
Abstract
Gut microbiota contribute to the metabolism of dietary polyphenols and affect the bioavailability of both the parent polyphenols and their metabolites. Although there is a large number of reports of specific polyphenol metabolites, relatively little is known regarding the chemistry and enzymology of the metabolic pathways utilized by specific microbial species and taxa, which is the focus of this review. Major classes of dietary polyphenols include monomeric and oligomeric catechins (proanthocyanidins), flavonols, flavanones, ellagitannins, and isoflavones. Gut microbial metabolism of representatives of these polyphenol classes can be classified as A- and C-ring cleavage (retro Claisen reactions), C-ring cleavage mediated by dioxygenases, dehydroxylations (decarboxylation or reduction reactions followed by release of H2O molecules), and hydrogenations of alkene moieties in polyphenols, such as resveratrol, curcumin, and isoflavones (mediated by NADPH-dependent reductases). The qualitative and quantitative metabolic output of the gut microbiota depends to a large extent on the metabolic capacity of individual taxa, which emphasizes the need for assessment of functional analysis in conjunction with determinations of gut microbiota compositions.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The existence of a relationship between diet and health has been known for centuries. Such relationships have resulted in the discovery of vitamins and minerals to prevent their respective deficiency diseases. In the past several decades, researchers have identified new roles for vitamins beyond prevention of diseases and have proposed doses to achieve optimum health. While dietary polyphenols are generally not recognized as essential components of the diet, epidemiological data do suggest a positive relationship between dietary exposure to polyphenols and health. Earlier work on the health benefits of polyphenols has focused on the hypothesis that polyphenols exert beneficial effects by acting as anti-oxidants in the sense that they scavenge free radical species. Support for the anti-oxidant hypothesis originated primarily from in vitro studies using standard anti-oxidant tests, such as the ORAC and FRAP assays (Cao and Prior 1998; Huang et al. 2005), at oftentimes supra-physiological concentrations. While these studies have accumulated valuable information on the chemical properties of polyphenols, they have not generated a satisfactory explanation for their beneficial effects in animal models of disease and in humans (Pompella et al. 2014). One well-recognized problem is the discrepancy between micromolar concentrations of polyphenols used in cell culture models to achieve a certain biological effect and the circulating concentrations in animals and humans in the nanomolar range resulting from efficacious doses of polyphenols. Even if polyphenols would reach micromolar concentrations in target cells, they would unlikely be able to compete with the two foremost small-molecule anti-oxidants, ascorbic acid and glutathione, which are both present in cells at low millimolar concentrations.
In addition, many orally administered polyphenols appear in the general circulation primarily as their glucuronides and sulfates, which further complicates the integration of pharmacological data with pharmacokinetic observations. For instance, in our own research on dose–effect relationships of xanthohumol, a prenylated flavonoid from the hops plant, we found poor correlations between circulating levels of xanthohumol and the primary endpoints, i.e., weight gain, fasting plasma glucose, and levels of dysfunctional fatty acid metabolism. However, the circulating levels of glucuronides of xanthohumol correlated well with dose and with the primary endpoints (Legette et al. 2012, 2013). Many researchers have made similar observations, which, collectively, have shifted the field of polyphenol research towards metabolism of polyphenols, interconversions of metabolites and biological activity of metabolites.
Mammalian metabolic transformations of polyphenols fall by and large into phase 1 and phase 2 metabolism, giving rise to hydroxylation of aromatic rings, O-methylation, O-demethylation, and conjugation of hydroxy groups to yield glucuronides and sulfates. Electrophilic moieties, such as quinones and α,β-unsaturated carbonyl compounds, are subject to conjugation with glutathione, the product of which is further metabolized to the N-acetylcysteine adduct via the mercapturic acid pathway. Polyphenol-derived quinone metabolites can be considered active metabolites in the sense that they have the ability to induce an adaptive stress response by activating the transcription factor Nuclear factor (erythroid-derived 2)-like 2 (Nrf2) (Lee-Hilz et al. 2006). Nrf2 is a key regulator that orchestrates the regulation of more than 100 anti-oxidant response element (ARE) genes that encode proteins with cytoprotective functions including heat shock proteins, anti-oxidant proteins, NADPH regenerating enzymes, detoxification enzymes and the principal enzymes involved in the biosynthesis of glutathione (Eggler et al. 2008; Kumar et al. 2014). Nrf2 is regulated by Kelch-like ECH-associated protein 1 (Keap1) protein, an unusually cysteine-rich protein that acts as a sensor for reactive oxidative and electrophilic species. Polyphenol-derived quinones function as Michael acceptors to modify distinct cysteine residues on Keap1 (Eggler et al. 2009). There is evidence that Keap1 modification by quinone-forming polyphenols leads to Nrf2 activation and expression of ARE genes (Kumar et al. 2014; Lee et al. 2011). Protocatechuic acid, a signature metabolite of gut microbial biotransformation of anthocyanins, contains a readily oxidizable 3,4-dihydroxy aromatic ring and is a known Nrf2 activator (Kumar et al. 2014; Varì et al. 2011). Caffeic acid derivatives are other examples of gut microbial catabolites that have Nrf2 activating properties (Gray et al. 2015; Ma et al. 2010; Rechner et al. 2004; Williamson and Clifford 2010).
In recent years, researchers have identified relationships between compositions of gut microbiota and health (Turnbaugh et al. 2009; Xu and Gordon 2003). Such observations raise the question whether dietary polyphenols exert their health effects by changing gut microbiota composition. Alternatively, gut microbiota may alter the pharmacokinetics of dietary polyphenols and may produce bioavailable metabolites whose pharmacological properties are different from those of the parent polyphenols. For example, xanthohumol does not exert demonstrable estrogenic effects in animals and in cell culture models, whereas its metabolite, 8-prenylnaringenin, which can be produced by gut microbiota (Possemiers et al. 2005, 2006), has greater estrogenic potency than the well-known phytoestrogen, genistein (Milligan et al. 2000). This and other examples cited in this review highlight the impact of gut microbial metabolism of polyphenols on host health.
Gut microbiota influence polyphenol bioavailability by hydrolytic release of aglycones from O-glycosides and hepatic O-glucuronides. Polyphenols occur primarily as O-glycosides in plants and foods. Quercetin and kaempferol, two of the most abundantly and ubiquitously present flavonoids, are predominantly found as their O-glycosides. The red, blue, and purple flavonoid pigments of berries are also predominantly found in glycoside form. A large proportion of these glycosides are hydrolytically converted into their corresponding aglycones in the acidic environment of the stomach, the mucosa of the small intestines, and by enzymes expressed by gut microbiota. After absorption in the small intestine, the aglycones reach the liver where they are enzymatically converted into their O-glucuronides and O-sulfates. A variable proportion of the polyphenol conjugates is excreted in the bile and enters the small intestine. Several gut microbes are known to have glucuronidase capacity and convert polyphenol glucuronides back into their aglycones, available for re-absorption. This enterohepatic recycling scenario demonstrates that gut microbes exerting glycosidase and glucuronidase activity would directly enhance bioavailability of polyphenol glycosides. Using germ-free (GF) and human microbiota-associated (HMA) rats to determine the influence of gut microbiota on polyphenol bioavailability, Hanske et al. (2010) found higher amounts of xanthohumol glucuronides excreted in the feces of GF rats given an oral dose of xanthohumol, indicating that human microbiota exert glucuronidase activity. The same research group performed a similar study with apigenin-7-O-glucoside and found higher levels of C-ring cleavage products of apigenin in the urine of HMA rats (Hanske et al. 2009). They also determined that cytosolic extracts of Eubacterium ramulus and Bacteroides distasonis enzymatically convert apigenin-7-O-glucoside into its aglycone. The authors conclude that gut microbiota contribute proportionally the most to metabolism of orally administered apigenin-7-O-glucoside (Hanske et al. 2009). Other researchers have made similar observations with other polyphenols, which generalizes the notion that gut microbiota affect polyphenol bioavailability in major ways.
