
Abstract
Thlaspi arvense, a wild species from the Brassicaceae family, was shown to have a higher level of freezing tolerance than either of its close relatives, the model plant Arabidopsis thaliana or the crop Brassica napus (canola). Over 600 clones were sequenced from a subtractive cDNA library generated from cold treated T. arvense tissue, establishing that T. arvense shared significant sequence identity with both A. thaliana and B. napus (90–92%). In light of the strong sequence similarity between T. arvense and A. thaliana and to exploit the available genomics resources for Arabidopsis, the efficacy of using long 70 mer oligonucleotide whole genome Arabidopsis microarrays was tested for T. arvense. Gene expression in T. arvense leaf tissue during the very early stages of cold acclimation (or cold stress) was assayed at three time points and compared to an untreated control. This analysis highlights some of the difficulties and benefits of using cross-species microarray analysis. The data suggested that T. arvense responds in a similar fashion to cold stress as the model plant A. thaliana. However, for a number of genes quantitative differences in the level and timing of expression were identified. One of the most notable differences suggested that sulphur assimilation leading to the increased production of the methyl donor S-adenosyl-methionine was playing a role in the response of T. arvense to cold stress.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Freezing temperatures represent a major environmental constraint affecting the performance and distribution of plant species (Boyer 1982). Many aspects of the physiology and biochemistry of plants can be affected by freezing stress depending on the plant species, environmental conditions and the extent of low temperature. Plants native to temperate climates can increase their ability to withstand the subsequent freezing temperatures in response to a period of low but non-freezing temperatures, a process known as cold acclimation (Levitt 1980; Guy 1990; Thomashow 1999). Cold acclimation requires many changes in cell biology and metabolism, involves many genes and a number of signalling pathways (Thomashow 1999; Xin and Browse 2000; Fowler and Thomashow 2002; Chinnusamy et al. 2004). The primary aim of cold acclimation research has been to identify cold responsive genes and understand their role in plant survival at low-temperatures. Arabidopsis thaliana has been widely employed as the model system to explore the mechanisms of cold acclimation and a large number of cold responsive genes and regulatory pathways, including the so-called CBF regulon, have been implicated in the cold acclimation process (Reviewed in Thomashow 1999). The over expression of genes within the CBF regulon, mediated through three transcription factors CBF1–3 (or DREB1B, DREB1C and DREB1A) has been shown to improve freezing tolerance in A. thaliana (Jaglo-Ottosen et al. 1998; Kasuga et al. 1999). However, there is limited experimental evidence to suggest the exact role that many of the cold induced genes play in conferring freezing tolerance and other potential cold regulatory pathways remain to be elucidated (Van Buskirk and Thomashow 2006).
The ability to cold acclimate and the length of time required to attain maximum freezing tolerance via cold acclimation varies greatly among plant species (Levitt 1980; Webb et al. 1994; Gilmour et al. 1998). The model plant A. thaliana is a chilling tolerant crucifer capable of cold acclimation, but to a lesser extent than its close relatives, the cultivated Brassica species (Laroche et al. 1992). Thlaspi arvense is a winter annual cruciferous weed, commonly known as stinkweed, which can survive Canadian Prairie winters where temperatures routinely drop to −30/40°C (Best and McIntyre 1975). T. arvense is phylogenetically closely related to both cultivated Brassica napus (2n = 38) and the model dicot A. thaliana (2n = 10) (Galloway et al. 1998). The different levels of freezing tolerance displayed by these three related species offers a useful basis for a comparative study to identify conserved and unique mechanisms by which the three species adapt to low temperatures.
Microarray technology has proven a powerful tool for identifying plant genes induced by environmental stimuli or stress and for analysing their expression profiles in response to such stresses (Chen et al. 2002; Fowler and Thomashow 2002; Kreps et al. 2002). Such studies have been particularly profitable in A. thaliana due to the availability of the whole genome sequence, which has allowed the development of microarrays where the majority of the annotated genes are represented and more recently culminated in the creation of high-density oligonucleotide-based arrays that span the whole-genome (Yamada et al. 2003). Previously, microarray analysis was limited in species such as T. arvense, where little or no sequence data was available. However, recent studies in B. napus, Arabidopsis halleri and Thellungiella halophila have suggested that cross-species microarray hybridisation can be an effective tool for exploring gene expression in close relatives of A. thaliana (Bacher et al. 2004; Lee et al. 2004; Taji et al. 2004; Gong et al. 2005). Additionally, the ability to compare responses to common stresses between species should allow genes with conserved and novel transcript profiles to be identified (Zhou and Gibson 2004). Such analysis, especially for complex traits, can facilitate the identification of candidate genes for further functional characterisation.
We initially developed a subtractive library for T. arvense that was enriched for transcripts expressed when plants are first exposed to low temperatures. A survey of over 600 expressed sequence tags (ESTs) established that a high level of similarity existed between the coding regions of the two species. In light of this, we tested the efficacy of using long oligonucleotide (70 mer) Arabidopsis whole genome arrays to perform transcript profiling in T. arvense during the early stages of cold acclimation (or cold stress). The results from these analyses are presented and contrasted to those previously published for A. thaliana. Although the results exposed some limitations of cross-species hybridisation experiments, they also identified commonalities between the two species in the qualitative nature of their response. In addition, some genes showed interesting differences in the timing and level of expression and warrant further study. The study also identified additional unpublished genes whose expression changes in response to low temperatures, adding to the obvious complexity of the plant’s response.
Materials and methods
Plant material and cold treatment
Thlaspi arvense seeds (provided by Dr. Ian McGregor, AAFC Saskatoon Research Centre) were imbibed on filter paper at 4°C for a period of 1 week prior to sowing in soil. Seedlings were grown at 18°C (day/night) with a 16-h photoperiod and 200 μE light intensity until the 4–5 true leaf stage and then subjected to a short photoperiod (12 h) for a period of 1 week. The plants were then transferred to a chamber set to cold acclimating conditions (3°C day/night, 12-h photoperiod and 150 μE light intensity) at virtual dawn, control tissues were harvested at this time. Samples for microarray analysis were collected at 1, 4 and 24 h after transferring plants to the cold chamber. It was anticipated that genes showing a circadian response would show an increase in transcript level at early sample points but would return to basal level after 12 h in the dark, at 24 h. A mixture of fully expanded and newly developed leaves were harvested, quickly frozen in liquid nitrogen and stored at −80°C until analysed.
