
Abstract
Purpose
Corticotroph tumor progression (CTP) or Nelson’s syndrome (NS) can occur in patients with Cushing’s disease (CD) following bilateral adrenalectomy. It has rarely been observed in patients treated with long-term medical therapy for persistent CD. Osilodrostat (LCI699) is a new steroidogenesis inhibitor of 11β-hydroxylase (CYP11β1) that induced remission of hypercortisolism in 86% of patients with refractory CD in the randomized placebo-controlled trial LINC-3 (NCT02180217).
Methods
A 40-year-old woman with persistent CD following transsphenoidal surgery was treated with osilodrostat in the LINC-3 trial and was followed with regular hormonal assessments and imaging of residual corticotroph tumor.
Results
Under oral therapy with osilodrostat 10 mg twice daily, urinary free cortisol (UFC) normalized and clinical signs of CD regressed during therapy. However after 4 years of treatment, ACTH levels increased from 73 to 500 pmol/L and corticotroph tumor size increased rapidly from 3 to 14 mm, while UFCs remained well controlled. Surgical resection of an atypical tumor with weak ACTH expression and increased proliferative index (Ki-67 ≥ 8%) resulted in current remission but will require close follow-up.
Conclusion
This case highlights the importance of monitoring ACTH and corticotroph tumor size in patients with persistent CD, either under effective treatment with steroidogenesis inhibitors or after bilateral adrenalectomy.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Cushing’s disease (CD) resulting from an ACTH secreting pituitary corticotroph tumor is the leading cause of endogenous hypercortisolism, accounting for 70% of cases [1]. Resection of the adenoma by transsphenoidal surgery is the first line therapy, with remission rates varying between 65 and 90%, but with long-term recurrence rates of up to 30% [2,3,4,5]. Management of refractory CD includes repeated pituitary surgery, radiotherapy, medical therapy and bilateral adrenalectomy [4, 5]. Nelson’s syndrome (NS) is characterized by corticotroph tumor progression (CTP) with high plasma ACTH levels and hyperpigmentation, which can occur as a serious complication following bilateral adrenalectomy. According to recent studies, the occurrence of CTP following bilateral adrenalectomy is estimated to be approximately 29% [6]. Only rare cases of mild CTP have been reported with medical therapies that lower cortisol levels such as ketoconazole and mitotane [7, 8].
Osilodrostat (LCI699) is a new steroidogenesis inhibitor that reduces cortisol production by inhibiting the enzyme 11β-hydroxylase (CYP11B1) [3]. Its efficacy was demonstrated in the randomized placebo-controlled trial LINC-3 (NCT02180217), where it achieved normalization of urinary free cortisol (UFC) in 86% of patients with refractory CD [9]. In this study, osilodrostat did not adversely affect the size of pituitary tumors over 48 weeks of follow-up, with small proportions of patients having either a decrease or an increase of 20% or more in tumor volume from baseline [9]. We now describe the case of a patient with persistent CD who participated in the LINC-3 trial and presented important CTP after 4 years of long-term effective treatment with osilodrostat.
Materials and methods
The investigation of this patient in the LINC-3 trial was approved by the ethics committee of Centre hospitalier de l’Université de Montréal (CHUM) and the patient provided informed signed consent to participate in the study and to publish these results. During the patient's participation in LINC-3 trial, biochemical and hormonal analyses were sent to the central laboratory designated by the study protocol (Q2 Solutions, Global Laboratory Services, Morrisville, NC, USA), where UFC was measured by liquid chromatography-tandem mass spectrometry (LC-MSMS; normal range 11–138 nmol/d) and plasma ACTH by immunoassay (Immulite 2000 ACTH kit; upper limit of normal (ULN) males = 11.1 pmol/L, females = 6.0 pmol/L). When samples were collected at CHUM before and after the LINC-3 trial, UFC was also measured by LC-MSMS coupled to on line solid phase extraction (ULN < 120 nmol/d) and ACTH was measured with an immunoradiometric assay (ELSA-ACTH, Cisbio Bioassays, Codolet, France) that was associated with a coefficient of variation (CV) of 8.7%. UFCs and ACTH blood levels were collected every 2–4 weeks during the study phase, then every 3 months during long-term treatment. Pituitary imaging was performed using a 3-T MRI with gadolinium enhancement and 2 mm slice thickness at baseline, week 24 and week 48, then every 6–12 months during extended LCI699 therapy. All the data was collected retrospectively using research files and digital records from the hospital.