In addition to enzymatic ability to cleave O-glycosides and O-glucuronides, gut microbes appear to have metabolic capacity to perform carbon–carbon cleavage of heterocyclic and aromatic rings in flavonoids, dehydroxylations, decarboxylations, and hydrogenation of alkene moieties. Such metabolic transformations do not occur at random but follow a sequence of reactions ruled by basic chemical principles. While there are numerous papers listing microbial metabolites of dietary polyphenols, few studies have focused on the actual metabolic pathways and underlying enzymology responsible for the production of these metabolites. For most of the major dietary polyphenols, the pattern of gut microbial metabolites has been reported. But, how useful is this information if we do not know which microbe species has capacity for what metabolic conversion? In other words, it is equally important to know ‘who is there’ as to know ‘what they do’ in the gut. If such knowledge would become available, one would be able to predict the pattern of microbial metabolites based on the composition of the gut microbiome and establish ‘enterotypes’. Knowledge of gut microbial pathways of polyphenol metabolism would also enable one to determine whether a metabolite detected in the host is of gut microbial origin.
This review does not intend to provide an exhaustive listing of microbial metabolites of individual polyphenols: such information can easily be obtained by structural searching of metabolites in combination with their parent polyphenols using databases such as SciFinder. What this review does provide is an overview of the chemical mechanisms of gut microbial transformations of the major classes of dietary polyphenols, information which is necessary to construct a plausible metabolic pathway for any given dietary polyphenol. The following gut microbe-mediated reactions are discussed next: (1) carbon–carbon cleavage reactions involving C- and A-rings, (2) dehydroxylations, and (3) hydrogenations.
Carbon–carbon cleavage reactions
Enzymatic cleavage of C–C bonds in flavonoids appears to be characteristic of gut microbiota. Cleavage can take place in the heterocyclic C-ring of flavonols, flavones, flavanones, and anthocyanidins, as well as in the aromatic ring A of flavanols. The resulting phenolic acids are usually detected in the general circulation or in the urine as major metabolites.
C-ring cleavage of anthocyanidins
Anthocyanins represent a class of flavonoid plant pigments, responsible for red, purple, and blue colors of flower petals, vegetables, berries and other fruits. Anthocyanins are the glycosylated forms of their corresponding aglycones, anthocyanidins. The estimated daily intake of anthocyanins by U.S. adults is estimated at 200 mg (Wang and Stoner 2008). While the glycosides are bioavailable (Bitsch et al. 2004; Bub et al. 2001; Czank et al. 2013; Felgines et al. 2003; Mertens-Talcott et al. 2008), they are subject to hydrolytic conversion into their corresponding anthocyanidins. Both the glycosides and the aglycones are found as glucuronides in the urine of animals and humans (Ichiyanagi et al. 2008; Kay et al. 2004). Alternatively, the anthocyanidins can be metabolized into protocatechuic acid (Aura et al. 2005). Using cyanidin as the prototypic anthocyanidin, its metabolism is initiated by opening of the heterocyclic flavylium ring at neutral or slightly basic conditions of the small intestine. Subsequent attack of the flavylium carbon at position 2 produces an instable hemi-ketal that forms a ketone. Through keto-enol tautomerism of the neighboring enol functionality, the resulting α-diketo moiety can be cleaved by gut microbiota to form protocatechuic acid (1.6, Fig. 1) and 2-(2,4,6-trihydroxyphenyl)acetic acid (1.7). The exact mechanism of the cleavage reaction remains to be elucidated, but it seems reasonable to propose that the C–C cleavage involves attack of either carbonyl by a peroxyl anion species, similar to the initial step in the dioxygenase-mediated conversion of α-ketoglutarate into succinate (Silverman 2002). Insertion of the resulting alkoxy oxygen between the original carbonyl carbons yields an anhydride, which forms the two phenolic acids upon hydrolysis (Fig. 1). This proposed mechanism is also similar to the oxidative conversion of benzoins into benzoic acids (Kang et al. 2011). After absorption from the intestinal tract and hepatic phase 2 metabolism, these phenolic acids are usually detected in the urine in unchanged form or as their O-glucuronides or O-methyl derivatives (Amin et al. 2015; Wang et al. 2012; Woodward et al. 2011).

Proposed mechanism for the conversion of cyanidin into protocatechuic acid (1.6) and 2-(2,4,6-trihydroxyphenyl)acetic acid (1.7). Steps: (i) hydrolytic attack of the flavylium carbon at position 2, (ii) conversion of the hemi-ketal into the keto form followed by keto-enol tautomerism of the neighboring enol yields an α-diketo species, (iii) microbial enzyme-mediated nucleophilic attack of one of the keto carbons by a peroxyl anion species, (iv) insertion of the alkoxy oxygen results in an acyl anhydride, (v) hydrolysis of anhydride releases two phenolic acids
C-ring cleavage of flavonols
Quercetin, the prototypic representative of the large group of flavonols, has a virtually ubiquitous distribution among flowering plants. It is present in most if not all fruits and vegetables, predominantly in the form of O-glycosides. Several studies of metabolism of quercetin or its glycosides by fecal microbiota have revealed the conversion of quercetin into protocatechuic acid (2.6) (Serra et al. 2012) and 2-(3,4-dihydroxy)-phenylacetic acid (4.4) (Hein et al. 2008; Peng et al. 2014; Schneider et al. 2000) as the major metabolites. These metabolites can be further transformed into their O-methyl metabolites (Chadwick et al. 1992). The enzymatic release of protocatechuic acid from quercetin can be mediated by fungal and bacterial quercetin dioxygenases, also referred to as quercetinases (Hirooka and Fujita 2010; Schaab et al. 2006) (Table 1). They may contain iron, copper, or manganese in their catalytic site (Sun et al. 2015). Several authors have proposed a mechanism for the reaction, which have in common that a metal ion chelates the oxygen atoms at positions 3 and 4 in quercetin and that the mechanism involves a superoxide or peroxyl species (Fetzner 2012; Hirooka and Fujita 2010). In addition, carbon 3 of quercetin is released as carbon monoxide.
The consensus mechanism of quercetinase is initiated by chelation of the 4-keto/2,3-enol moiety by a divalent cation in the enzyme, which appears to favor the 3,4-diketo tautomer. The metal ion-bound flavonol complex sets the hydrogen atom at position 2 up for abstraction, conceivably via a semi-quinone species formed from a B-ring phenoxy radical. The carbon 2-centered radical then reacts with superoxide to form a five-membered endo-peroxide (Fig. 2, steps i and ii). In steps iii and iv, rearrangement of the endo-peroxide, initiated by conversion of the 3-keto into an acylium group, results in an ester linking the two aromatic rings, a carboxylic acid group at ring A, and release of carbon monoxide. While quercetin dioxygenases yield 2-protocatechuoyl-phloroglucinol carboxylic acid (Fig. 2, compound 2.5) in enzyme incubations or in incubations with microbiota, it has not been reported as a metabolite of quercetin in animals or humans, conceivably due to hydrolytic cleavage of the ester into protocatechuic acid (2.6) and 2,4,6-trihydroxybenzoic acid (2.7). Furthermore, there are no reports of the degradation product 2.7 as a metabolite of quercetin, but decarboxylation would explain the formation of the known gut microbial metabolite of quercetin, phloroglucinol (Ulbrich et al. 2015).

a A mechanism for quercetinase-mediated conversion of quercetin (2.1) into 2-protocatechuoylphloroglucinolcarboxylic acid (2.5) adapted from (Schaab et al. 2006). Decarboxylation of 1,3,5-trihydroxybenzoic acid (2.7) into phloroglucinol (2.8) is not mediated by quercetinase. b The co-crystal structure of Aspergillus japonicus quercetinase 2,3 dioxygenase complexed with the natural substrate quercetin under anaerobic condition is available (pdb 1H1I). The X-ray structure shows that the flavonol coordinates to the copper ion through the C-ring OH group at position 3 (Steiner et al. 2002)
The dependence on molecular oxygen in quercetinase- and other dioxygenase-mediated metabolic conversions raises the question how these microbial reactions can occur in the virtually anoxic environment of the gut. The gut epithelium separates the near-anoxic gut lumen from the highly vascularized and oxygen-rich sub-epithelium (Heinken and Thiele 2015; Taylor and Colgan 2007). Blood vessels in sub-epithelial tissue of the intestine facilitate efficient nutrient uptake, but they also supply oxygen to the epithelium and indirectly to the gut lumen by diffusion. The resulting steep oxygen gradient across the epithelial cell layer poses stress on both the epithelium and on obligate anaerobic micro-organisms in the gut lumen. Facultative anaerobic micro-organisms may survive or even thrive at this oxic/anoxic interface without interference from obligate anaerobes. Previously documented as an aerobic soil microbe, Bacillus subtilis has gained a new status as a facultative anaerobe (Hoffmann et al. 1995). Moreover, its isolation from and behavior in the human intestinal tract led some researchers to consider B. subtilis as a normal gut commensal in humans (Hong et al. 2009). Quercetinase from B. subtilis has been overexpressed in E. coli, isolated and biochemically characterized (Bowater et al. 2004; Schaab et al. 2006). Therefore, B. subtilis is a possible quercetinase-dependent producer of protocatechuic acid from dietary quercetin in the human gut.