Freeze tests
Thlaspi arvense, B. napus (DH12075, provided by Drs Gerhard Rakow and Ginnette Séguin-Swartz, AAFC Saskatoon Research Centre) and A. thaliana (ecotype Columbia, COL4, acquired from the Arabidopsis Biological Resource Centre, http://www.arabidopsis.org) were grown as described above. At zero time and at 7, 14 and 21 days after moving to low-temperature conditions, the level of freezing tolerance was determined by measuring changes in electrical conductivity using a method based on that described by Webb et al. (1994). Briefly, a leaf disc borer was used to excise uniform tissue samples from T. arvense and B. napus, and whole leaves of uniform size were taken from Arabidopsis plants. These tissue samples were placed on wet 3 MM Whatman paper in glass tubes which were placed in a refrigerated circulating ethylene glycol freezing bath (RTE 740, Thermo NESLAB, Portsmouth, NH, USA). After incubating at −2°C for 1 h, ice nucleation was induced with ice-shavings, the samples were then frozen by lowering the temperature at a rate of 1°C h−1. Three replicate samples were removed every hour and incubated at 4°C overnight before being equilibrated using deionised water for 4 h with constant agitation. Conductivity measurements were taken using a conductivity meter (Conductivity cell 011050, Orion Research Inc., Beverly, MA, USA) at room temperature both before and after freezing overnight to −80°C. The LT50 value for each sample was determined as the temperature at which 50% of the total conductivity was measured (Griffith and McIntyre 1993). All calculated LT50 were expressed as the mean from three independently replicated experiments.
Construction of suppression subtraction hybridisation cDNA library
Suppression subtractive hybridisation (SSH) was performed according to the manufacturer’s instructions using the polymerase chain reaction (PCR)-based cDNA subtraction kit (Clontech, Mountain View, CA, USA). T. arvense was grown on 0.5 × MS media at 18°C until the 3–4 leaf stage. The aerial tissue from half of the seedlings was used to prepare RNA to represent untreated tissue (driver) and half were treated at 3°C for 4 h before RNA extraction (tester). Briefly, tester and driver cDNA was generated from 2 μg of poly(A) RNA derived from the treated and untreated tissue samples, respectively, and the cDNA digested with Rsa I. The tester cDNA was separated into two fractions and ligated to different sets of adapters. Two rounds of hybridisation with the driver cDNA at 68°C was followed by two rounds of PCR amplification using nested primers specific to the annealed adapters to enrich for the desired differentially expressed sequences. The PCR products were ligated into the pGEM-T easy vector (Promega, ON, Canada) to generate the subtracted cDNA library.
Plasmid DNA extraction, sequencing and sequence analysis
Plasmid DNA extraction was carried out according to the manufacturer’s instructions using R.E.A.L. Prep 96 Plasmid Kit (Qiagen, Mississauga, ON, Canada). Sequencing reactions were performed using an ABI BigDye™ v2 Terminator cycle sequencing kit according to the instructions of the manufacturer and subsequently the reactions were analysed using an automated ABI3700 DNA Sequencer (Applied Biosystems, Foster City, CA, USA). The sequence traces were quality checked using PHRED (score greater than 20), trimmed for vector sequence using Sequencher (Gene Codes Corp., Ann Arbor, MI, USA) and then compared to the A. thaliana genomic sequence using BLASTN (http://ww.ncbi.nlm.nih.gov/) set with the default values for cost (mismatch cost = −3.0), reward (match reward = 1.0), wordsize (11 bp), default gap opening penalty (5.0) and the gap extension penalty (2.0). The repeat filtering option was switched off when comparing the T. arvense ESTs with the 70 mer oligonucleotide sequences.
Probe labelling and microarray hybridisation
The microarrays used in the experiments were spotted with the Operon Arabidopsis Genome Oligo Set Version V1 (http://www.operon.com) at the Microarray and Proteomics Facility at the University of Alberta, Edmonton, AL, Canada.
Total RNA was isolated from each sample using the RNEasy kit (Qiagen) according to the manufacturer’s instructions, and quantified with a Nanodrop-1000 Spectrophotometer (NanoDrop Technologies, Wilmington, DE, USA). The quality and quantification of the RNA was confirmed using gel electrophoresis. Each hybridisation experiment used 75 μg of total RNA, which was reverse transcribed to cDNA using the Amersham CyScribe Post-Labelling Kit (GE Healthcare, Piscataway, NJ, USA) according to the manufacturer’s instructions. The kit uses a two-step process to first incorporate a chemically reactive nucleotide analog (aminoallyl-dUTP) into the cDNA, subsequently the cDNA is then ‘post-labelled’ with reactive forms of Cy3- or Cy5-NHS esters, which bind to the modified nucleotides. The cDNA samples from two treatments were labelled with different fluorescent dyes (cyanine-3 and cyanine-5) and purified using the Amersham CyScribe GFX purification kit (GE Healthcare). The efficiency of probe labelling was confirmed using a Nanodrop-1000 Spectrophotometer. Two equivalent, differentially labelled (cyanine-3 and cyanine-5) cDNA samples were combined and applied onto the microarray surface under a cover slip and incubated for 16–18 h at 37°C in a hybridisation chamber. The degree of hybridisation was assessed by simultaneous scanning for Cy3 and Cy5 using a Virtex, Chipreader® Version 2.3 at a resolution of 10 μm.
Total RNA collected from non-acclimated control plants (0 h) was reverse transcribed to cDNA and co-hybridised independently with cDNA copies derived from cold acclimated samples collected at 1, 4 or 24 h. Each of the three sets of microarray experiments were replicated a total of four times including dye swaps.
