
Abstract
Certain pituitary imaging abnormalities are a specific indicator of hypopituitarism. The objective of this study is to compare phenotypical features with radiological findings in patients with congenital growth hormone deficiency (GHD). Magnetic Resonance imaging (MRI) was performed in 103 patients [72 with Isolated GHD (IGHD) and 31 with Combined Pituitary Hormone Deficiency (CPHD)]. Images were assessed for the following abnormalities: (1) small/absent anterior pituitary, (2) thin or interrupted pituitary stalk (PSA), and (3) Ectopic posterior pituitary (EPP), and (4) others. Radiological findings were correlated with the clinical and biochemical parameters. MRI abnormalities were observed in 48.6% patients with IGHD, 93.5% with CPHD. Jaundice, hypoxia, hypoglycemia and breech deliveries were more common in EPP/PSA group. EPP/PSA was observed in 87.1% patients with severe GHD (peak GH < 3 μg/L) as compared to 12.9% with mild to moderate GHD (peak GH: 3–10 μg/L). Amongst CPHD, EPP/PSA was present in 80% of subjects with associated hypocortisolism ± hypothyroidism as compared to 18.2% of subjects with hypogonadism. Over a mean follow up period of 4.5 years, 5.4% of subjects with IGHD and abnormal MRI progressed to CPHD while none of those with normal MRI progressed. This study emphasizes a significant clinico-radiological correlation in Asian Indian GHD patients. MRI abnormalities in the hypothalamic pituitary area, especially EPP/PSA are more common in patients with CPHD and severe GHD. Among CPHD, EPP/PSA predicts association with hypothyroidism or hypocortisolism. IGHD with MRI abnormality may evolve into CPHD.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Morphological alterations of the hypothalamic-pituitary region on magnetic resonance imaging (MRI), such as pituitary stalk interruption and ectopic posterior pituitary (EPP), have been associated with isolated growth hormone deficiency (IGHD) or combined pituitary hormone deficiency (CPHD) [1, 2].
Literature search reveals varied results of phenotype- radiological association in subjects with growth hormone deficiency (GHD). The presence of EPP has been shown to be a useful predictor of GHD in a subset of patients with short stature [3]. Bozzola et al. in a study of 93 patients with GHD concluded that the IGHD patients showing EPP inevitably develop additional hormone deficiencies, while IGHD subjects having no MRI abnormalities develop no additional hormonal deficiencies [4]. Similar results have been published by Maghnie et al. [5].
Fujisawa and Kikuchi et al. reported stalk transection and EPP by MRI in patients with hypopituitarism who had a history of perinatal insults [6]. In these studies the proposed pathophysiological mechanism for MRI abnormalities was birth trauma, owing to the high frequency of obstetric complications and perinatal asphyxia observed in these children.
In contrast, some studies suggest disordered embryogenesis as the cause of EPP and stalk transection [7]. Scotti et al. [1] postulated that, in some cases, perinatal abnormalities could be the result, rather than the cause, of hypopituitarism.
Although a genetic cause is likely for EPP, the concerned genes have not been identified [8]. Isolated reports of mutations in the HESX1 [9] and LHX4 [10] genes and abnormalities in dosage of SOX3 [11] have been associated with the development of an EPP.
Argyropoulou et al. [12] and Bozzola et al. [13] reported a higher prevalence of EPP in CPHD than IGHD while exact opposite results have been reported by Kikuchi et al. [6] and Dutta et al. [14]. Thus amidst these conflicting reports we have prospectively evaluated the correlation between phenotype and radiological findings in Asian Indian patients with congenital GHD. The aim of this study was to evaluate the association of EPP and PSA with severity and other associated anterior pituitary hormonal deficiencies in Asian Indian patients with congenital growth hormone deficiency.
Subjects and methods
Study design
Written informed parental consent, patient assent, and approval by the Hospital Ethics Committee were obtained before enrolling the study subjects. 103 patients who fulfilled the criteria of GHD namely, height less than -3 SD for age and sex, failure of GH stimulation by insulin tolerance tests (stimulated peak GH < 10 μg/L) and growth velocity <3 cm/year were analyzed and subjected to MRI of the hypothalamic-pituitary region. According to the hormonal axis involvement, patients were classified either into IGHD or CPHD. These children were consecutive patients following in a single tertiary care center over 10 year period from 2000 to 2010.