Initial evaluation of the patient
A 40-year old white female was referred to our service for suspected Cushing’s syndrome. During the previous year, she had gained 20 kg despite attempting to reduce caloric intake and exercising. She reported fatigue, insomnia, difficulty concentrating and depressive symptoms. Hypertension was diagnosed two years earlier and was poorly controlled by a thiazide; recent onset diabetes was treated with metformin, with an HbA1C of 6.4%. Her previous medical history was also significant for a deep vein thrombosis and pulmonary embolism at age 37 treated with warfarin. She denied alcohol intake or the use of corticosteroids. There was no family history of diabetes, hypertension or other endocrinopathies. On physical examination, central obesity, round face, plethora and enlarged supraclavicular fat pads were noted. Blood pressure was 150/100, weight 125 kg and BMI 44 kg/m2. Mild proximal muscle weakness was found, but there was no oedema, hirsutism or bruising. Basal blood biochemistry values were unremarkable and showed no hypokalemia. Following overnight 1 mg dexamethasone suppression test (DST), plasma cortisol was elevated at 235 nmol/L (N < 50 nmol/L). Late-night salivary cortisol values were also elevated ranging from 13 to 17 nmol/L (N < 7 nmol/L) and 24-h UFCs varied between 114 and 405 nmol/d (N < 120 nmol/d). Plasma ACTH was not suppressed at 9.3 pmol/L (N = 2.0–11.0 pmol/L). A high-dose DST (4 mg IV) showed a reduction in cortisol and ACTH, followed by a rebound the next morning, compatible with Cushing’s disease (Table 1) [10]. Pituitary MRI revealed a 3.3 mm right lesion with mild cerebral atrophy (Fig. 1a). CRH and desmopressin stimulation tests showed increases in ACTH of 250% and 206%, and cortisol of 40% and 48% respectively (Tables 2 and 3). Inferior petrosal sinus sampling demonstrated a central to peripheral ACTH gradient of 58 at baseline and 244 following desmopressin stimulation with a right side lateralization, confirming the pituitary source of ACTH excess. Transsphenoidal surgery was performed after placement of an inferior vena cava filter, as a second episode of deep vein thrombosis and pulmonary embolism had occurred three weeks pre-operatively. The neurosurgeon identified a tumor on the left side of the pituitary with semi-solid, milky consistency that was resected. Pathology studies identified no distinct adenoma tissue, but only Crooke’s cell changes. Immediate post-operative evaluation included a morning cortisol of 422 nmol/L and diabetes insipidus, which became permanent. Hydrocortisone supplementation was discontinued a few days after the surgery and UFCs remained elevated at ≥ 3 × ULN. Treatment with ketoconazole was rapidly initiated and second line therapeutic options were discussed with the patient and at the institutional multidisciplinary pituitary tumor board. Considering post-operative MRI findings of uncertain residual tumor or scar tissue and the occurrence of two recent thromboembolic events, medical therapy with a drug that could offer the highest likelihood of remission for hypercortisolism was recommended. Since preliminary results from the osildrostat phase II study showed a nearly 90% control rate of hypercortisolism, it was proposed to the patient to be enrolled in the phase III LINC-3 trial assessing the efficacy of LCI699 in refractory Cushing’s disease.