Data analysis
Hybridisation signals were quantified using image analysis software (TIGR Spotfinder; TIGR Institute, http://www.tigr.org). Gene expression values measured as absolute intensities were normalised by block (local normalisation) using the LOWESS function (locally weighted scatter plot smoothing) in TIGR MIDAS (http://www.tigr.org).
To identify differentially expressed genes between time points, significance analysis of microarrays (SAM analysis; http://www-stat.stanford.edu/∼tibs/SAM/) was performed. Normalised data were log2 transformed and scaled by subtracting the median of the log2 intensity values from individual log2 transformed intensities. Data for the three-hybridisation experiments were analysed separately using the paired SAM model. Significant genes, with an estimated false discovery rate (FDR) of less than 10%, were mapped to the Arabidopsis gene database (http://www.arabidopsis.org) to obtain annotation information. Significant genes were grouped into different functional categories based on gene ontology (GO) provided at http://www.arabidopsis.org.
Results
Freezing tolerance of Arabidopsis thaliana, Brassica napus and Thlaspi arvense
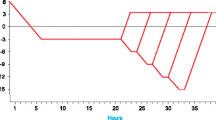
The plasma membrane is thought to be the primary site of injury during freezing and the most commonly used method for assaying freezing tolerance tests the level of membrane damage by quantifying electrolyte leakage from freeze injured tissues (Levitt 1980; Griffith and McIntyre 1993; Webb et al. 1994). Using this method, the freezing tolerance of T. arvense was shown to increase from an LT50 of −5 to −16.8°C after 3 weeks of cold acclimation (Fig. 1). As reflected by the percent electrolyte leakage curve, the increase in freezing tolerance was rapid during the first 2 weeks of acclimation with only a gradual increase observed in the last week. Over an equivalent acclimation period, under identical growth conditions, A. thaliana displayed an increase in freezing tolerance from −3.8 to −9.1°C and B. napus showed an increase from −3.8 to −12°C (Fig. 1). T. arvense exhibited a significantly higher freezing tolerance compared to closely related species, the model crucifer A. thaliana and cultivated Brassica species, suggesting T. arvense could be a useful comparative tool for studying the mechanisms by which plants attain freezing tolerance.

Acquisition of freezing tolerance in Arabidopsis thaliana, Brassica napus and Thlaspi arvense during cold acclimation. The temperature at which 50% cell death occurs (LT50), as measured by electrolyte leakage, is shown for the three species at zero time and after 1, 2 or 3 weeks growth at 3°C
Analysis of SSH Thlaspi arvense cDNA library
Since there were no ESTs available for T. arvense and only a small number of genomic sequences, a small sequencing project was initiated to establish the degree of similarity between T. arvense and A. thaliana. A subtractive library was constructed that was enriched for sequences expressed after exposure to low-temperatures for 4 h. A total of 646 high quality, trimmed, single pass sequences, with an average length of 309 bp (median = 280 bp) were generated from this library. The EST sequences were compared to each other and the annotated nuclear and plastid transcripts from A. thaliana (v5) using BLASTN, this analysis assembled the clones into non-redundant contigs and identified their closest homologous sequence from A. thaliana (Supplemental Table 1). Due to the use of an enzyme with a 4 bp recognition site (Rsa I) during the library construction some of the clones are fairly short, which de facto gives lower significance hits. In addition, a number of the sequences were derived from the 3′ end of transcripts, which are generally more divergent. To allow for this, a low-stringency Expect value of 1 × E-05 was used as the cut-off to identify homology between T. arvense and A. thaliana. However, only 53 of the clones were matched with an E-value greater than 1 × E-10. The majority of the clones, 594, identified 295 A. thaliana nuclear genes. A further 16 clones identified four annotated genes within the chloroplast genome and two clones identified a single unannotated region of the chloroplast genome. The remaining 64 clones were assembled into 11 contigs (24 clones) and 40 singletons and shared no significant homology with A. thaliana sequence.
The T. arvense sequences displayed, on average, 90% identity with the A. thaliana sequence, suggesting a greater homology between these two species than between B. napus and A. thaliana, which have previously been shown to share on average 86% sequence identity (Parkin et al. 2005). Thirty-three (11%) of the identified A. thaliana genes were functionally classified, based on GO annotation, as being involved in a plant’s stress response and included a number of previously defined COR genes, for example COR47, KIN1, ERD15, LOS2 and LEA14. The T. arvense ESTs were also compared against a database of 66,218 B. napus EST sequences (http://www.brassica.ca) using BLASTN and an E value cut off of 1 × E-07. Brassica napus and T. arvense demonstrated an average sequence identity of 91.6% across all aligned highest scoring pairs (HSPs). The sequence identity demonstrated across the three species correlated well with previous phylogenetic studies, which placed T. arvense in the same clade with B. oleracea separate from, but closely related to a clade containing A. thaliana (Galloway et al. 1998). Approximately 20% of the B. napus EST sequences were derived from leaves of cold treated plants. The T. arvense sequences identified multiple sequences within the B. napus database; however, there were significantly more hits found with sequences derived from the cold treated B. napus EST libraries (p < 0.001).