Clinical evaluation
Height was measured with a Harpenden stadiometer and height SD score (SDS) was calculated using Indian reference standards [15]. Bone age was determined by the standards of Greulich and Pyle and pubertal development was rated using Tanner stages [16–18]. Micropenis was defined as a penile length shorter than −2.5 SD [19].
Hormonal assays
Basal T3, T4, thyroid stimulating hormone (TSH), cortisol, follicle stimulating hormone (FSH), leutinizing hormone (LH), testosterone, insulin like growth factor 1 (IGF1) and prolactin were measured. Growth hormone (GH) was measured before and 0, 30, 60, 90, and 120 min during insulin tolerance test (ITT) after overnight fast with appropriate priming when required. Post ITT cortisol was measured at 60 min after insulin administration. GHD was considered when GH peak after ITT was less than 10 μg/L in children and 3 μg/L in adults. Those children having peak GH of less than 3 μg/L were classified as severe GHD. Cortisol response to hypoglycemia was considered normal when peak was 504 nmol/L or greater. Hypothyroidism was diagnosed if total T4 values were less than the lower limit of the normal for that age in the setting of low or normal TSH [20]. Free T4 is not routinely available at our institution, hence total T4 was used instead of FT4 for diagnosis of central hypothyroidism. Hypogonadism was defined by the absence of physical signs of pubertal development at advanced bone age (>12 years in girls and >13.5 years in boys) and confirmed by biochemical data (prepubertal baseline serum gonadotropin concentrations). The diagnosis of hypogonadism in some was made on follow up. Hyperprolactinemia was defined as prolactin levels more than 1,086.95 pmol/L.
All hormonal measurements were carried out by chemiluminescence assay (Immulite 1,000, Siemens, Los Angeles USA). Intraassay and interassay coefficients of variation were less than 8 and 10%, respectively, for all hormonal evaluation.
Pituitary MRI
MRI scans were performed in a 1.5 Tesla unit using T1- weighted sagital and coronal scans using gadolinium contrast. Maximal height of the pituitary gland was measured perpendicular to the sella turcica and considered hypoplastic when less than -2 SD, compared with normal controls [21].
Lack of the normal posterior lobe hyperintense signal in the sella turcica and presence of a hyperintense nodule in an ectopic location was classified as EPP. The authors considered a normal pituitary stalk when they identified a normal diameter from the level of the optic chiasm to its insertion on the pituitary gland. The stalk was considered thin when it had a continuous but extremely thin appearance and its proximal and distal diameter size were below normal [22].
Optic nerves and midline structures, such as septum pellucidum and corpus callosum, were examined. MRI was performed and reported by a single radiologist experienced in reading pediatric MRI.
Statistical analysis
Data are presented as percentage or mean ± SD as appropriate. Groups with and without PSA/EPP were compared by student’s t tests. A P value of <0·05 was considered significant. SPSS version 16 was used for analysis.
Results
Demographic characteristics
Seventy-two patients had IGHD and 31 patients had CPHD. The mean age at presentation was 12.88 years (range: 4 months–43 years) and male to female ratio was 1.94: 1. Mean height SDS at presentation was −4.85 (range: −2.07 to −9.33). Family history of GHD in first degree relatives was present in 22.33% of the subjects.
Radiological characteristics
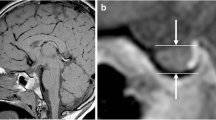
MRI abnormality was more common in CPHD (93.5%) than IGHD (48.6%). The abnormalities included hypoplastic anterior pituitary (HAP) (Fig. 1a), ectopic posterior pituitary (EPP) and pituitary stalk abnormality (PSA) (Fig. 1b). The triad of HAP, EPP and PSA was seen in five subjects of IGHD and twelve of CPHD. Four patients with EPP had evidence of Septo-optic dysplasia (SOD) (Fig. 1c). All four of them were CPHD. The comparison of MRI features of IGHD and CPHD is given in Table 1.