Pituitary MRI before and after initial transsphenoidal surgery. a Coronal T1-weighted post-gadolinium image shows a 3 mm hypoenhancing focus involving the right aspect of the pituitary gland suggestive of a microadenoma (arrow). b Coronal T1-weighted post-gadolinium image performed 3 months post-operatively reveals a 3 mm hypoenhancing focus involving the right aspect of the pituitary gland suggestive of a residual microadenoma (arrow)
Medical therapy with osilodrostat for persistent CD
According to the study protocol, osilodrostat was initiated at the dose of 2 mg twice daily with increments every two weeks to 5 mg then 10 mg twice daily, based on efficacy and tolerability until UFC was normalized. At baseline, the patient’s UFCs varied between 2.47 and 2.90 × ULN, ACTH was 12.9 pmol/L and pituitary MRI indicated the presence of a right 3 mm tumor residue or scar tissue (Fig. 1b). After only 6 weeks of osilodrostat, UFC had normalized and the patient remained on a stable dose of 10 mg twice daily until the end of the study. She noted progressive improvement in neuropsychiatric symptoms including fatigue, sleep pattern and emotional lability; physical signs of Cushing’s also improved as she lost 5 kg during the treatment. The patient had no significant adverse event, particularly no adrenal insufficiency, hirsutism or hypokalemia. During the first three years of osilodrostat therapy, plasma ACTH remained between 20 and 40 pmol/L and MRI performed every 6 months showed a stable 3–3.5 mm pituitary adenoma. After 3 years (156 weeks) of treatment, ACTH began to increase from 24.9 to 72.6 pmol/L, while UFCs remained in the normal range with the same dose of osilodrostat (Fig. 2). Pituitary MRI performed at 3.5 years revealed that the tumor had grown to 5 mm (Fig. 3a). Thereafter, ACTH and tumor size continued to increase; after 4 years of treatment, ACTH had gone up to 159 pmol/L and tumor had nearly tripled in size to 14 × 6 × 12 mm, abutting 25% of the cavernous carotid, without chiasmatic compression (Fig. 3b). The patient presented no headache, visual field deficit or hyperpigmentation. Considering the rapid corticotroph tumor progression and elevated ACTH levels, the multidisciplinary institutional pituitary tumor board recommended a second pituitary surgery. Osilodrostat was stopped after a total of 4 years and 3 months (222 weeks) since its initiation. On the morning of the second transsphenoidal surgery, ACTH had reached 500 pmol/L. Despite the presence of substantial amount of scar tissue, the neurosurgeon was able to remove a pinkish semi-liquid tumor on the right side of the gland. Pathology examination identified a pituitary adenoma characterized by loss of acinary architecture, mitoses and strongly basophilic cells; there was weak ACTH expression and an elevated proliferation index (Ki-67 ≥ 8%) (Fig. 4). Following surgery, morning cortisol on post-op day three was 33 nmol/L. A 10 mcg desmopressin (dDAVP) stimulation test performed two months after the surgery induced no rise in cortisol levels but showed a significant ACTH increase, suggesting possible residual tumor cells (Table 4) [11]. At last follow-up 6 months after the surgery, the patient was still on hydrocortisone tapering with a low morning cortisol (< 16 nmol/L) and normal ACTH (8.0 pmol/L); her antihypertensive medication had been reduced by half and all of her oral hypoglycemic agents were stopped with normal glucose levels. MRI showed complete resection without any sign of residual tumor (Fig. 3c).

Evolution of urinary free cortisol (a; normal 11–138 nmol/d), plasma ACTH (b; normal < 6.0 pmol/L) and morning plasma cortisol (c; normal 127–567 nmol/L) before and after initiating osilodrostat therapy (week 0). Baseline cortisolurias were very erratic, varying from normal range (shaded area) to 405 nmol/d, concordant with cyclical Cushing’s syndrome. After a first failed transsphenoidal surgery (week-20, left arrow), ketoconazole was briefly initiated then stopped 4 weeks before beginning osilodrostat at the dose of 2 mg bid; it was then increased every 2 weeks to 5 mg bid, then 10 mg bid until UFC was normalized. After 6 weeks, UFC remained in the normal range with the same dose of 10 mg bid throughout the treatment period. Plasma ACTH was initially stable but showed a first sharp increase at 156 weeks (24.9–72.6 nmol/L) and continued to rise, reaching 500 pmol/L on the day of the second transsphenoidal surgery for corticotroph tumor progression (right arrow); osilodrostat was stopped at week 222. A low post-operative morning plasma cortisol (33 nmol/L) indicated remission of hypercortisolism

Pituitary MRI under treatment with osilodrostat. a Coronal T1-weighted post-gadolinium image after 3 years of LCI699 therapy shows interval growth to 5 mm of the right-sided recurrent adenoma (arrow). b Coronal T1-weighted post-gadolinium image performed after 4 years of osilodrostat therapy demonstrates substantial growth of the right-sided adenoma (arrow) now reaching 14 mm in maximal diameter with extension toward the cavernous sinus (Knosp grade 1). c Coronal T1-weighted post-gadolinium image following the second surgical resection demonstrates gross total resection of the adenoma