It has been suggested that greater than 75% similarity over a 50 bp window and as little as 25 bp of contiguous complementarity can result in cross-hybridisation to probe sequences on oligonucleotide microarrays (Kane et al. 2000; Chou et al. 2004). Although the homology between A. thaliana and T. arvense is sufficient across the coding regions to ensure cross-species hybridisation, the 70 mer oligonucleotides found on the Arabidopsis array were predominantly designed to the 3′ end of A. thaliana transcripts. The variability present in such regions could reduce the efficiency of hybridisation. To validate the use of the microarray, the oligonucleotide sequences were compared to the available T. arvense EST sequences. Although a number of the EST sequences were only partially over-lapping with the 70 mer sequences, it was possible to align with confidence 159 of the ESTs to 104 different oligonucleotide sequences, with an average alignment length of 43.5 bp and an average identity of 90.7%. The majority of the oligonucleotide probes (82) were matched to an EST, which showed the most significant homology to the gene for which the probe was designed. However, 32 of the oligonucleotide probes showed sufficient homology based on the above criteria to allow cross-hybridisation between genes. In almost all instances the cross-hybridisation was between multiple members of gene families, for example: between At3g25520, 60S ribosomal protein L5 (RPL5A) and At5g40130, 60S ribosomal protein L5 (RPL5C); between At1g12010, 1-aminocyclopropane-1-carboxylate oxidase (ACC oxidase) and At1g62380 also an ACC oxidase; and between five tandemly repeated auxin responsive genes (At1g18010, At1g18020, At1g18050, At1g18060 and At1g18080). However, although over a small region of identity, the oligoncucleotide target (A016732_01) for At5g57650, an annotated eukaryotic translation initiation factor, showed 94% sequence similarity over 34 bp with an EST showing strongest homology to At5g02840, which encodes a putative myb family transcription factor.
The results indicated that although the oligonucleotide arrays should be suitable for cross-species analysis between A. thaliana and T. arvense, caution should be used when inferring the expression profiles of members of gene families, since competition between paralogues could obfuscate the results.
Cross-species microarray analysis
Notwithstanding the apparent close homology between A. thaliana and T. arvense, the limitations of cross-species microarray analysis were apparent from the efficiency of hybridisation across the slides. On average, 31.1 ± 7.9% (8,186 detectable spots from 26,282 possible probes on the slide, not including controls) of the probes, across all time points and all replicates, gave appreciable fluorescence after filtering for spot quality, background noise, and subsequent LOWESS normalisation. In order to limit the numbers of falsely identified differentially regulated genes, only those target probes, which showed significant hybridisation in at least three of the four replicates were considered. This resulted in 4,158, 3,858 and 4,547 data points being available for each of the time-point comparisons 0/1, 0/4 and 0/24 h, respectively.
Significance analysis of microarrays developed by Tusher et al. (2001) has been widely adopted as a tool for analysing transcriptome data (Larsson et al. 2005). The software uses a modified t-test to identify genes with statistically significant changes in expression, based on the observed change in expression for each gene relative to the standard deviation of replicated measurements. The SAM analysis from the three time-point comparisons (0/1, 0/4 and 0/24 h) revealed in total 590 genes which demonstrated a significant change in expression, either up-regulated or down-regulated, with an estimated FDR of less than 10% (Supplemental data Table 2).
The variability in the hybridisations was also reflected in the proportion of genes for which fully replicated data was available in all time points studied. Data was available for all three time points for only 1,678 targets on the arrays, this meant that most of the observed changes were time-point specific; however, a small fraction of the significant genes were consistently up- or down-regulated over the course of the experiment. Only 10 genes were found to be consistently up- or down-regulated across all three sets of experiments while 90 genes (50 up-regulated and 40 down-regulated) were observed in at least two of three sampling times (1, 4 and 24 h) (Table 1).
Comparison with expression data from Arabidopsis thaliana
The genes identified as responding to cold stress in T. arvense were compared with cold-regulated gene sets previously identified in five separate microarray analysis studies of A. thaliana, all based on hybridisation to Affymetrix genechips (Fowler and Thomashow 2002; Kreps et al. 2002; Lee et al. 2005; Vogel et al. 2005; http://affymetrix.arabidopsis.info/). Of 590 genes identified as cold regulated in T. arvense, 233 were also found to be cold regulated in one or more of the microarray experiments using A. thaliana. For the remaining genes, there was either no target represented on the array or there was no significant difference in gene expression observed (Supplemental data, Table 2). It is to be expected that there will be inconsistencies in the genes identified, since the experiments employed a diverse range of growing conditions. However, the identification of responsive genes in more than one data set affords greater confidence that those genes are regulated by low-temperature stress. In general the observed change in expression was in the same direction across data sets, although there were a number of obvious exceptions, which appeared to be species specific and could not be explained by variations in either growth conditions or duration of cold treatments (Supplemental data Table 1).
We did not observe some of the previously reported A. thaliana cold responsive genes, which could be attributed to the low-hybridisation efficiency between the A. thaliana oligomers and the T. arvense cDNA. For some genes, such as transcription factors like CBF, no hybridisation signal was detected, while in other cases the signal intensity was simply not large enough for a change to be called significant. Because of the relatively short length of the 70-mer oligonucleotides, each representing a single gene, hybridisation intensities (signals) are often low (Wang et al. 2003). The low sensitivity of oligoarrays in detecting low-abundance genes in a given sample combined with relatively weaker hybridisation signals due to sequence divergence between A. thaliana and T. arvense might have rendered a cold responsive gene non-significant. In addition, because of the inherent genome duplication present within A. thaliana and the unknown level of gene/genome duplication present within T. arvense, there is an unquantifiable subset of the cold responsive genes which may be the result of changes in transcript abundance of paralogues (or duplicated copies) of the identified A. thaliana gene.
Functional classification of differentially expressed genes
The cold responsive genes from T. arvense were classified based on GO, which had previously been determined for the Arabidopsis genes present on the array (http://www.arabidopsis.org). This assigned genes into potential functional categories and to different cellular compartments. Based on the functional categorisation of all the annotated genes within the Arabidopsis genome and their distribution within each of the GO-Slim categories, it was possible to calculate the number of genes expected to be found within each of these categories for the 302 up-regulated and 293 down-regulated genes observed in T. arvense across all time points (Fig. 2). This identified a number of categories that were significantly over-represented for either up- or down-regulated genes, which assisted in dissecting the plant’s response to cold stress and in narrowing down interesting candidates for further analysis (Fig. 2). As might be expected within the up-regulated genes, those predicted to be involved in the plant’s response to abiotic or biotic stress were over-represented. In addition, the ‘transferase activity’ class and the ‘other binding’ class also showed a higher number of up-regulated genes than expected. These classes included several novel cytochrome P450 proteins, numerous representatives of the protease inhibitor/lipid transfer family of proteins and proteins potentially involved in cell wall conformation, such as cellulose synthases and xyloglucan endotransglucosylases. The down-regulated genes were again non-randomly distributed with classes including ‘other enzyme activity’, ‘transporter activity’ and ‘structural molecules’ over-represented. As has been shown very recently, the involvement of hormones other than ABA in controlling the plant’s response was clearly demonstrated, with auxin transport and metabolism proteins being over-represented within the down-regulated genes (Lee et al. 2005). There were numerous ribosomal proteins, aquaporins and ATPases observed among the down-regulated genes.