As a major gut microbial metabolite of anthocyanidins and quercetin, protocatechuic acid contributes to health effects of fruits and vegetables rich in these flavonoids due to its reported anti-inflammatory (Kakkar and Bais 2014; Masella et al. 2012), anti-glycemic (Harini and Pugalendi 2010), and cancer chemopreventive effects (Lin et al. 2011; Peiffer et al. 2014; Tsao et al. 2014; Yin et al. 2009).
A-ring cleavage of catechin: reverse Claisen reactions
Catechin and its 3-epimer, epicatechin, are flavan-3-ol constituents of many fruits, such as apples and grapes. The gallic acid ester of epigallocatechin at position 3, epigallocatechin-3-O-gallate (EGCG), is an abundant flavanol in green tea. In addition, (epi)catechin forms oligomers, termed proanthocyanidins or condensed tannins, which give many fruits and especially unripe fruits, an astringent taste. Monomeric and oligomeric catechins constitute a prominent group of dietary flavonoids, and their total daily intake has been estimated to exceed 50 mg for adults. The major sources of dietary proanthocyanidins are apples, chocolate, and grapes in the U.S. (Gu et al. 2004). While the monomeric, dimeric, and trimeric catechins are absorbed from the small intestine to some extent, the higher oligomers are very poorly or not bioavailable (Rasmussen et al. 2005). Spencer et al. (2000) have demonstrated that proanthocyanidins undergo partial acid-catalyzed cleavage into their monomeric flavan-3-ol units in the gastric milieu. However, others have disputed the contribution of gastric depolymerization to the bioavailability of the constituent catechin units of proanthocyanidins (Donovan et al. 2002).
The major gut microbial metabolites of monomeric and oligomeric catechins appear to be 3-hydroxyphenylacetic acid, 3-hydroxyphenylpropionic acid, 3,4-dihydroxyphenylacetic acid (4.4), and 3-hydroxyphenyl-γ-valerolactone (3.13) (Appeldoorn et al. 2009). The latter metabolite is an example of an A-ring cleavage achieved by two consecutive reverse Claisen reactions (Fig. 3, steps iv through viii). Opening of the C-ring, possibly involving a quinone-methide intermediate (3.2), sets the stage for retro Claisen cleavage. At this point in the catabolism of catechin, there are two possibilities: (1) reduction of the quinone carbonyl yields the corresponding p-hydroxy metabolite, which is subject to dehydroxylation (Fig. 3, step iii), and (2) the quinone carbonyl is not reduced and hydride attack at the methide carbon yields a 3,4-dihydroxyphenyl metabolite which would eventually give rise to 3,4-dihydroxyphenyl-γ-valerolactone. Several authors have reported the latter compound as a product of gut microbial metabolism (Barroso et al. 2013; Goodrich and Neilson 2014; Jimenez-Giron et al. 2015; Margalef et al. 2014; Mulek and Hogger 2015; Takagaki and Nanjo 2013).

Gut microbial conversion of (epi)catechin (3.1) into 3-hydroxyphenyl-γ-valerolactone (3.13) by reverse Claisen-driven degradation of the A-ring (steps iv–viii). D = hydride donor, B = basic amino acid residue
C-ring cleavage of catechin
The metabolic conversion of monomeric catechins into 3-hydroxyphenyl acetic acid and 3,4-dihydroxyphenyl acetic acid, as observed by Gonthier et al. (Gonthier et al. 2003), must involve cleavage of the C3–C4 bond and resembles the cleavage of the terminal C–C bond of 2-oxoglutaric acid in α-ketoglutarate dependent dioxygenases (Silverman 2002). In catechin, keto-enol tautomerism yields a keto group at position 3 (Fig. 4, intermediate 4.1), which forms a complex with peroxyl-iron of the dioxygenase followed by a 5-membered transition state. Subsequently, the A-ring p-hydroxy group initiates the C3–C4 cleavage in catechin, thereby producing a quinone methide species, and iron-oxo species, and 3,4-dihydroxyphenyl acetic acid (4.4). The A-ring derived quinone methide has not been reported as a metabolite of catechin; this product is expected to undergo further catabolism by reverse Claisen reactions as shown in Fig. 3.

Proposed mechanism for gut microbial conversion of (epi)catechin into 2-(3,4-dihydroxyphenyl)acetic acid (4.4). Iron is linked to a dioxygenase via one or more amino acids, indicated as Ln
C–C cleavage of dihydrochalcones
An example of a C–C cleavage that is not mediated by dioxygenases is the cleavage of the C1′-CO bond in dihydrochalcones by the anaerobic gut bacterium Eubacterium ramulus. Blaut and co-workers identified a hydrolase from E. ramulus that catalyzes the conversion of phloretin (5.1) into 3-(4-hydroxyphenyl)propanoic acid (5.3) and phloroglucinol (5.4), and named it Phy (Schoefer et al. 2004). They cloned the gene encoding the enzyme which they biochemically characterized. Its sequence suggested that Phy has similarity to PhIG, a 2,4-diacetylphloroglucinol hydrolase from Pseudomonas fluorescens (Bottiglieri and Keel 2006; He et al. 2010; Schnider-Keel et al. 2000). The mechanism of the Phy-catalyzed hydrolytic C–C cleavage can be characterized as a reverse Claisen cleavage reaction facilitated by a 6-membered transition state (5.2, Fig. 5). Although phloretin and its O-glycosides are a well-known dihydrochalcones from apples, phloretin can also be derived from gut microbial conversion from naringenin via chalconaringenin. In this pathway, the conversion of naringenin into chalconaringenin is mediated by bacterial chalcone isomerase and the conversion of chalconaringenin into phloretin by enoate reductase (Gall et al. 2014; Herles et al. 2004; Thomsen et al. 2015).

Reverse Claisen type mechanism for the conversion of phloretin (5.1) into 3-(4-hydroxyphenyl)propanoic acid (5.3) and phloroglucinol (5.4)
Dehydroxylation of polyphenols
The removal of a p-hydroxy group from an aromatic ring is energetically unfavorable and requires an activated precursor. The best described example is the metabolic conversion of 4-hydroxybenzoic acid into benzoic acid by anaerobic bacteria. In this reaction, the carboxyl group is first ‘activated’ as its coenzyme A thioester by 4-hydroxybenzyl CoA ligase (Carmona et al. 2009; McInerney and Gieg 2004). Next, the thioester forms the intermediate quinone methide tautomer, which is reduced by a ferredoxin. The resulting semi-hydroquinone sets the stage for the dehydration step to form benzoyl-CoA, which yields benzoic acid upon hydrolysis (Fig. 6).

Carboxyl group-driven dehydroxylations of polyphenols are illustrated by the example of the gut microbial conversion of ellagic acid into urolithins. Strawberries, pomegranate juice, and walnuts are good sources of dietary ellagic acid (Gonzalez-Sarrias et al. 2010; Nunez-Sanchez et al. 2014; Tomas-Barberan et al. 2014; Truchado et al. 2012), which is formed by C–C coupling of two molecules of gallic acid followed by intramolecular condensation to form the di-lactone. Urolithins are arguably the main urinary biomarkers of nut consumption (Tulipani et al. 2012). In the metabolic pathway from ellagic acid to urolithins, one of the two lactone moieties undergoes hydrolysis, and the quinone methide tautomer of the resulting carboxylic acid is reduced, similar to the ferredoxin-mediated reduction of 4-hydroxybenzoic acid shown in Fig. 6. The result is a semi-hydroquinone, of which the p-hydroxy group leaves as a water molecule following decarboxylation (Fig. 7, step iii). Subsequent dehydroxylations can occur, all via reduction of quinone-methide tautomers, to form urolithins C, A, and B (Fig. 7).