Histology of corticotroph tumor progression. a Strongly staining basophilic cells (200X). b Weak ACTH expression (400X). c High proliferation index (Ki-67 ≥ 8%) suggesting agressive tumoral behavior (400X)
Discussion
This patient is the first described case of corticotroph tumor progression requiring surgical intervention after long-term treatment with osilodrostat for persistent CD. CTP, or NS, is defined as the enlargement of an ACTH-secreting pituitary tumor that can occur following bilateral adrenalectomy. The classic clinical triad is characterized by a rapidly progressive pituitary mass that can lead to invasion and cranial nerves compression, accompanied by high ACTH and cutaneous hyperpigmentation. Diagnostic criteria for NS vary in the literature, depending on which imaging modality was used at the time to describe the cases; the first case series used plain radiographs to detect sella turcica deformations, which were much less sensitive than MRI. Although some studies used criterions like size (≥ 2 mm progression in ≥ 1 dimension on MRI) or ACTH blood level, a recent expert consensus (Corticotroph tumor progression, “Munich Expert Consensus Recommendations”, manuscript in revision 2020) reviewed 80 publications on NS and established that "significant" progression or occurrence of a new pituitary tumor on MRI post-adrenalectomy was sufficient for diagnosis. No cut-off value for ACTH allowed accurate diagnosis, as results in NS series ranged from 133 to 899 pg/mL (29.3–198 pmol/L, mean 86.1 pmol/L) and assay methods varied among centers. Hyperpigmentation did not appear as a reliable criterion either, because it is difficult to quantify and varies according to ethnicity, sun exposure, polymorphisms in melanocytes MC1R receptors and possibly tumoral secretion of a mutated or inactive form of ACTH. Therefore, ACTH level and hyperpigmentation are considered secondary criteria, while clinically significant tumor growth or appearance of a new lesion on pituitary MRI constitutes the primary criteria to diagnose CTP/NS after bilateral adrenalectomy. Its incidence varies once again depending on imaging technique and diagnostic criteria applied in the studies. While older publications reported a mean incidence of 29% (8–53%) after adrenalectomy, availability of MRI and use of more consistent diagnostic criteria since 2007 have raised the incidence to 42% (24–53%). The time delay between adrenalectomy and CTP when using modern imaging is on average 2.5 years (0.2–8.0) and most patients had their tumor progression within the first 3 years after surgery in one of the largest recent series [6]. Risk factors for CTP/NS include high ACTH levels (> 200 pg/mL or > 44 pmol/L) in the first year after adrenalectomy—especially if there is a rapid increase; a documented pituitary tumor on MRI at the time of adrenalectomy and young age (< 35 years old). Pituitary radiotherapy was reported to have a protective effect on the incidence of CTP/NS in small retrospective studies, but results are conflicting and prospective randomized data are lacking (Munich consensus).
Pathophysiology of CTP/NS is not fully understood. Controversy exists as whether tumor progression is caused by loss of cortisol negative feedback, or whether it is driven by genetic mechanisms implicated in corticotroph tumor development, implying that CTP/NS is a part of the natural history of Cushing’s disease. It is well known that corticotroph tumor cells are partially resistant to glucocorticoid feedback, as shown by abnormal dexamethasone suppression tests [10]. Thus, for the same level of cortisol, corticotroph tumor cells would proliferate and secrete more ACTH than normal corticotrophs; this could be exacerbated in situations where cortisol is low or absent, such as bilateral adrenalectomy and medical therapies that reduce cortisol or inhibits its actions. Mutations have been identified in corticotroph tumor cells, but no molecular marker or histologic feature has been found to predict the risk of developing CTP/NS when examining corticotroph adenoma specimens before adrenalectomy. Mutations of ubiquitin-specific protease 8 (USP8), a protein implicated in degradation of EGF receptors, have been found in 23–60% cases of Cushing’s disease, causing overexpression of EGFR at corticotrophs cell surface, which increases POMC expression and ACTH secretion [3, 12]. However, USP8 mutations are found at a similar frequency in Cushing's disease and CTP/NS, so they do not represent a distinctive pathophysiological feature. Nelson's syndrome tumors are typically more aggressive and have a higher proliferative index (Ki-67) than typical CD corticotroph tumors; they also have low p27 expression, a protein that inhibits CDKs (cyclin-dependent kinases) that regulate cell cycle, thereby promoting cell proliferation [13].