Selected GO-Slim classification of Arabidopsis genes, which are either (A) up-regulated or (B) down-regulated in Thlaspi arvense in response to cold stress. The black and white bars indicate the observed and expected number of genes for each functional class, respectively. The asterisks indicate the significance of the resulting χ2 test for each class: *p < 0.05, **p < 0.01 and ***p < 0.001. The white asterisks indicate those tests with less reliability due to the low number of genes present within the classes
The detected changes in gene regulation were overlaid onto known metabolic pathways in A. thaliana using the Pathway Tools Omics Viewer at TAIR (http://www.arabidopsis.org) to further assist in identifying key processes that were altered in response to cold stress. Several pathways involved in stress responses, specifically those involved with oxidative stress were found to be up-regulated, including the phenylpropanoid pathway, the flavanoid biosynthesis pathway and the ascorbate glutathione pathway. Conversely, the control of de novo nucleotide biosynthesis appeared to be down-regulated. Strikingly, eight genes of the sulphate assimilation pathway were found to be up-regulated, representing six of eight steps leading to the generation of S-adenosyl methionine (Fig. 3). This pathway has not been shown previously to play a role in cold stress and none of the genes were found to up-regulated among the five A. thaliana microarray experiments.

Up-regulation of the sulphate assimilation pathway in response to cold stress in Thlaspi arvense. (A) Table listing the genes identified as significantly up-regulated from the microarray analysis with the respective time point at which the difference was observed and the corresponding fold change and q-value (probability that the differential expression occurs by chance). (B) Diagram of the sulphate assimilation pathway. For each of the identified Arabidopsis thaliana genes where we had an available cDNA clone (underlined), the corresponding transcript profile in response to cold treatment based on northern analysis in Thlaspi arvense is shown adjacent to the gene
Confirmation of microarray results
Common approaches used to confirm microarray data are in silico comparative analysis and quantification of transcript abundance through either northern analysis or reverse transcription polymerase chain reaction (RT-PCR). The comparison with the publicly available A. thaliana microarray data provided confirmatory evidence that a significant proportion of the identified genes were changing in response to cold stress. However, those genes, which demonstrated a potentially disparate expression pattern between the two species and novel genes identified in this analysis warrant further study. Initial confirmation was carried out using semi-quantitative RT-PCR for ten genes, nine up-regulated and one down-regulated in T. arvense. Eight of these genes exhibited similar expression profiles in A. thaliana from the in silico comparison, while two, At3g02750 (a protein phosphatase, PP2C) and At1g25440 (a CONSTANS domain Zn finger family protein) showed no significant difference in gene expression (Fig. 4). In general, the expected expression patterns in both species were detected, including the two genes that had not been shown to be cold regulated in A. thaliana, although it did appear that PP2C (At3g02750) was up-regulated in A. thaliana, similar to T. arvense. The genes showed similar trends for RT-PCR amplification across the two species, although three of the genes, namely At2g17840 (senescence associated protein), At4g11600 (glutathione peroxidase-like protein) and At1g09070 (SRC2-like gene) showed markedly lower expression under control conditions and were up-regulated more rapidly in T. arvense compared to A. thaliana (Fig. 4).

RT-PCR analysis of the expression patterns of ten cold responsive genes in Arabidopsis thaliana and Thlaspi arvense. RT-PCR analysis was carried out for each gene using RNA extracted from plants grown as described in the Materials and methods, at zero time and after moving plants to a 3°C chamber for the time points shown. Changes in gene expression were estimated with reference to the level of the housekeeping gene, β-tubulin, which was amplified at the same time (one example shown)
It was not always possible to design functional primers for both A. thaliana and T. arvense, due to a lack of available sequence data and divergence between the two species. Cross-species northern blot analysis was carried out for further genes for which there was an available T. arvense or B. napus homologous EST sequence, allowing the expression profiles to be assayed in all three species. The results for three genes are shown in Fig. 5, these included At4g03210 (putative xyloglucan xyloglucosyl transferase, B. napus EST clone, RL1216), At4g08950 (phosphate-responsive protein, B. napus EST clone, CL4103) and At2g36880 (putative S-adenosylmethionine synthetase, T. arvense EST clone, TA150). Each of these genes were expected to be up-regulated in T. arvense but based on the public microarray data were predicted to be either down-regulated or to show no response in A. thaliana. Homologues of At4g03210 were found to be significantly up-regulated in both B. napus and T. arvense within hours of moving the plants to low temperatures, but no change was observed in A. thaliana. A similar trend was observed for At4g08950 with the most obvious increase being observed for B. napus. In the case of At2g36880 the transcript only appeared to cold responsive in T. arvense. The cold responsive expression profiles of an additional 14 T. arvense genes was confirmed by northern analysis and included six of the genes identified as cold responsive from the sulphate assimilation pathway (Fig. 3 and data not shown).

Northern blot analysis for three genes identified as up-regulated in Thlaspi arvense in response to cold stress. The three panels show the response to cold stress (growth at 3°C) for the times indicated in Arabidopsis thaliana, Brassica napus and Thlaspi arvense, respectively. Ethidium bromide stained total RNA is shown to demonstrate evenness of loading
Discussion
Arabidopsis thaliana has long been used as a model for the acclimation process in plants and numerous A. thaliana mutants affected in their innate ability to cold acclimate have been identified (reviewed in Xin and Browse 2000). Further, numerous genes have been demonstrated to respond to low temperatures. Such cold-regulated genes, including the key regulatory transcription factor CBF, have been found and are similarly responsive in a diverse range of plant species, each of which vary in their ability to cold acclimate. T. arvense was shown to have a greater ability to withstand freezing temperatures than either the model crucifer A. thaliana or the cultivated crop species B. napus. Although it is possible that T. arvense has evolved divergent mechanisms for surviving in its harsh indigenous environment, it is probable that T. arvense has adapted the existing mechanisms found within its closely related Brassicaceae species.