a MRI showing Hypoplastic anterior pituitary. b MRI showing Ectopic posterior pituitary and absent stalk. c MRI of a patient with Septo-optic dysplasia
Perinatal characteristics
Thirty-four subjects had history of perinatal insults in the form of hypoglycemia (n: 9), jaundice (n: 13), and hypoxia (n: 12). Jaundice and hypoglycemia was significantly more in EPP/PSA group than non EPP/PSA (P = 0.007 and 0.022, respectively). History of breech delivery was positive in 41.9% of subjects with EPP/PSA as compared to 16.7% without EPP/PSA (P = 0.005). Micropenis was also significantly more common in those subjects with EPP/PSA (P = 0.001). Also those with CPHD had higher prevalence of breech delivery, jaundice and micropenis as compared to subjects with IGHD (P = 0.05, 0.04 and 0.017, respectively).
Hormonal characteristics
The demographic and hormonal characteristics of 25 patients with EPP is summarized in Table 2. 58.8% of patients with EPP/PSA were CPHD as compared to 18.1% of those without these abnormalities (P = 0.004). CPHD patients with EPP and/or PSA were more commonly associated with deficiencies of TSH or ACTH or both. Those with CPHD but normal posterior pituitary and stalk had hypogonadism with GHD more frequently than cortisol or thyroid dysfunction. The comparison of phenotype between patients with EPP and/or PSA and those with normal stalk and posterior pituitary is shown in Table 3.
The mean prolactin level in those with stalk abnormality was significantly higher than in those with normal stalk (803.1 pmol/L and 412.2 pmol/L, respectively, P = 0.002). Also four out of 31 (12.9%) patients with EPP/PSA had hyperprolactinemia as compared to two out of 72 (2.8%) patients without EPP/PSA.
The mean follow up of the entire cohort is 4.5 years ranging from 1 to 10 years. Among 31 subjects of CPHD the hormonal deficiency patterns are as given in Fig. 2. Gonadal status was not assessed in five as the age was in prepubertal range. One of the patients had diabetes insipidus. He had panhypopituitarism (GH, TSH, ACTH and GNRH deficiencies). He had SOD with EPP on MRI.

Hormone deficiency patterns in CPHD
Two out of 37 patients who had initially presented as IGHD but had abnormal MRI progressed to CPHD while none of those with IGHD and normal MRI progressed.
Severity of GHD
Peak GH during ITT varied from undetectable to 9.05 μg/L with mean of 1.7 μg/L. A higher frequency of MRI abnormalities was found in those with peak GH level less than 3 μg/L, irrespective of the number of anterior pituitary hormone deficits. A significantly greater prevalence of EPP was determined in patients with peak GH level less than 3 μg/L compared with those whose peak GH level was 3 μg/L or greater. Higher percentage of EPP/PSA subjects had severe GHD than those with normal posterior pituitary and stalk. There was no significant difference in the mean peak GH of IGHD and CPHD subjects. Severe GHD was present in 66.66 and 61.29% of IGHD and CPHD subjects, respectively.
Discussion
To the best of our knowledge this is the largest prospective study in Indian subcontinent describing phenotype and radiological correlation in patients with congenital GHD. We describe a total of 103 patients including the data of 44 patients which was published earlier [23].
Around 22% of our subjects have a positive family history of GHD. This number is higher than in western literature. It may be because of the different ethnicity of Indian population and higher frequency of consanguineous marriages in India than in West.
Patients with IGHD had a more preserved hypothalamic pituitary region on MRI than those with CPHD, and therefore, the presence of more than one hormonal deficiency could be attributed to more severe abnormalities of the pituitary gland, as has been also previously observed [3, 4].