Gut microbial conversion of ellagic acid into urolithins A, B, and C. In this metabolic pathway, dehydration reactions (iii, v, x) are driven by decarboxylation (iii) and reduction reactions (ii, vii, ix). Other steps: hydrolysis (i) and keto-enol tautomerism (i, iv, vi, viii). D = hydride donor, B = basic amino acid residue
In the microbial pathway to the urolithins, only the first dehydroxylation is driven by decarboxylation. Subsequent dehydroxylations involve a staged reduction of a quinone, in which keto-enol tautomerism first produces a secondary alcohol and subsequent hydride attack of the quinone drives the dehydration step. Analogous dehydrations are evident in the dehydroxylation of catechin (see Fig. 3), leading to the formation of 3-hydroxyphenyl-γ-valerolactone. In addition, the pathway described for dehydroxylation of 4-hydroxybenzoic acid would explain the formation of 3-hydroxybenzoic acid from protocatechuic acid derived from C-ring cleavage of cyanidin or quercetin (Figs. 1 and 2).
Hydrogenations of enone and keto moieties
Biotransformation of curcumin into tetrahydrocurcumin
The prototypic dietary polyphenols containing alkene moieties are curcumin and resveratrol. Numerous metabolites of curcumin, a yellow pigment from turmeric, have been identified in animal and human studies, including hydrogenated (dihydro, tetrahydro, hexahydro, and octahydro metabolites), desmethyl, glucuronide, and sulfate metabolites (Holder et al. 1978; Ireson et al. 2002). Hassaninasab and co-workers identified an NADPH-dependent reductase in E.coli isolated from human feces that is capable of stepwise reduction of curcumin (Hassaninasab et al. 2011) (Fig. 8). The enzyme, which they named NADPH-dependent curcumin/dihydrocurcumin reductase or CurA, showed selectivity for curcumin as the substrate and did not further reduce tetrahydrocurcumin to the corresponding secondary alcohols. The corresponding gene, curA, was identified as a member of the medium-chain dehydrogenase/reductase superfamily (Hassaninasab et al. 2011).

Stepwise reduction of curcumin by the E. coli enzyme, CurA (Hassaninasab et al. 2011)
Conversion of resveratrol to dihydroresveratrol
Resveratrol, a stilbene found in wine and grapes, has received much attention in the past decade due to its wide spectrum of health claims. A thorough study conducted by Bode et al. (Bode et al. 2013) demonstrated that the main gut microbial metabolites of resveratrol are dihydroresveratrol and the m-deoxy metabolites of both resveratrol and dihydroresveratrol (Fig. 9). Using a panel of gut microbes, they identified Slackia equolifaciens and Adlercreutzia equolifaciens as dihydroresveratrol producers.

Gut microbial metabolism of resveratrol (Bode et al. 2013)
Upon incubation of resveratrol with fecal samples, the kinetic profiles of resveratrol and its metabolites indicated that both dihydroresveratrol and 3,4′-dihydroxystilbene can serve as intermediates for the formation of lunularin (Fig. 9) (Bode et al. 2013).
Conversion of daidzein to equol
The main dietary sources of isoflavones are legumes and soy-based foods. Daidzin is one of the most abundantly present isoflavone glycosides in soy and soy products. Isoflavones are among the most potent phytoestrogens found in plants. Isoflavones bind to the estrogen receptor and affect the conformational dynamics in ways distinct from the endogenous ligand estrogen. Therefore isoflavones have been classified as selective estrogen receptor modulators (SERMs) (Brzezinski and Debi 1999). Metabolism and health promoting properties of dietary soy isoflavones have been recently reviewed by Yuan et al. (2007) and Rafii (2015).
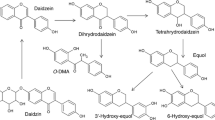
Enzymatic hydrolysis of the isoflavone glucosides in the small intestine by mammalian β-glycosidases promotes absorption (Bowey et al. 2003). The aglycone of daidzin, daidzein, is further metabolized in the liver and transformed into more water-soluble metabolites by O-glucuronidation and O-sulfation (Legette et al. 2014). The intestinal microbial transformation of daidzein entails sequential reduction or hydrogenation reactions and results in the formation of the following metabolites: dihydrodaidzein (DHD, 4′,7-dihydroxyisoflavone), tetrahydrodaidzein (THD, 4′,7-dihydroxyisoflavan-4-ol), and equol (4′,7-dihydroxyisoflavan). An alternative metabolic route produces O-desmethylangolensin (O-DMA) (Fig. 10) (Bowey et al. 2003; Heinonen et al. 2003; Joannou et al. 1995).

Gut microbial biotransformation of daidzein into the phytoestrogens equol and O-desmethylangolensin (O-DMA)
Inter-individual differences in the composition of the gut microbiome and associated biotransformation capacity govern the metabolism of isoflavone and their biological activities (Rafii 2015; Yuan et al. 2007). The biotransformation of daidzein into equol is dependent on the composition of the gut microbiome. However, the identification of the bacterial species or communities responsible for the conversion of daidzein to equol remains a challenge due to the vast diversity of the microbial communities with an ever-changing composition modifiable by diet, lifestyle, health status and age (van Duynhoven et al. 2011).
Only a few genera of equol-producing bacteria have been isolated and identified from the human intestinal microbiota. Some of the strains host the complete ensemble of enzymes that is capable of facilitating the metabolic conversion of daidzein to equol, whereas other strains are limited in their biotransformation capabilities. Among the genetically best characterized genera of equol-producing bacteria are Eggerthella species. For instance, the Eggerthella sp. strain YY7918 metabolizes daidzein to equol (Yokoyama et al. 2011). Maruo et al. (Maruo et al. 2008) isolated seven strains that were capable of metabolizing daidzein via dihydrodaidzein to equol. Although they found >90 % 16S rRNA gene similarity to Eggerthella species, there were dissimilarities in cell wall peptidoglycan types and therefore they proposed a new genus and species, Adlercreutzia equolifaciens (Maruo et al. 2008). A strain with a more restricted enzymatic capability, for instance, is the Eggerthella sp. strain Julong 732 which is capable of converting dihydrodaidzein, but not daidzein, into equol (Kim et al. 2009, 2010).
Other equol-producing bacteria isolated from human fecal material have been identified as taxa of the genus Slackia. Examples of genetically and biochemically well-characterized equol-producers are Slackia sp. strain NATTS (Tsuji et al. 2010, 2012) and S. isoflavoniconvertens (Matthies et al. 2012; Schroder et al. 2013).
Uchiyama and co-workers were the first to isolate an equol-producing lactic acid bacterium from human feces that they identified by 16S rRNA analysis as Lactococcus sp. strain 20–92 which belongs to the species Lactococcus garvieae (Uchiyama et al. 2013). Subsequently, Shimada and co-workers reported a detailed analysis of the enzyme machinery responsible for the biotransformation of daidzein into equol in L. garvieae strain 20-92 (Shimada et al. 2010, 2011, 2012).
Chemistry and enzyme systems for daidzein to equol conversion
The biosynthetic pathway of (S)-equol is interesting because the transformation begins with the achiral substrate daidzein but all subsequent reaction steps proceed stereospecifically with chiral intermediates. The enzymatic reduction of the Δ2,3 double bond in daidzein results in the formation of dihydrodaidzein with a chiral center at the C-3 position (Kim et al. 2010). Subsequent reductive transformations result in the formation of (3R,4S)-tetrahydrodaidzein and (S)-equol (Kim et al. 2010). Human intestinal microbiota produce exclusively (S)-equol (Setchell et al. 2005).
