynthesis. Curr Opin Plant Biol 8:292–300
Laemmli UK (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680–685
Levêque D, Wihlm J, Jehl F (1996). Pharmacology of Catharanthus alkaloids. Bull Cancer 83:176–186
Lichtenthaler HK, Schwender J, Disch A, Rohmer M (1997) Biosynthesis of isoprenoids in higher plant chloroplasts proceeds via a mevalonate-independent pathway. FEBS Lett 400:271–274
Lichtenthaler HK (1999) The 1-Deoxy-d-xylulose 5-phosphate pathway of isoprenoid biosynthesis in plants. Annu Rev Plant Physiol Plant Mol Biol 50:47–65
Mahroug S, Burlat V, St-Pierre B (2006a). Cellular and sub-cellular organization of the monoterpenoid indole alkaloid pathway in Catharanthus roseus. Phytochem Rev (in press). DOI 10.1007/s11101–006–9017–1
Mahroug S, Courdavault V, Thiersault M, St-Pierre B, Burlat V (2006b). Epidermis is a pivotal site of at least four secondary metabolic pathways in Catharanthus roseus aerial organs. Planta 223:1191–1200
Menke F, Champion A, Kijne J, Memelink J (1999a) A novel jasmonate- and elicitor-responsive element in the periwinkle secondary metabolite biosynthetic gene Str interacts with a jasmonate- and elicitor-inducible AP2-domain transcription factor, ORCA2. EMBO J 18:4455–4463
Menke F, Parchamnn S, Mueller MJ, Kijne J, Memelink J (1999b) Involvement of the octadenoid pathway and protein phosphorylation in fungal elicitor-induced expression of terpenoid indole alkaloid biostynthetic genes in Catharanthus roseus. Plant Physiol 119:1289–1296
Murata J, De Luca V (2005) Localization of tabersonine 16-hydroxylase and 16-OH tabersonine-16-O-methyltransferase to leaf epidermal cells defines them as a major site of precursor biosynthesis in the vindoline pathway in Catharanthus roseus. Plant J 44:581–594
Nagata N, Suzuki M, Yoshida S, Muranaka T (2002) Mevalonic acid partially restores chloroplast and etioplast development in Arabidopsis lacking the non-mevalonate pathway. Planta 216:345–350
Oudin A, Courtois M, Rideau M, Clastre M (2007) The iridoid pathway in Catharanthus roseus alkaloid biosynthesis Phytochem Rev (in press). DOI 10.1007/s11101–006–9054–9
Oudin A, Hamdi S, Ouélhazi L, Chénieux JC, Rideau M, Clastre M (1999) Induction of a novel cytochrome P450 (CYP96 family) in periwinkle (Catharanthus roseus) cells induced for terpenoid indole alkaloid production. Plant Sci 149:105–113
Papon N, Bremer J, Vansiri A, Andreu F, Rideau M, Crèche J (2005) Cytokinin and ethylene control indole alkaloid production at the level of the MEP/terpenoid pathway in Catharanthus roseus suspension cells. Planta Med 71:572–574
Querol J, Campos N, Imperial S, Boronat A, Rodríguez-Concepción M (2002) Functional analysis of the Arabidopsis thaliana GCPE protein involved in plastid isoprenoid biosynthesis. FEBS Lett 514:343–346
Rischer H, Oresic M, Seppanen-Laakso T, Katajamaa M, Lammertyn F, Ardiles-Diaz W, Van Montagu MC, Inzé D, Oksman-Caldentev KM, Goossens A (2006) Gene-to-metabolite networks for terpenoid indole alkaloid biosynthesis in Catharanthus roseus cells. Proc Natl Acad Sci USA 103:5614–5619
Rodríguez-Concepción M (2006) Early steps in isoprenoid biosynthesis: Multilevel regulation of the supply of common precursors in plant cells. Phytochem Rev 5:1–15
Rodríguez-Concepción M, Boronat A (2002) Elucidation of the methylerythritol phosphate pathway for isoprenoids biosynthesis in bacteria and plastids. A metabolic milestone achieved through genomics. Plant Physiol 130:1079–1089