The incidence of breech delivery in our patients with GHD was 16.85%, compared with 3% in the general population [24]. In our series, breech presentation occurred in 41.9% of subjects with EPP/PSA as compared to 16.7% without EPP/PSA (P = 0.005). Our study confirms that ectopic posterior pituitary and stalk abnormality is more common in breech delivery but EPP and PSA were absent in 40% of the study subjects who had history of breech delivery. Previously it was thought that adverse antenatal factors may be important in the pathogenesis of an EPP. The absence of such history in a significant number of cases suggests that for some patients it is unlikely that the perinatal insult has caused the pituitary lesion. It is postulated that the hypopituitarism leads to the abnormal birth through mechanisms which are not yet understood. Since the fetal endocrine system is an important trigger in the induction of labour, it is possible that the hypothalamo-hypophyseal abnormalities may lead to complications during birth, viz the breech position [25].
Compared to those with IGHD, CPHD subjects had significantly higher frequency of breech delivery, pituitary stalk interruption, EPP, and pituitary gland hypoplasia, pointing towards a more severe hypothalamic damage in this group.
In our study the prevalence of CPHD was 58.1 and 18.1% in those with and without EPP, respectively. The frequency of presence of CPHD amongst those with EPP in studies published previously ranges from 44 to 100%. Those with eutopic posterior pituitary are less likely to be CPHD. This fact has been reiterated in all studies (Table 4) except one [14]. In this Indian study Dutta et al. reported 80% prevalence of CPHD amongst those with eutopic posterior pituitary as compared to 50% in those with EPP. Also EPP subjects those who had a coexisting PSA were more likely to be CPHD than those with normal stalk. These results suggest that in GHD associated with EPP, patients with PSA present a more severe form of the disease associated with CPHD, whereas presence of normal stalk suggested IGHD.
Two of our 37 patients with IGHD and abnormal MRI with a follow up of 4.5 years progressed to CPHD. In a study by Bozzola et al. [4], follow-up of 93 patients with GHD over a mean period of about 6 years showed no additional hormone deficiencies in 55 out of 60 patients who were initially classified as having IGHD with a normal (15 cases) or reduced (40 cases) pituitary gland size, without other MRI abnormalities. The remaining five children, who had initially shown an apparently IGHD with PSA and EPP, developed CPHD over time. Other follow up studies have also suggested that children with both PSA and EPP, and apparent IGHD will develop additional pituitary hormone deficiencies over time. These children need repeated reassessment of pituitary function when CPHD is not demonstrated at the first evaluation, as progression to complete anterior pituitary deficiency may occur progressively, even during second or third decade of life.
We found that EPP correlates with severity of GHD. Bozzola et al. [13] reported 82.4% of those with EPP to be severe GHD as compared to only 40% of those with normal posterior pituitary. Similar observations have been made by Maghnie et al. [5] and Hamilton et al. [2]. We feel that the patients present to us towards the severe end of spectrum, hence the prevalence of severe GHD overall is high in our study as well as in the other from India [14].
The prevalence of ACTH, TSH and gonadotropin deficiency amongst EPP subjects in our study is 40, 44 and 24%, respectively. In previously available studies the prevalence of ACTH, TSH and gonadotropin deficiency amongst EPP subjects varies from 18 to 43%, 44 to 68% and 0 to 81%, respectively (Table 5). The novel point in our study was the significant correlation of posterior pituitary and stalk abnormality in CPHD with presence of hypocortisolism and/or hypothyroidism with GHD than with normal thyroid and cortisol axes. The exact reason for this finding is not known but it can be hypothesized that those with EPP or PSA have more severe disease. This would imply that in a CPHD with these MRI abnormalities a close follow up with assessment of the cortisol and thyroid axes should be done if not already manifested.
Mean prolactin levels were significantly higher in patients with stalk and posterior pituitary abnormalities (P = 0.002). However hyperprolactinemia was observed in only four patients with EPP. The cause of hyperprolactinemia is loss of dopaminergic inhibition of the PRL secretion.
Four patients with EPP/PSA had septo-optic dysplasia (SOD), characterized by hypoplasia of the optic nerve, various types of forebrain defects and hormonal deficiencies. All of them presented as CPHD before 2 years of age. One also had diabetes insipidus. Three of them had corpus callosum agenesis (Fig. 1c) and two had optic nerve hypoplasia.