A microarray approach was taken to address possible differences in gene regulation in response to cold stress in T. arvense. Presently, there are few endeavours to provide genomics resources for wild plant species that may harbour some of the more interesting allelic variation that could be exploited for crop improvement. Without access to genomics resources for a species of interest it is becoming commonplace to attempt to exploit information and technologies developed for model species, such as A. thaliana. A small EST sequencing project was initiated to assess the level of homology between A. thaliana and T. arvense, this established an average sequence identity of 90% shared between the two species. The level of similarity was sufficient to warrant testing the efficiency of utilising the Arabidopsis whole genome long oligomer microarrays that allow the expression of over 26,000 genes to be assayed at one time. The arrays were used to query three early time points in the response of T. arvense to cold stress. The application of stringent analysis criteria meant that on average only 15.9% of the target sequences could be consistently studied at each time point comparison. This is similar to results from an experiment conducted at the Nottingham Arabidopsis Stock Centre (http://affymetrix.arabidopsis.info/), where Arabidopsis Affymetrix arrays were used to assay gene expression in T. arvense resulting in 14.3% of the targets being reliably detected across five replicates. Although the low-hybridisation efficiency may limit the depth of the analysis, this still allowed the expression of at least 4,000 genes to be queried with confidence, in an uncharacterised crucifer species, of which, nearly 600 genes were shown to be responding to cold stress in T. arvense.
Microarray expression data are generally validated through experimental determination of transcript abundance, and RT-PCR and northern analysis were used to confirm some of the observed changes in gene expression in T. arvense. However, with the plethora of publicly available microarray data, particularly data studying the response of A. thaliana to low temperatures, a comparison with these available data sets allowed more extensive validation of the changes in the T. arvense transcriptome. Five microarray experiments conducted using A. thaliana were considered, each of which employed a different set of growth conditions, with plants grown hydroponically, on plates or in soil, with a range in light intensities and day lengths, the only constant being the cold stress treatment (Fowler and Thomashow 2002; Kreps et al. 2002; Lee et al. 2005; Vogel et al. 2005; http://affymetrix.arabidopsis.info/). It was anticipated that reproducible changes between these experiments and between the two species would represent conserved cold responsive genes. Over 200 of the differentially regulated genes in T. arvense were also identified as cold responsive in one or more of the A. thaliana experiments. Interestingly, the majority of these genes, 131 were only identified in one of five experiments, while the remainder were identified as cold regulated in two or more of the different A. thaliana expression studies. The lack of corroboration across the A. thaliana studies, could be the result of the variable growing conditions, or could reflect the complexity and subtlety of the low-temperature response.
Almost 50% of the cold responsive genes in T. arvense were found to be similarly regulated by cold in A. thaliana suggesting the wild crucifer reacts to low-temperature stress in a manner similar to A. thaliana. However, functional characterisation through GO analysis and studying contributing metabolic pathways for the genes identified in this study has uncovered novel responses to the low temperature stress some of which appear to be unique to T. arvense.
A number of the down-regulated genes appeared to contribute to the control of water balance within the cell. It has previously been shown that the majority of the plasma membrane aquaporins are down-regulated in response to cold in A. thaliana (Jang et al. 2004). Similarly, three members of this gene family were identified as down-regulated among the T. arvense data. Genes implicated in controlling plant growth were also found to be down-regulated. This included members of the auxin induced family of proteins (IAA9, IAA18, IAA7), the auxin transporter PIN3 and GASA4 that responds to gibberellic acid. A significant number of ribosomal proteins were also down-regulated. Previously, a mutant with reduced expression of one of these genes At1g79850 (30S ribosomal protein S17) was shown to exhibit reduced leaf growth and delayed leaf senescence, which was associated with increased membrane integrity and reduced chlorophyll fluorescence over the life span of the line (Woo et al. 2002). This would be consistent with the observation that of five senescence associated genes found to respond to cold stress, four were down-regulated in our analysis. Reduction in plant growth is a common feature of cold stress. Additionally, loss of membrane integrity and photoinhibition caused by an imbalance between the light reactions of photosystem (PS)II and the dark reactions of PSI leads to cell damage within cold sensitive plants. It will be interesting to determine if one of the controlling elements in a plant’s capacity to withstand cold stress is the ability to tightly regulate what might appear to be inconsequential house-keeping genes.