Detailed description of the genes and biochemical characterizations of the major daidzein-converting enzymes are available for three equol-forming bacterial strains, i.e., Slackia sp. strain NATTS, Slackia isoflavoniconvertens, and Lactococcus sp. strain 20-92 (Schroder et al. 2013; Shimada et al. 2010, 2011, 2012; Tsuji et al. 2012). The organization of the gene cluster involved in the biosynthetic transformation of daidzein is similar in all three bacterial strains with the open reading frames oriented in the same direction in the respective genomes (Fig. 11). Cloning and functional characterization of the gene products have consistently shown that the biochemical conversion of daidzein into (S)-equol involves the following enzymes: Daidzein reductase (DZNR); dihydrodaidzein reductase (DHDR) and tetrahydrodaidzein reductase (THDR). The amino acid sequences of these proteins showed high homology for the three daidzein-to-equol converting bacteria, i.e., S. isoflavoniconvertens, Slackia sp. strain NATTS, and Lactococcus sp. strain 20-92 (Table 2). In addition to the three reductases, Schroder et al. and Shimada et al. described an auxiliary enzyme, a dihydrodaidzein racemase, which is capable of catalyzing the conversion of (R)-dihydrodaidzein to (S)-dihydrodaidzein (Schroder et al. 2013; Shimada et al. 2012).

Gene organization maps of the gene cluster encoding enzymes involved in the metabolic conversion of daidzein to S-equol in three equol-producing bacterial strains: a S. isoflavoniconvertens (Schroder et al. 2013), b Slackia sp. strain NATTS (Tsuji et al. 2012), and c Lactococcus strain 20-92 (Shimada et al. 2011, 2012)
Bioinformatic analysis of the encoding genes and amino acid sequences of the proteins associated with the daidzein-to-equol conversion allowed the classification of the enzymes based on sequence homology and identification of consensus sequences associated with cofactor binding and active site architectures (Table 3) (Schroder, Matthies, Engst et al. 2013; Tsuji et al. 2012). The first enzyme of the conversion cascade, DZNR showed similarities to the old yellow enzyme (OYE) family (Karplus et al. 1995). Based on consensus sequences for an extended dinuculeotide binding motif, a cysteine-rich sequence region that identifies 4Fe-4S cluster binding and a FMN binding motif DZNR was classified as belonging to the NAD(P)H:flavin oxidoreductase family. The sequence of DHDR showed the typical active site tetrad, YX3K, for classical short-chain dehydrogenase/reductases (SDRs) and a conserved dinucleotide binding motif (Schroder et al. 2013; Tsuji et al. 2012). Tetrahydrodaidzein reductase (THDR) was classified as a member of the glycyl radical enzyme family based on the RVXG motif and the presence of a dinucleotide binding motif near the N-terminus (Buckel and Golding 2006; Schroder et al. 2013; Shisler and Broderick 2014). A radical-based reduction mechanism for the conversion of (3S,4R)-tetrahydrodaidzein to (S)-equol is consistent with the proposed mechanism by Kim et al. (2010), Fig. 12).

a Stereospecific conversion of (3S,4R)-tetrahydrodaidzein (THD) to (S)-equol and b the proposed mechanistic pathway of (3S,4R)-THD to (S)-equol catalyzed by tetrahydrodaidzein reductase (THDR) according to Kim et al. (2010)
In addition to the three reductases, Shimada et al. described a fourth enzyme encoded in the gene cluster for daidzein-metabolizing enzymes in Lactococcus sp. strain 20-92 (Shimada et al. 2012). Based on sequence homology to bacterial methylmalonyl coenzyme A (methylmalonyl-CoA) epimerases from Chlorobium spp. and subsequent functional characterization of the recombinant Lactococcus protein, this enzyme was identified as dihydrodaidzein racemase (DDRC). The enzyme is capable of converting each of the dihydrodaidzein enantiomers into the other to yield the racemate. It is conceivable that the enzymatic racemization proceeds via enolization and epimerization (Fig. 13). The respective putative proteins in Eggerthella sp. strain YY7918 and S. isoflavoniconvertens show high sequence similarities with 99 and 81.5 % identity, respectively. Biochemical experiments seem to indicate that DHDR prefers (S)-dihydrodaidzein as substrate. Efficient conversion of daidzein to (S)-equol was observed using a mixture of the four recombinant Lactococcus proteins DZNR, DHDR, THDR, and DDRC (Shimada et al. 2012).

Racemization of dihydrodaidzein catalyzed by dihydrodaidzein racemase
Stereospecific transformation of (3S,4R)-tetrahydrodaidzein to (S)-equol
The stereochemistry of the enzymatic conversion of intermediates involved in the biosynthetic route to (S)-equol have been studied by Kim et al. using partially characterized enzymes from Eggerthella sp. strain Julong 732 (Kim et al. 2010). Kim et al. used deuterium-labeled tetrahydrodaidzein to conduct mechanistic studies of the stereospecific transformation of (3S,4R)-tetrahydrodaidzein to (S)-equol. Their observations allowed proposing a radical-based mechanism in analogy to ribonucleotide reductases. In this model (Fig. 12), biotransformation of (3S,4R)- tetrahydrodaidzein is initiated by abstraction of hydrogen from the C-3 position triggered by an enzyme-based radical, X., thereby yielding a relatively stable benzylic radical. It is conceivable that the radical at C-3 induces loss of the hydroxyl group at position 4 under generation of a radical cation. Subsequent reduction of this intermediate by a hydride donor can then occur at C-4 with retention of the stereochemistry in accord with the observed deuterium labeling pattern. Finally, the biotransformation is completed by the addition of hydrogen to C-3 under preservation of the correct stereochemistry and formation of (3S)-equol. The authors note that tetrahydrodaidzein reductase (THDR) activity is only observed under strictly anaerobic conditions which would be consistent with that THDR is possibly a radical enzyme belonging to the glycyl radical enzyme family (Buckel and Golding 2006; Schroder et al. 2013; Shisler and Broderick 2014).
Conversion of daidzein to O-desmethylangolensin
The other major metabolite of daidzein is O-desmethylangolensin. Formation of O-desmethylangolensin involves cleavage of the C-ring, and so far only a few bacterial strains have been described that have the genetic make-up to catalyze this biotransformation which include Eubacterium ramulus (Schoefer et al. 2002), E. ramulus strain Julong 601 (Wang et al. 2004) and Clostridium sp. strain HGH 136 (Hur et al. 2002).
Early studies of human urinary metabolites suggested that the metabolic transformation of daidzein to O-desmethylangolensin proceeds via 2-dehydro-O-desmethylangolensin (Fig. 14) (Bowey et al. 2003; Heinonen et al. 2003; Joannou et al. 1995). However, the enzymes responsible for the C-ring fission and reduction have not been characterized yet.

a Intestinal microbial transformation of daidzein to O-desmethylangolensin (O-DMA) according to (Joannou et al. 1995). b Structure of (–)–(R)–O–desmethylangolensin
O-Desmethylangolensin has a chiral center which poses the question which of the two enantiomers is predominately produced in vivo. Gardana et al. showed that the metabolism of daidzein in anaerobic batch cultures inoculated with mixed fecal bacteria produces (–)–(R)–O–desmethylangolensin as the predominant product with an enantiomeric excess (EE) of 91 % (Gardana et al. 2014). Combined chiroptical studies with time-dependent density functional theory calculation supported a stereospecific biosynthetic route that proceeds predominantly via (–)–(S)–dihydrodaidzein to yield the urinary metabolite (–)–(R)–O–desmethylangolensin (Kim and Han 2014).
Conclusion and perspective
The systemic health benefits of dietary polyphenols cannot be satisfactorily explained by their oral bioavailability and host metabolism. Knowledge of the composition of gut microbiota and their functional capacity, obtained by metabolomics analysis in conjunction with bioinformatics analysis of microbial genes, may allow the characterization of ‘enterotypes’ or ‘nutritional phenotypes’, thereby providing a link between bioactive metabolites and the health benefits of dietary polyphenols (van Duynhoven et al. 2011). The empty entries in Table 1 exemplify that much work on the enzymology and identification of genes coding for metabolic enzymes remains to be completed before ‘enterotypes’ can be predicted on the basis of gene sequencing (Knights et al. 2014).