The limitation of the study was that the normative data for pituitary height and stalk thickness on MRI was derived from Caucasian population as no Indian data is available for the same.
Conclusions
There is strong correlation between phenotype and radiological features in Asian Indian GHD patients. CPHD subjects are more likely to have abnormal MRI findings (especially EPP and PSA) than IGHD. Those with CPHD and EPP/PSA are more likely to have cortisol and or thyroid insufficiency. EPP and PSA are associated with severe GHD. IGHD with MRI abnormality may evolve into CPHD.
References
Scotti G, Triulzi F, Chiumello G, Di Natale B et al (1989) New imaging techniques in endocrinology: magnetic resonance of the pituitary gland and sella turcica. Acta Paediatr Scand 356:5–14
Hamilton J, Blaser S, Daneman D et al (1998) MR imaging in idiopathic growth hormone deficiency. Am J Neuroradiol 19:1609–1615
Tillmann V, Tang VW, Price DA et al (2000) Magnetic resonance imaging of the hypothalamic pituitary axis in the diagnosis of growth hormone deficiency. J Pediatr Endocrinol Metab 13:1577–1583
Bozzola M, Mengarda F, Sartirana P et al (2000) Long-term follow-up evaluation of magnetic resonance imaging in the prognosis of permanent GH deficiency. Eur J Endocrinol 143:493–496
Maghnie M, Strigazzi C, Tinelli C, Autelli M et al (1999) Growth hormone (GH) deficiency (GHD) of childhood onset: reassessment of GH status and evaluation of the predictive criteria for permanent GHD in young adults. J Clin Endocrinol Metab 84:1324–1328
Kikuchi K, Fujisawa I, Momoi T et al (1988) Hypothalamic-pituitary function in growth hormone deficient patients with pituitary stalk transection. J Clin Endocrinol Metab 67:817–823
di Natale B, Pellini C, Ackermann S et al (1994) Persisting functional connection in growth-hormone-deficient patients with a transected stalk. Horm Res 41:193–196
Sloop KW, Walvoord EC, Showalter AD et al (2000) Molecular analysis of LHX3 and PROP-1 in pituitary hormone deficiency patients with posterior pituitary ectopia. J Clin Endocrinol Metab 85:2701–2708
Caravalho LR, Woods KS, Mendonca BB et al (2003) A homozygous mutation in HESX1 is associated with evolving hypopituitarism due to impaired repressor–corepressor interaction. J Clin Invest 112:1192–1201
Machinis K (2001) Syndromic short stature in patients with a germline mutation in the LIM homeobox LHX4. Am J Hum Genet 69:961–968
Woods KS, Cundall M, Turton J et al (2005) Over- and underdosage of SOX3 is associated with infundibular hypoplasia and hypopituitarism. Am J Hum Genet 76:833–849
Argyropoulou M, Perignon F, Brauner R et al (1992) Magnetic resonance imaging in the diagnosis of growth hormone deficiency. J Pediatr 120:886–891
Bozzola M, Adamsbaum C, Biscaldi I et al (1996) Role of magnetic resonance imaging in the diagnosis and prognosis of growth hormone deficiency. Clin Endocrinol 45:21–26
Dutta P, Bhansali A, Singh P et al (2009) Congenital hypopituitarism: clinico-radiological correlation. J Pediatr Endocrinol Metab 22(10):921–928
Agarwal DK, Agarwal KN, Upadhyay SK et al (1992) Physical and sexual growth pattern of affluent Indian children from 6 to 18 years of age. Indian Pediatr 29:1082–1203
Greulich WN, Pyle SI (1959) Radiographic atlas of skeletal development of the hand and wrist, 2nd edn. Stanford University Press, Stanford
Marshall WA, Tanner JM (1969) Variation in the pattern of pubertal changes in girls. Arch Dis Child 44:291–303
Marshall WA, Tanner JM (1970) Variation in the pattern of pubertal changes in boys. Arch Dis Child 45:13–23
Lee PA, Mazur T, Danish R et al (1980) Micropenis I: criteria, etiologies and classification. Johns Hopkins Med J 146:156–163