The genes demonstrated to be up-regulated in T. arvense also produced some interesting candidates for further analysis and suggested that novel pathways may be induced under cold stress conditions. The most striking observation was the up-regulation of genes in the pathway controlling sulphate assimilation (Fig. 3). None of the known sulphate transporters appeared to be up-regulated which suggested that the response was not reflecting sulphate nutrition. The ability of the plant to control the level of oxidative stress, which results from cold stress, has been shown to be a factor in a plant’s innate freezing tolerance (Thomashow 1999; Kocsy et al. 2000). It could be speculated that the increase in sulphate assimilation is due to a requirement for larger pools of glutathione, which is generated from cysteine. However, at least three of the steps subsequent to cysteine, leading to the accumulation of S-adenosyl-methionine (AdoMet), are up-regulated in the pathway. This indicates that increasing AdoMet may be an important factor in T. arvense’s response to cold stress. AdoMet is an important methyl-donor, which is involved in the production of numerous secondary metabolites, including phenylpropanoids, polyamines and the phytohormone ethylene (Ravanel et al. 1998). The potential role of polyamines in the cold response of plants has been previously suggested but it is possible that AdoMet may have additional roles in this process (Nadeau et al. 1987; Mo and Pua 2002; Kasukabe et al. 2004). Four annotated methyl-transferases were found among the significantly up-regulated genes in T. arvense, two of which (At1g48600 and At5g54160) are known to be AdoMet dependent and are involved in choline and flavanoid biosynthesis, respectively. Further, AdoMet is essential for vitamin E biosynthesis and VTE1, one of the key genes in this pathway is also up-regulated in T. arvense. Vitamin E has recently been shown to serve two important roles during cold stress, protecting PSII from photo-inhibition and limiting photo-oxidative damage of lipid membranes (Havaux et al. 2005). AdoMet also may play a role in altering cell wall composition during cold stress through its role in lignin biosynthesis; recent work in tomato has suggested that lignification of the xylem during salt stress assists the plant in maintaining water flow under negative pressure (Sánchez-Aguayo et al. 2004). The recent release of a large collection of ESTs developed from salt, drought and cold treated libraries of the ‘extremophile’ Thellungiella salsuginea (Wong et al. 2005) suggested the up-regulation of sulphate assimilation may not be restricted to the crucifer T. arvense. The available T. salsuginea EST sequences were downloaded from NCBI and compared to the A. thaliana coding sequences using BLASTN only. By considering the most significant hit in A. thaliana to be the primary homologue, 6,529 EST sequences were found to identify 3,209 A. thaliana gene codes at the nucleotide level, while 317 had no detectable homologue. The ESTs whose primary homologue was a gene within the sulphate assimilation pathway were identified and based on a comparison with the distribution of contig sizes across all the EST sequences, it appeared that the genes within this pathway were over-represented within the T. salsuginea stress treated libraries.
While gene expression analysis using microarrays has been widely adopted for studying a plant’s response to a multitude of environmental stimuli, it has been predominantly limited to the study of either model species or those species with large EST collections. The ability to exploit the extensive resources developed for the model crucifer A. thaliana in the study of its wild relatives would provide an avenue for harnessing the wide spectrum of variation found within the Crucifer family. The present study has identified a number of novel genes, some of which are differentially expressed between A. thaliana and T. arvense and represent good candidates for further study. This work indicates that the widely available A. thaliana genome oligoarrays can be used to study gene regulation in related species.
References
Bacher M, Talke IN, Krall L, Kramer U (2004) Cross species microarray transcript profiling reveals high constitutive expression of metal homeostasis genes in shoots of the zinc hyperaccumulator Arabidopsis halleri. Plant J 37:251–268
Best KF, McIntyre GI (1975) The biology of Canadian weeds, 9. Thlapsi arvense L. Can J Plant Sci 55:279–292
Boyer JS (1982) Plant productivity and environment. Science 281:443–448
Chen W, Provart NJ, Glazebrook J, Katagiri F, Chang HS, Eulgem T, Mauch F, Luan S, Zou G, Whitham SA, Budworth PR, Tao Y, Xie Z, Chen X, Lam S, Kreps JA, Harper JF, Si-Ammour A, Mauch-Mani B, Heinlein M, Kobayashi K, Hohn T, Dangl JL, Wang X, Zhu T (2002) Expression profile matrix of Arabidopsis transcription factor genes suggests their putative functions in response to environmental stresses. Plant Cell 14:559–574
Chinnusamy V, Schumaker K, Zhu JK (2004) Molecular genetic perspectives on cross-talk and specificity in abiotic stress signalling in plants. J Exp Bot 55:225–236
Chou C-C, Chen C-H, Lee T-T, Peck K (2004) Optimisation of probe length and the number of probes per gene for optimal microarray analysis of gene expression. Nucl Acids Res 32:e99
Fowler S, Thomashow MF (2002) Arabidopsis transcriptome profiling indicates that multiple regulatory pathways are activated during cold acclimation in addition to the CBF cold response pathway. Plant Cell 14:1675–1690
Galloway GL, Malmberg RL, Price RA (1998) Phylogenetic utility of the nuclear gene arginine decarboxylase: an example from Brassicaceae. Mol Biol Evol 15:1312–1320
Gilmour SJ, Hajela RK, Thomashow MF (1998) Cold acclimation of Arabidopsis thaliana. Plant Physiol 879:745–750
Gong Q, Li P, Ma S, Indu Rupassara S, Bohnert HJ (2005) Salinity stress adaptation competence in the extremophile Thellungiella halophila in comparison with its relative Arabidopsis thaliana. Plant J 44:826–839
Griffith M, McIntyre HCH (1993) The interrelationship between growth and frost tolerance in winter rye. Physiol Plant 87:335–344
Guy CL (1990) Cold acclimation and freezing tolerance: role of protein metabolism. Ann Rev Plant Physiol Mol Biol 41:187–223