References
Amin HP, Czank C, Raheem S et al (2015) Anthocyanins and their physiologically relevant metabolites alter the expression of IL-6 and VCAM-1 in CD40L and oxidized LDL challenged vascular endothelial cells. Mol Nutr Food Res 59:1095–1106
Appeldoorn MM, Vincken JP, Aura AM et al (2009) Procyanidin dimers are metabolized by human microbiota with 2-(3,4-dihydroxyphenyl)acetic acid and 5-(3,4-dihydroxyphenyl)-gamma-valerolactone as the major metabolites. J Agric Food Chem 57:1084–1092
Aura AM, Martin-Lopez P, O’Leary KA et al (2005) In vitro metabolism of anthocyanins by human gut microflora. Eur J Nutr 44:133–142
Barroso E, Sanchez-Patan F, Martin-Alvarez PJ et al (2013) Lactobacillus plantarum IFPL935 favors the initial metabolism of red wine polyphenols when added to a colonic microbiota. J Agric Food Chem 61:10163–10172
Bitsch I, Janssen M, Netzel M et al (2004) Bioavailability of anthocyanidin-3-glycosides following consumption of elderberry extract and blackcurrant juice. Int J Clin Pharmacol Ther 42:293–300
Bode LM, Bunzel D, Huch M et al (2013) In vivo and in vitro metabolism of trans-resveratrol by human gut microbiota. Am J Clin Nutr 97:295–309
Bottiglieri M, Keel C (2006) Characterization of PhlG, a hydrolase that specifically degrades the antifungal compound 2,4-diacetylphloroglucinol in the biocontrol agent Pseudomonas fluorescens CHA0. Appl Environ Microbiol 72:418–427
Bowater L, Fairhurst SA, Just VJ et al (2004) Bacillus subtilis YxaG is a novel Fe-containing quercetin 2,3-dioxygenase. FEBS Lett 557:45–48
Bowey E, Adlercreutz H, Rowland I (2003) Metabolism of isoflavones and lignans by the gut microflora: a study in germ-free and human flora associated rats. Food Chem Toxicol 41:631–636
Brzezinski A, Debi A (1999) Phytoestrogens: the “natural” selective estrogen receptor modulators? Eur J Obstet Gynecol Reprod Biol 85:47–51
Bub A, Watzl B, Heeb D et al (2001) Malvidin-3-glucoside bioavailability in humans after ingestion of red wine, dealcoholized red wine and red grape juice. Eur J Nutr 40:113–120
Buckel W, Golding BT (2006) Radical enzymes in anaerobes. Annu Rev Microbiol 60:27–49
Cao G, Prior RL (1998) Comparison of different analytical methods for assessing total antioxidant capacity of human serum. Clin Chem 44:1309–1315
Carmona M, Zamarro MT, Blazquez B et al (2009) Anaerobic Catabolism of Aromatic Compounds: a Genetic and Genomic View. Microbiol Mol Biol Rev 73:71
Chadwick RW, George SE, Claxton LD (1992) Role of the gastrointestinal mucosa and microflora in the bioactivation of dietary and environmental mutagens or carcinogens. Drug Metab Rev 24:425–492
Czank C, Cassidy A, Zhang Q et al (2013) Human metabolism and elimination of the anthocyanin, cyanidin-3-glucoside: a (13) C-tracer study. Am J Clin Nutr 97:995–1003
Donovan JL, Manach C, Rios L et al (2002) Procyanidins are not bioavailable in rats fed a single meal containing a grapeseed extract or the procyanidin dimer B3. Br J Nutr 87:299–306
Eggler AL, Gay KA, Mesecar AD (2008) Molecular mechanisms of natural products in chemoprevention: induction of cytoprotective enzymes by Nrf2. Mol Nutr Food Res 52:S84–S94
Eggler AL, Small E, Hannink M et al (2009) Cul3-mediated Nrf2 ubiquitination and antioxidant response element (ARE) activation are dependent on the partial molar volume at position 151 of Keap1. Biochem J 422:171–180
Felgines C, Talavera S, Gonthier MP et al (2003) Strawberry anthocyanins are recovered in urine as glucuro- and sulfoconjugates in humans. J Nutr 133:1296–1301
Fetzner S (2012) Ring-cleaving dioxygenases with a cupin fold. Appl Environ Microbiol 78:2505–2514
Gall M, Thomsen M, Peters C et al (2014) Enzymatic conversion of flavonoids using bacterial chalcone isomerase and enoate reductase. Angew Chem Int Ed Engl 53:1439–1442
Garcia-Villalba R, Beltran D, Espin JC et al (2013) Time course production of urolithins from ellagic acid by human gut microbiota. J Agric Food Chem 61:8797–8806
Gardana C, Canzi E, Simonetti P (2014) R(–)–O–desmethylangolensin is the main enantiomeric form of daidzein metabolite produced by human in vitro and in vivo. J Chromatogr B Analyt Technol Biomed Life Sci 953–954:30–37
Gonthier MP, Cheynier V, Donovan JL et al (2003) Microbial aromatic acid metabolites formed in the gut account for a major fraction of the polyphenols excreted in urine of rats fed red wine polyphenols. J Nutr 133:461–467
Gonzalez-Sarrias A, Gimenez-Bastida JA, Garcia-Conesa MT et al (2010) Occurrence of urolithins, gut microbiota ellagic acid metabolites and proliferation markers expression response in the human prostate gland upon consumption of walnuts and pomegranate juice. Mol Nutr Food Res 54:311–322
Goodrich KM, Neilson AP (2014) Simultaneous UPLC-MS/MS analysis of native catechins and procyanidins and their microbial metabolites in intestinal contents and tissues of male Wistar Furth inbred rats. J Chromatogr B Analyt Technol Biomed Life Sci 958:63–74
Gray NE, Sampath H, Zweig JA et al (2015) Centella asiatica attenuates amyloid-beta-induced oxidative stress and mitochondrial dysfunction. J Alzheimers Dis 45:933–946
Gu L, Kelm MA, Hammerstone JF et al (2004) Concentrations of proanthocyanidins in common foods and estimations of normal consumption. J Nutr 134:613–617
Hanske L, Loh G, Sczesny S et al (2009) The bioavailability of apigenin-7-glucoside is influenced by human intestinal microbiota in rats. J Nutr 139:1095–1102
Hanske L, Loh G, Sczesny S et al (2010) Recovery and metabolism of xanthohumol in germ-free and human microbiota-associated rats. Mol Nutr Food Res 54:1405–1413
Harini R, Pugalendi KV (2010) Antihyperglycemic effect of protocatechuic acid on streptozotocin-diabetic rats. J Basic Clin Physiol Pharmacol 21:79–91
Hassaninasab A, Hashimoto Y, Tomita-Yokotani K et al (2011) Discovery of the curcumin metabolic pathway involving a unique enzyme in an intestinal microorganism. Proc Natl Acad Sci USA 108:6615–6620
He YX, Huang L, Xue Y et al (2010) Crystal structure and computational analyses provide insights into the catalytic mechanism of 2,4-diacetylphloroglucinol hydrolase PhlG from Pseudomonas fluorescens. J Biol Chem 285:4603–4611
Hein EM, Rose K, van’t Slot G et al (2008) Deconjugation and degradation of flavonol glycosides by pig cecal microbiota characterized by Fluorescence in situ hybridization (FISH). J Agric Food Chem 56:2281–2290
Heinken A, Thiele I (2015) Anoxic conditions promote species-specific mutualism between gut microbes in silico. Appl Environ Microbiol 81:4049–4061
Heinonen S-M, Hoikkala A, Wähälä K et al (2003) Metabolism of the soy isoflavones daidzein, genistein and glycitein in human subjects: identification of new metabolites having an intact isoflavonoid skeleton. J Steroid Biochem Mol Biol 87:285–299
Herles C, Braune A, Blaut M (2004) First bacterial chalcone isomerase isolated from Eubacterium ramulus. Arch Microbiol 181:428–434
Hirooka K, Fujita Y (2010) excess production of bacillus subtilis quercetin 2,3-dioxygenase affects cell viability in the presence of quercetin. Biosci Biotechnol Biochem 74:1030–1038
Hoffmann T, Troup B, Szabo A et al (1995) The anaerobic life of Bacillus subtilis: cloning of the genes encoding the respiratory nitrate reductase system. FEMS Microbiol Lett 131:219–225