LaFranchi S (2008) Disorders of the thyroid gland. In: Kliegman RM (ed) Nelson textbook of pediatrics. W.B.Saunders, Philadelphia, pp 2316–2340
Argyropoulou M, Perignon F, Brunelle F et al (1991) Height of normal pituitary gland as a function of age evaluated by magnetic resonance imaging in children. Pediatr Radiol 21:247–249
Simmons GE, Suchnicki JE, Rak KM, Damiano TR (1992) MR imaging of the pituitary stalk: size, shape, and enhancement pattern. Am J Roentgenol 159:375–377
Acharya SV, Gopal RA, Lila AR et al (2011) Phenotype and radiological correlation in patients with growth hormone deficiency. Indian J Pediatr 78:49–54
Dutta DC (2006) Malpresentation and malposition. In: Textbook of obstetrics, 6th edn. New Central Book Agency, Kolkata, pp 375–381
Murray PG, Hague C, Fafoula O et al (2008) Associations with multiple pituitary hormone deficiency in patients with an ectopic posterior pituitary gland. Clin Endocrinol 69:597–602
Arrigo T, De Luca F, Maghnie M et al (1998) Relationships between neuroradiological and clinical features in apparently idiopathic hypopituitarism. Eur J Endocrinol 139:84–88
Bordallo MA, Tellerman LD, Bosignoli R et al (2004) Neuroradiological investigation in patients with idiopathic growth hormone deficiency. Jornal de Pediatria 80(3):223–228
Nagel BHP, Palmbach M, Petersen D et al (1997) Magnetic resonance images of 91 children with different causes of short stature: pituitary size reflects growth hormone secretion. Eur J Pediatr 156:758–763
Kornreich L, Horev G, Lazar L et al (1997) MR findings in hereditary isolated growth hormone deficiency. Am J Neuroradiol 18:1743–1747
Marcu M, Trivin C, Souberbielle JC et al (2008) Factors influencing the growth hormone peak and plasma insulin-like growth factor I in young adults with pituitary stalk interruption syndrome. BMC Endocrine Disorders 8:7
Tauber M, Chevrel J, Diene G et al (2005) Long-term evolution of endocrine disorders and effect of GH therapy in 35 patients with pituitary stalk interruption syndrome. Horm Res 64:266–273
Pinto G, Netchine I, Sobrier M et al (1997) Pituitary stalk interruption syndrome: a clinical- biological-genetic assessment of its pathogenesis. J Clin Endocrinol Metab 82:3450–3454
Chen S, Léger J, Garel C et al (1999) Growth hormone deficiency with ectopic neurohypophysis: anatomical variations and relationship between the visibility of the pituitary stalk asserted by magnetic resonance imaging and anterior pituitary function. J Clin Endocrinol Metab 84:2408–2413
Le′ger J, Danner S, Simon D et al (2005) Do all patients with childhood-onset growth hormone deficiency (GHD) and ectopic neurohypophysis have persistent GHD in adulthood? J Clin Endocrinol Metab 90:650–656
Louvel M, Marcu M, Trivin C et al (2009) Diagnosis of growth hormone (GH) deficiency: comparison of pituitary stalk interruption syndrome and transient GH deficiency. BMC Pediatr 9:29
Rottembourg D, Linglart A, Adamsbaum C et al (2008) Gonadotrophic status in adolescents with pituitary stalk interruption syndrome. Clin Endocrinol 69:105–111
Cacciari E, Zucchini S, Carla G et al (1990) Endocrine function and morphological findings in patients with disorders of hypothalamo-pituitary area: a study with magnetic resonance. Arch Dis Child 65:1199–1202
Conflicting interests and financial disclosure
None.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Jagtap, V.S., Acharya, S.V., Sarathi, V. et al. Ectopic posterior pituitary and stalk abnormality predicts severity and coexisting hormone deficiencies in patients with congenital growth hormone deficiency. Pituitary 15, 243–250 (2012). https://doi.org/10.1007/s11102-011-0321-4
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11102-011-0321-4