Nelson’s syndrome is classically associated with surgical bilateral adrenalectomy, which is the most drastic intervention to suppress endogenous cortisol production. Similar occurrence of corticotroph tumor progression has been described – to a lesser degree of severity—with medical therapies used for the treatment of hypercortisolism in Cushing's disease. Among 38 CD patients treated with ketoconazole, a new pituitary tumor appeared after 12–30 months in 5 out of 15 patients who had no visible tumor at baseline [7]. However, none of these patients had previous transsphenoidal surgery or radiotherapy. All these cases of CTP were then treated and cured by pituitary surgery. Authors mentioned that “no Nelson’s syndrome was observed in [their] study, although secondary visualization of previously non-visible lesions might be considered as an equivalent of NS in terms of mechanism.” In another study, 76 CD patients were treated with mitotane either as a first- or second-line therapy; among 48 subjects with no pituitary adenoma at baseline, 12 of them developed a new tumor after a median time of 10.9 months of treatment [8]. Only two out of these 12 patients had previous transsphenoidal surgery; all other subjects received mitotane as first-line therapy. Corticotroph tumor progression was also observed with the glucocorticoid receptor antagonist mifepristone in a prospective trial including 43 CD patients, of whom 42 were treated by surgery and 18 with radiotherapy [2]. Four cases of CTP (≥ 2 mm in any dimension) were documented: 3 macroadenomas (one of them being a large 4.6 cm silent corticotroph tumor that grew to 5.2 cm after 2.5 months) and 1 microadenoma (4 mm, occurring at 25 months of treatment in a patient who had no prior tumor at baseline). No risk factors for progression, such as baseline Ki-67, were identified and ACTH levels under mifepristone therapy were not predictive of CTP.
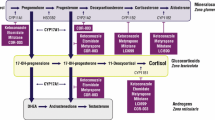
Osilodrostat (LCI699) is a new steroidogenesis inhibitor that blocks the enzyme 11β-hydroxylase (CYP11β1) at the final step of cortisol synthesis [3]. It also inhibits the 18-hydroxylase (CYP11β2) or aldosterone synthase, and it was first discovered as an inhibitor of aldosterone production [14]. It was initially studied in the treatment of hypertension and heart failure until 2013, after which its cortisol-lowering properties were more investigated. In vitro, LCI699 is significantly more potent than ketoconazole and metyrapone to reduce cortisol production in human adrenocortical cells [14]. In the phase II pilot study LINC-2 (NCT01331239), osilodrostat normalized UFC in 78.9% of 19 CD patients at 22 weeks [15]. The larger phase III randomized placebo-controlled trial LINC-3 (NCT02180217) included 137 patients with refractory Cushing’s disease who had a median UFC of 3.5 × ULN; 88% had previous transsphenoidal surgery and 15% had radiotherapy that failed to cure their hypercortisolism [9]. After 26 weeks of dose titration and 8 weeks of randomization to either continue LCI699 or placebo, significantly more osilodrostat receivers maintained a normal UFC (86.1% versus 29.4%, OR 13.7, p < 0.001). Ninety-six percent of enrolled patients had a UFC ≤ ULN at some point during the study after a median period of 41 days. Improvements were also observed in weight, blood pressure, diabetes and quality of life. Adverse events included nausea, headaches, fatigue and adrenal insufficiency (27.7%), especially during the first 12 weeks of LCI699 titration, and authors mentioned that a more gradual dose augmentation could be done in practice. Adverse effects like hypokalemia or hirsutism described in previous studies related to adrenal precursors accumulation, such as 11-deoxycorticosterone and androgens, were not significantly reported. Most of the adverse events were mild and easily controlled by dose reduction or corticosteroid supplementation, and only 2.9% of patients had to discontinue osilodrostat because of hypocortisolism.
Corticotroph tumor progression was also documented; among 137 patients included, 69 had a microadenoma and 13 had a macroadenoma at the beginning of the study. After 48 weeks of treatment, a similar proportion of patients had either ≥ 20% increase or ≥ 20% decrease in tumor volume (32.8% and 37.5% respectively). Among patients with a macroadenoma at baseline, 1.5% had ≥ 20% increase and 0.7% had ≥ 20% decrease in size. LCI699 was stopped in 2.9% of participants because of significant tumor enlargement. Following results of the LINC-3 trial, osilodrostat was approved in Europe and USA in January and March 2020 for the treatment of Cushing’s disease, and another long-term five years study LINC-4 (NCT02697734) is currently ongoing [14].