Rodríguez-Concepción M, Querol J, Lois LM, Imperial S, Boronat A (2003) Bioinformatic and molecular analysis of hydroxymethylbutenyl diphosphate synthase (GCPE) gene expression during carotenoid accumulation in ripening tomato fruit. Planta 217:476–482
Sambrook J, Fritsch EF, Maniatis T (1989) Molecular cloning: A laboratory manual, 2nd edn. Cold Spring Harbor Laboratory Press, Cold Spring Habour, NY
Sauret-Güeto S, Botella-Pavia P, Flores-Perez U, Martinez-Garcia JF, San Roman C, Leon P, Boronat A, Rodríguez-Concepción M (2006) Plastid cues post-transcriptionally regulate the accumulation of key enzymes of the methylerythritol phosphate pathway in Arabidopsis. Plant Physiol 141:75–84
Seemann M, Wegner P, Schunemann V, Bui BT, Wolff M, Marquet A, Trautwein AX, Rohmer M (2005) Isoprenoid biosynthesis in chloroplasts via the methylerythritol phosphate pathway: the (E)-4-hydroxy-3-methylbut-2-enyl diphosphate synthase (GcpE) from Arabidopsis thaliana is a [4Fe-4S] protein. J Biol Inorg Chem 10:131–137
Silverstone AL, Chang CW, Krol E, Sun TP (1997) Developmental regulation of the gibberellin biosynthetic gene GA1 in Arabidopsis thaliana. Plant J 12:9–19
St-Pierre B, Vazquez-Flota FA, De Luca V (1999) Multicellular compartmentation of Catharanthus roseus alkaloid biosynthesis predicts intercellular translocation of a pathway intermediate. Plant Cell 11:887–900
Thompson JD, Higgins DG, Gibson TJ (1994) CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, positions-specific gap penalties and weight matrix choice. Nucl Ac Res 22:4673–4680
Van der Fits L, Memelink J (2000) ORCA3, a jasmonate-responsive transcriptional regulator of plant primary and secondary metabolism. Science 289:295–297
Van der Heijden R, Jacobs D, Snoeijer W, Hallard D, Verpoorte R (2004) The Catharanthus Alkaloids: Pharmacognosy and Biotechnology. Curr Med Chem 11:607–628
Veau B, Courtois M, Oudin A, Chénieux JC, Rideau M, Clastre M (2000) Cloning and expression of cDNAs encoding two enzymes of the MEP pathway in Catharanthus roseus. Biochim Biophys Acta 1517:159–163
Yahia A, Kevers C, Gaspar T, Chénieux JC, Rideau M, Crèche J (1998) Cytokinins and ethylene stimulate indole alkaloids accumulation in cell suspension cultures of Catharanthus roseus by two distinct mechanisms. Plant Sci 133:9–15
Yamaguchi S, Kamiya Y, Sun TP (2001) Distinct cell-specific expression patterns of early and late gibberellin biosynthetic genes during Arabidopsis seed germination. Plant J 28:443–453
Acknowledgements
This research was financially supported by the Ministère de l’Education Nationale, de la Recherche et de la Technologie (MENRT, France), by Biotechnocentre, by the Conseil Régional du Centre, by the Ligue contre le Cancer (comité d’Indre et Loire and comité de l’Indre) and by the Spanish Ministerio de Educacion y Ciencia and FEDER (grant BIO2005–00367 to MR-C). We thank Dr. J. Memelink (University of Leiden, the Netherlands) who kindly provided C. roseus G10H cDNA and the oriented cDNA library, and Dr. N. Campos (Universitat de Barcelona) for the EcAB3-3 strain. We also thank Dr. B. Arbeille and the staff of the Electron Microscopy Platform (CHU Bretonneau, Tours, France) for providing excellent working conditions for ultramicrotomy and TEM studies.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Oudin, A., Mahroug, S., Courdavault, V. et al. Spatial distribution and hormonal regulation of gene products from methyl erythritol phosphate and monoterpene-secoiridoid pathways in Catharanthus roseus . Plant Mol Biol 65, 13–30 (2007). https://doi.org/10.1007/s11103-007-9190-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11103-007-9190-7