Havaux M, Eymery F, Porfirova S, Rey P, Dormann P (2005) Vitamin E protects against photoinhibition and photooxidative stress in Arabidopsis thaliana. Plant Cell 17:3451–3469
Jaglo-Ottosen KR, Gilmour SJ, Zarka DG, Schabenberger O, Thomashow MF (1998) Arabidopsis CBF1 overexpression induces COR genes and enhances freezing tolerance. Science 280:104–106
Jang JY, Kim DG, Kim YO, Kim JS, Kang H (2004) An expression analysis of a gene family encoding plasma membrane aquaporins in response to abiotic stresses in Arabidopsis thaliana. Plant Mol Biol 54:713–725
Kane MD, Jatkoe TA, Stumpf CR, Lu J, Thomas JD, Madore SJ (2000) Assessment of the sensitivity and specificity of oligonucleotide (50mer) microarrays. Nucl Acids Res 28:4552–4557
Kasuga M, Liu Q, Miura S, Yamaguchi-Shinozaki K, Shinozaki K (1999) Improving plant drought, salt, and freezing tolerance by gene transfer of a single stress-inducible transcription factor. Nat Biotechnol 17:287–291
Kasukabe Y, He L, Nada K, Misawa S, Ihara I, Tachibana S (2004) Overexpression of spermidine synthase enhances tolerance to multiple environmental stresses and up-regulates the expression of various stress-regulated genes in transgenic Arabidopsis thaliana. Plant Cell Physiol 45:712–722
Kocsy G, Szalai G, Vágújfalvi A, Stéhli L, Orosz G, Galiba G (2000) Genetic study of glutathione accumulation during cold hardening in wheat. Planta 210:295–301
Kreps JA, Wu Y, Chang HS, Zhu T, Wang X, Harper JF (2002) Transcriptome changes for Arabidopsis in response to salt, osmotic, and cold stress. Plant Physiol 230:2129–2141
Laroche A, Geng X-M, Singh J (1992) Differentiation of freezing tolerance and vernalization responses in Cruciferae exposed to low temperatures. Plant Cell Environ 15:439–445
Larsson O, Wahlestedt C, Timmons JA (2005) Considerations when using the significance analysis of microarrays (SAM) algorithm. BMC Bioinformatics 6:129
Lee H-S, Wang J, Tian L, Jiang H, Black MA, Madlung A, Watson B, Lukens L, Pires JC, Wang JJ, Comai L, Osborn TC, Doerge RW, Chen ZJ (2004) Sensitivity of oligonucleotides and cDNAs for microarray analysis of gene expression in Arabidopsis and its related species. Plant Biotech J 2:55–57
Lee BH, Henderson DA, Zhu JK (2005) The Arabidopsis cold-responsive transcriptome and its regulation by ICE1. Plant Cell 17:3155–3175
Levitt J (1980) Response of plants to environmental stresses, 2nd edn. Academic Press, New York
Mo H, Pua EC (2002) Up-regulation of arginine decarboxylase gene expression and accumulation of polyamines in mustard (Brassica juncea) in response to stress. Physiol Plant 114:439–449
Nadeau P, Delaney S, Chouinard L (1987) Effects of cold hardening on the regulation of polyamine levels in wheat (Triticum aestivum L.) and Alfalfa (Medicago sativa L.). Plant Physiol 84:73–77
Parkin IAP, Gulden SM, Sharpe AG, Lukens L, Trick M, Osborn TC, Lydiate DJ (2005) Segmental structure of the Brassica napus genome based on comparative analysis with Arabidopsis thaliana. Genetics 171:765–781
Ravanel S, Gakiere B, Job D, Douce R (1998) The specific features of methionine biosynthesis and metabolism in plants. Proc Natl Acad Sci USA 95:7805–7812
Sánchez-Aguayo I, Rodríguez-Galán JM, García R, Torreblanca J, Pardo JM (2004) Salt stress enhances xylem development and expression of S-adenosyl-L-methionine synthase in lignifying tissues of tomato plants. Planta 220:278–285
Taji T, Seki M, Satou M, Sakurai T, Kobayashi M, Ishiyama K, Narusaka Y, Narusaka M, Zhu JK, Shinozaki K (2004) Comparative genomics in salt tolerance between Arabidopsis and Arabidopsis-related halophyte salt cress using Arabidopsis microarray. Plant Physiol 135:1697–1709
Thomashow MF (1999) Plant cold acclimation: freezing tolerance genes and regulatory mechanisms. Ann Rev Plant Physiol Plant Mol Biol 50:571–599
Tusher VG, Tibshirani R, Chu G (2001) Significance analysis of microarrays applied to the ionizing radiation response. Proc Natl Acad Sci USA 98:5116–5121
Van Buskirk HA, Thomashow MF (2006) Arabidopsis transcription factors regulating cold acclimation. Physiol Plant 126:72–80
Vogel JT, Zarka DG, Van Buskirk HA, Fowler SG, Thomashow MF (2005) Roles of the CBF2 and ZAT12 transcription factors in configuring the low temperature transcriptome of Arabidopsis. Plant J 41:195–211
Wang HY, Malek RL, Kwitek AE, Greene AS, Luu TV, Behbahani B, Frank B, Quackenbush J, Lee NH (2003) Assessing unmodified 70-mer oligonucleotide performance on glass-slide microarrays. Genome Biol 4(1):R5
Webb MS, Uemura M, Steponkus PL (1994) A comparison of freezing injury in oat and rye: two cereals at the extremes of freezing tolerance. Plant Physiol 104:467–478
Wong CE, Li Y, Whitty BR, Diaz-Camino C, Akhter SR, Brandle JE, Golding GB, Weretilnyk EA, Moffatt BA, Griffith M (2005) Expressed sequence tags from the Yukon ecotype of Thellungiella reveal that gene expression in response to cold, drought and salinity shows little overlap. Plant Mol Biol 58:561–574
Woo HR, Goh CH, Park JH, Teyssendier de la Serve B, Kim JH, Park YI, Nam HG (2002) Extended leaf longevity in the ore4-1 mutant of Arabidopsis with a reduced expression of a plastid ribosomal protein gene. Plant J 31:331–340
Xin Z, Browse J (2000) Cold comfort farm: the acclimation of plants to freezing temperatures. Plant Cell Environ 23:893–902
Yamada K, Lim J, Dale JM, Chen H, Shinn P, Palm CJ, Southwick AM, Wu HC, Kim C, Nguyen M et al (2003) Empirical analysis of transcriptional activity in the Arabidopsis genome. Science 302:842–846
Zhou XJ, Gibson G (2004) Cross-species comparison of genome-wide expression patterns. Genome Biol 5:232
Acknowledgements
The authors would like to thank Drs Steve Robinson and Dwayne Hegedus for critical reading of this manuscript and Drs Branimir Gjetvaj and Andrew Sharpe for assistance with EST development. This work was funded in part by the Saskatchewan Agricultural Development Fund and in part by the Genome Prairie project ‘Functional Genomics of Abiotic Stress’.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Rights and permissions
About this article
Cite this article
Sharma, N., Cram, D., Huebert, T. et al. Exploiting the wild crucifer Thlaspi arvense to identify conserved and novel genes expressed during a plant’s response to cold stress. Plant Mol Biol 63, 171–184 (2007). https://doi.org/10.1007/s11103-006-9080-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11103-006-9080-4