The patient described here developed a significant corticotroph tumor progression after 4 years of osilodrostat therapy, as tumor size nearly quadrupled to 14 mm coming in contact with the cavernous carotid artery, while ACTH increased from 72.6 to 500 pmol/L. This case illustrates that CTP can happen later during LCI699 treatment, even if there was no detectable change in tumor size after the first years of administration. Since hypercortisolism and clinical signs of Cushing’s were well controlled under osilodrostat, the only way to detect CTP was by monitoring pituitary MRI and plasma ACTH. As CTP/NS tumors can be aggressive and rapidly invasive, this highlights the importance of regular radiologic and biochemical follow-up for CD patients who are well controlled with osilodrostat. It also shows that a Nelson’s syndrome-like phenomenon is possible when hypercortisolism is inhibited under effective medical therapy, even without previous bilateral adrenalectomy. Previous studies have reported that medical therapy could induce corticotroph tumor growth in CD patients who had no visible adenoma at baseline, allowing surgical treatment and cure [7, 8]. However, we cannot conclude that CTP was solely due to osilodrostat therapy; as in other cases of Nelson’s, it is impossible to draw conclusions on the exact underlying mechanisms and we cannot exclude that CTP was a result of the natural history of this patient’s specific corticotroph tumor genotype. Long-term surveillance studies will be needed to evaluate the impact of osilodrostat on pituitary tumor size in large number of patients with CD. No genetic studies were performed on the pathology specimen of our case, as no particular mutation or molecular marker are known to be specifically related to CTP/NS in corticotroph tumors analysed before and after adrenalectomy [13]. However, Ki-67 index was elevated (≥ 8%), concordant with the more dedifferentiated and aggressive tumors found in this syndrome.
There are no guidelines for the follow-up of corticotroph tumor progression under osilodrostat therapy. The Munich Expert Consensus (manuscript submitted) proposed the following recommendations for the surveillance of CTP/NS after bilateral adrenalectomy: pituitary MRI (1–2 mm slice thickness) should be done 3 months after surgery, then every 12 months for the first three years. Thereafter, clinical assessment should be conducted annually if tumor size is stable. The Endocrine Society recommends lifelong follow-up for CD patients treated with bilateral adrenalectomy, including evaluation for hyperpigmentation, ACTH level and pituitary MRI [4]. As a precaution, similar measures could apply to CD patients treated with osilodrostat or other steroidogenesis inhibitors, but longer follow-up studies are needed to document the evolution of pituitary tumor size with these medications and to establish clear recommendations. The case we report here demonstrates that corticotroph tumor progression can happen years after initiating osilodrostat treatment. Long-term follow-up with pituitary MRI and plasma ACTH should be considered, whether under such an effective medical therapy or after bilateral adrenalectomy in patients with persistent Cushing’s disease.
Data availability
Available from authors upon request.