Holder GM, Plummer JL, Ryan AJ (1978) The metabolism and excretion of curcumin (1,7-bis-(4-hydroxy-3-methoxyphenyl)-1,6-heptadiene-3,5-dione) in the rat. Xenobiotica 8:761–768
Hong HA, Khaneja R, Tam NM et al (2009) Bacillus subtilis isolated from the human gastrointestinal tract. Res Microbiol 160:134–143
Huang D, Ou B, Prior RL (2005) The chemistry behind antioxidant capacity assays. J Agric Food Chem 53:1841–1856
Hur HG, Beger RD, Heinze TM et al (2002) Isolation of an anaerobic intestinal bacterium capable of cleaving the C-ring of the isoflavonoid daidzein. Arch Microbiol 178:8–12
Ichiyanagi T, Shida Y, Rahman MM et al (2008) Effect on both aglycone and sugar moiety towards Phase II metabolism of anthocyanins. Food Chem 110:493–500
Ireson CR, Jones DJ, Orr S et al (2002) Metabolism of the cancer chemopreventive agent curcumin in human and rat intestine. Cancer Epidemiol Biomark Prev 11:105–111
Jimenez-Giron A, Ibanez C, Cifuentes A et al (2015) Faecal metabolomic fingerprint after moderate consumption of red wine by healthy subjects. J Proteome Res 14:897–905
Joannou GE, Kelly GE, Reeder AY et al (1995) A urinary profile study of dietary phytoestrogens. The identification and mode of metabolism of new isoflavonoids. J Steroid Biochem Mol Biol 54:167–184
Kakkar S, Bais S (2014) A review on protocatechuic Acid and its pharmacological potential. ISRN Pharmacol 2014:952943
Kang S, Joo C, Kim SM et al (2011) Oxidation of benzoins to benzoic acids using sodium hydride under oxygen atmosphere. Tetrahedron Lett 52:502–504
Karplus PA, Fox KM, Massey V (1995) Flavoprotein structure and mechanism. 8. Structure-function relations for old yellow enzyme. FASEB J 9:1518–1526
Kavanagh KL, Jornvall H, Persson B et al (2008) Medium- and short-chain dehydrogenase/reductase gene and protein families: the SDR superfamily: functional and structural diversity within a family of metabolic and regulatory enzymes. Cell Mol Life Sci 65:3895–3906
Kay CD, Mazza G, Holub BJ et al (2004) Anthocyanin metabolites in human urine and serum. Br J Nutr 91:933–942
Kim M, Han J (2014) Chiroptical study and absolute configuration of (–)–O–DMA produced from daidzein metabolism. Chirality 26:434–437
Kim M, Kim SI, Han J et al (2009) Stereospecific biotransformation of dihydrodaidzein into (3S)-equol by the human intestinal bacterium Eggerthella strain Julong 732. Appl Environ Microbiol 75:3062–3068
Kim M, Marsh EN, Kim SU et al (2010) Conversion of (3S,4R)-tetrahydrodaidzein to (3S)-equol by THD reductase: proposed mechanism involving a radical intermediate. Biochemistry 49:5582–5587
Knights D, Ward TL, McKinlay CE et al (2014) Rethinking “enterotypes”. Cell Host Microbe 16:433–437
Kumar H, Kim I-S, More SV et al (2014) Natural product-derived pharmacological modulators of Nrf2/ARE pathway for chronic diseases. Nat Prod Rep 31:109–139
Lee I-S, Lim J, Gal J et al (2011) Anti-inflammatory activity of xanthohumol involves heme oxygenase-1 induction via NRF2-ARE signaling in microglial BV2 cells. Neurochem Int 58:153–160
Lee-Hilz YY, Boerboom AM, Westphal AH et al (2006) Pro-oxidant activity of flavonoids induces EpRE-mediated gene expression. Chem Res Toxicol 19:1499–1505
Legette L, Ma L, Reed RL et al (2012) Pharmacokinetics of xanthohumol and metabolites in rats after oral and intravenous administration. Mol Nutr Food Res 56:466–474
Legette LL, Luna AY, Reed RL et al (2013) Xanthohumol lowers body weight and fasting plasma glucose in obese male Zucker fa/fa rats. Phytochemistry 91:236–241
Legette LL, Prasain J, King J et al (2014) Pharmacokinetics of equol, a soy isoflavone metabolite, changes with the form of equol (dietary versus intestinal production) in ovariectomized rats. J Agric Food Chem 62:1294–1300
Lin HH, Chen JH, Chou FP et al (2011) Protocatechuic acid inhibits cancer cell metastasis involving the down-regulation of Ras/Akt/NF-kappaB pathway and MMP-2 production by targeting RhoB activation. Br J Pharmacol 162:237–254
Ma Z-C, Hong Q, Wang Y-G et al (2010) ferulic acid protects human umbilical vein endothelial cells from radiation induced oxidative stress by phosphatidylinositol 3-kinase and extracellular signal-regulated kinase pathways. Biol Pharm Bull 33:29–34
Margalef M, Pons Z, Muguerza B et al (2014) A rapid method to determine colonic microbial metabolites derived from grape flavanols in rat plasma by liquid chromatography-tandem mass spectrometry. J Agric Food Chem 62:7698–7706
Maruo T, Sakamoto M, Ito C et al (2008) Adlercreutzia equolifaciens gen. nov., sp. nov., an equol-producing bacterium isolated from human faeces, and emended description of the genus Eggerthella. Int J Syst Evol Microbiol 58:1221–1227
Masella R, Santangelo C, D’Archivio M et al (2012) Protocatechuic acid and human disease prevention: biological activities and molecular mechanisms. Curr Med Chem 19:2901–2917
Matthies A, Loh G, Blaut M et al (2012) Daidzein and genistein are converted to equol and 5-hydroxy-equol by human intestinal Slackia isoflavoniconvertens in gnotobiotic rats. J Nutr 142:40–46
McInerney MJ, Gieg LM (2004) An overview of anaerobic metabolism. In: Nakano MM, Zuber P (eds) Strict and facultatie anaerobes: medical and environmental aspects. Horiz Biosci, Norfolk, pp 27–66
Mertens-Talcott SU, Rios J, Jilma-Stohlawetz P et al (2008) Pharmacokinetics of anthocyanins and antioxidant effects after the consumption of anthocyanin-rich acai juice and pulp (Euterpe oleracea Mart.) in human healthy volunteers. J Agric Food Chem 56:7796–7802
Milligan SR, Kalita JC, Pocock V et al (2000) The endocrine activities of 8-prenylnaringenin and related hop (Humulus lupulus L.) flavonoids. J Clin Endocrinol Metab 85:4912–4915
Mulek M, Hogger P (2015) Highly sensitive analysis of polyphenols and their metabolites in human blood cells using dispersive SPE extraction and LC-MS/MS. Anal Bioanal Chem 407:1885–1899
Nunez-Sanchez MA, Garcia-Villalba R, Monedero-Saiz T et al (2014) Targeted metabolic profiling of pomegranate polyphenols and urolithins in plasma, urine and colon tissues from colorectal cancer patients. Mol Nutr Food Res 58:1199–1211
Oppermann U, Filling C, Hult M et al (2003) Short-chain dehydrogenases/reductases (SDR): the 2002 update. Chem Biol Interact 143–144:247–253
Peiffer DS, Zimmerman NP, Wang LS et al (2014) Chemoprevention of esophageal cancer with black raspberries, their component anthocyanins, and a major anthocyanin metabolite, protocatechuic acid. Cancer Prev Res (Phila) 7:574–584
Peng X, Zhang Z, Zhang N et al (2014) In vitro catabolism of quercetin by human fecal bacteria and the antioxidant capacity of its catabolites. Food Nutr Res 58:23406
Pompella A, Sies H, Wacker R et al (2014) The use of total antioxidant capacity as surrogate marker for food quality and its effect on health is to be discouraged. Nutrition 30:791–793
Possemiers S, Heyerick A, Robbens V et al (2005) Activation of proestrogens from hops (Humulus lupulus L.) by intestinal microbiota; conversion of isoxanthohumol into 8-prenylnaringenin. J Agric Food Chem 53:6281–6288