References
Lacroix A, Feelders RA, Stratakis CA, Nieman LK (2015) Cushing’s syndrome. Lancet 386(9996):913–927. https://doi.org/10.1016/s0140-6736(14)61375-1
Fleseriu M, Findling JW, Koch CA, Schlaffer SM, Buchfelder M, Gross C (2014) Changes in plasma ACTH levels and corticotroph tumor size in patients with Cushing’s disease during long-term treatment with the glucocorticoid receptor antagonist mifepristone. J Clin Endocrinol Metab 99(10):3718–3727. https://doi.org/10.1210/jc.2014-1843
Feelders RA, Newell-Price J, Pivonello R, Nieman LK, Hofland LJ, Lacroix A (2019) Advances in the medical treatment of Cushing’s syndrome. Lancet Diabetes Endocrinol 7(4):300–312. https://doi.org/10.1016/s2213-8587(18)30155-4
Nieman LK, Biller BM, Findling JW, Murad MH, Newell-Price J, Savage MO, Tabarin A (2015) Treatment of cushing’s syndrome: an endocrine society clinical practice guideline. J Clin Endocrinol Metab 100(8):2807–2831. https://doi.org/10.1210/jc.2015-1818
Pivonello R, De Leo M, Cozzolino A, Colao A (2015) The treatment of cushing’s disease. Endocr Rev 36(4):385–486. https://doi.org/10.1210/er.2013-1048
Assié G, Bahurel H, Coste J, Silvera S, Kujas M, Dugué MA, Karray F, Dousset B, Bertherat J, Legmann P, Bertagna X (2007) Corticotroph tumor progression after adrenalectomy in Cushing’s Disease: A reappraisal of Nelson’s Syndrome. J Clin Endocrinol Metab 92(1):172–179. https://doi.org/10.1210/jc.2006-1328
Castinetti F, Morange I, Jaquet P, Conte-Devolx B, Brue T (2008) Ketoconazole revisited: a preoperative or postoperative treatment in Cushing’s disease. Eur J Endocrinol 158(1):91–99. https://doi.org/10.1530/eje-07-0514
Baudry C, Coste J, Bou Khalil R, Silvera S, Guignat L, Guibourdenche J, Abbas H, Legmann P, Bertagna X, Bertherat J (2012) Efficiency and tolerance of mitotane in Cushing’s disease in 76 patients from a single center. Eur J Endocrinol 167(4):473–481. https://doi.org/10.1530/eje-12-0358
Pivonello R, Fleseriu M, Newell-Price J, Bertagna X, Findling J, Shimatsu A, Gu F, Auchus R, Leelawattana R, Lee EJ, Kim JH, Lacroix A, Laplanche A, O’Connell P, Tauchmanova L, Pedroncelli AM, Biller BMK (2020) Efficacy and safety of osilodrostat in patients with Cushing’s disease (LINC 3): a multicentre phase III study with a doubleblind, randomised withdrawal phase. Lancet Diabetes Endocrinol. https://doi.org/10.1016/S2213-8587(20)30240-0
Jung C, Alford FP, Topliss DJ, Burgess JR, Long F, Gome JJ, Stockigt JR, Inder WJ (2010) The 4-mg intravenous dexamethasone suppression test in the diagnosis of Cushing’s syndrome. Clin Endocrinol (Oxf) 73(1):78–84. https://doi.org/10.1111/j.1365-2265.2009.03756.x
Vassiliadi DA, Tsagarakis S (2018) DIAgnosis of endocrine disease: the role of the desmopressin test in the diagnosis and follow-up of Cushing’s syndrome. Eur J Endocrinol 178(5):R201-r214. https://doi.org/10.1530/eje-18-0007
Nishioka H, Yamada S (2019) Cushing’s disease. J Clin Med. https://doi.org/10.3390/jcm8111951
Grossman AB (2017) The molecular pathology of cushing disease: are we nearly there? J Endocr Soc 1(2):144–148. https://doi.org/10.1210/js.2017-00036
Duggan S (2020) Osilodrostat: first approval. Drugs 80(5):495–500. https://doi.org/10.1007/s40265-020-01277-0
Fleseriu M, Pivonello R, Young J, Hamrahian AH, Molitch ME, Shimizu C, Tanaka T, Shimatsu A, White T, Hilliard A, Tian C, Sauter N, Biller BM, Bertagna X (2016) Osilodrostat, a potent oral 11β-hydroxylase inhibitor: 22-week, prospective, Phase II study in Cushing’s disease. Pituitary 19(2):138–148. https://doi.org/10.1007/s11102-015-0692-z
Funding
No specific funding for this publication.
Author information
Authors and Affiliations
Contributions
CFS and AL designed the analysis, data collection and wrote the manuscript; LLG analyzed pituitary MRI imaging, RAM performed the pituitary surgeries and FB the pathology studies. All authors revised and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
AL received funding as an investigator for participation in the LINC-3 trial from Novartis.
Ethical approval
LINC-3 study was approved by the CHUM ethics committee.
Informed consent
Patient provided informed signed consent to participate in the study and to publish results.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Fontaine-Sylvestre, C., Létourneau-Guillon, L., Moumdjian, R.A. et al. Corticotroph tumor progression during long-term therapy with osilodrostat in a patient with persistent Cushing’s disease. Pituitary 24, 207–215 (2021). https://doi.org/10.1007/s11102-020-01097-1
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11102-020-01097-1