Possemiers S, Bolca S, Grootaert C et al (2006) The prenylflavonoid isoxanthohumol from hops (Humulus lupulus L.) is activated into the potent phytoestrogen 8-prenylnaringenin in vitro and in the human intestine. J Nutr 136:1862–1867
Rafii F (2015) The role of colonic bacteria in the metabolism of the natural isoflavone daidzin to equol. Metabolites 5:56–73
Rasmussen SE, Frederiksen H, Struntze Krogholm K et al (2005) Dietary proanthocyanidins: occurrence, dietary intake, bioavailability, and protection against cardiovascular disease. Mol Nutr Food Res 49:159–174
Rechner AR, Smith MA, Kuhnle G et al (2004) Colonic metabolism of dietary polyphenols: influence of structure on microbial fermentation products. Free Radic Biol Med 36:212–225
Schaab MR, Barney BM, Francisco WA (2006) Kinetic and spectroscopic studies on the quercetin 2,3-dioxygenase from Bacillus subtilis. Biochemistry 45:1009–1016
Schneider H, Simmering R, Hartmann L et al (2000) Degradation of quercetin-3-glucoside in gnotobiotic rats associated with human intestinal bacteria. J Appl Microbiol 89:1027–1037
Schnider-Keel U, Seematter A, Maurhofer M et al (2000) Autoinduction of 2,4-diacetylphloroglucinol biosynthesis in the biocontrol agent Pseudomonas fluorescens CHA0 and repression by the bacterial metabolites salicylate and pyoluteorin. J Bacteriol 182:1215–1225
Schoefer L, Mohan R, Braune A et al (2002) Anaerobic C-ring cleavage of genistein and daidzein by Eubacterium ramulus. FEMS Microbiol Lett 208:197–202
Schoefer L, Braune A, Blaut M (2004) Cloning and expression of a phloretin hydrolase gene from Eubacterium ramulus and characterization of the recombinant enzyme. Appl Environ Microbiol 70:6131–6137
Schroder C, Matthies A, Engst W et al (2013) Identification and expression of genes involved in the conversion of daidzein and genistein by the equol-forming bacterium Slackia isoflavoniconvertens. Appl Environ Microbiol 79:3494–3502
Selma MV, Beltran D, Garcia-Villalba R et al (2014a) Description of urolithin production capacity from ellagic acid of two human intestinal Gordonibacter species. Food Funct 5:1779–1784
Selma MV, Tomas-Barberan FA, Beltran D et al (2014b) Gordonibacter urolithinfaciens sp. nov., a urolithin-producing bacterium isolated from the human gut. Int J Syst Evol Microbiol 64:2346–2352
Serra A, Macia A, Romero MP et al (2012) Metabolic pathways of the colonic metabolism of flavonoids (flavonols, flavones and flavanones) and phenolic acids. Food Chem 130:383–393
Setchell KD, Clerici C, Lephart ED et al (2005) S-equol, a potent ligand for estrogen receptor beta, is the exclusive enantiomeric form of the soy isoflavone metabolite produced by human intestinal bacterial flora. Am J Clin Nutr 81:1072–1079
Shimada Y, Yasuda S, Takahashi M et al (2010) Cloning and expression of a novel NADP(H)-dependent daidzein reductase, an enzyme involved in the metabolism of daidzein, from equol-producing Lactococcus strain 20-92. Appl Environ Microbiol 76:5892–5901
Shimada Y, Takahashi M, Miyazawa N et al (2011) Identification of two novel reductases involved in equol biosynthesis in Lactococcus strain 20-92. J Mol Microbiol Biotechnol 21:160–172
Shimada Y, Takahashi M, Miyazawa N et al (2012) Identification of a novel dihydrodaidzein racemase essential for biosynthesis of equol from daidzein in Lactococcus sp. strain 20-92. Appl Environ Microbiol 78:4902–4907
Shisler KA, Broderick JB (2014) Glycyl radical activating enzymes: structure, mechanism, and substrate interactions. Arch Biochem Biophys 546:64–71
Silverman RB (2002) The organic chemistry of enzyme-catalyzed reactions. Academic Press, London
Spencer JP, Chaudry F, Pannala AS et al (2000) Decomposition of cocoa procyanidins in the gastric milieu. Biochem Biophys Res Commun 272:236–241
Steiner RA, Kalk KH, Dijkstra BW (2002) Anaerobic enzyme·substrate structures provide insight into the reaction mechanism of the copper-dependent quercetin 2,3-dioxygenase. Proc Natl Acad Sci 99:16625–16630
Sun YJ, Huang QQ, Li P et al (2015) Catalytic dioxygenation of flavonol by M-complexes (M=Mn, Fe, Co, Ni, Cu and Zn)—mimicking the M-substituted quercetin 2,3-dioxygenase. Dalton Trans 44:13926–13938
Takagaki A, Nanjo F (2013) Catabolism of (+)-catechin and (−)-epicatechin by rat intestinal microbiota. J Agric Food Chem 61:4927–4935
Taylor CT, Colgan SP (2007) Hypoxia and gastrointestinal disease. J Mol Med (Berl) 85:1295–1300
Thomsen M, Tuukkanen A, Dickerhoff J et al (2015) Structure and catalytic mechanism of the evolutionarily unique bacterial chalcone isomerase. Acta Crystallogr D Biol Crystallogr 71:907–917
Toh H, Oshima K, Suzuki T et al (2013) Complete genome sequence of the equol-producing bacterium adlercreutzia equolifaciens DSM 19450T. Genome Announc 1:e00742
Tomas-Barberan FA, Garcia-Villalba R, Gonzalez-Sarrias A et al (2014) Ellagic acid metabolism by human gut microbiota: consistent observation of three urolithin phenotypes in intervention trials, independent of food source, age, and health status. J Agric Food Chem 62:6535–6538
Truchado P, Larrosa M, Garcia-Conesa MT et al (2012) Strawberry processing does not affect the production and urinary excretion of urolithins, ellagic acid metabolites, in humans. J Agric Food Chem 60:5749–5754
Tsao SM, Hsia TC, Yin MC (2014) Protocatechuic acid inhibits lung cancer cells by modulating FAK, MAPK, and NF-kappaB pathways. Nutr Cancer 66:1331–1341
Tsuji H, Moriyama K, Nomoto K et al (2010) Isolation and characterization of the equol-producing bacterium Slackia sp. strain NATTS. Arch Microbiol 192:279–287
Tsuji H, Moriyama K, Nomoto K et al (2012) Identification of an enzyme system for daidzein-to-equol conversion in Slackia sp. strain NATTS. Appl Environ Microbiol 78:1228–1236
Tulipani S, Urpi-Sarda M, Garcia-Villalba R et al (2012) Urolithins are the main urinary microbial-derived phenolic metabolites discriminating a moderate consumption of nuts in free-living subjects with diagnosed metabolic syndrome. J Agric Food Chem 60:8930–8940
Turnbaugh PJ, Ridaura VK, Faith JJ et al (2009) The effect of diet on the human gut microbiome: a metagenomic analysis in humanized gnotobiotic mice. Sci Transl Med 1:6ra14
Uchiyama S, Ueno T, Suzuki T (2013) Equol-producing lactic acid bacteria-containing composition. PCT/JP2004/009484
Ulbrich K, Reichardt N, Braune A et al (2015) The microbial degradation of onion flavonol glucosides and their roasting products by the human gut bacteria Eubacterium ramulus and Flavonifractor plautii. Food Res Int 67:349–355
van Duynhoven J, Vaughan EE, Jacobs DM et al (2011) Metabolic fate of polyphenols in the human superorganism. Proc Natl Acad Sci USA 108(Suppl 1):4531–4538
Varì R, D’Archivio M, Filesi C et al (2011) Protocatechuic acid induces antioxidant/detoxifying enzyme expression through JNK-mediated Nrf2 activation in murine macrophages. J Nutr Biochem 22:409–417
Wang LS, Stoner GD (2008) Anthocyanins and their role in cancer prevention. Cancer Lett 269:281–290
Wang XL, Kim KT, Lee JH et al (2004) C-ring cleavage of isoflavones daidzein and genistein by a newly-isolated human intestinal bacterium Eubacterium ramulus Julong 601. J Microbiol Biotechnol 14:766–771
Wang D, Xia M, Yan X et al (2012) Gut microbiota metabolism of anthocyanin promotes reverse cholesterol transport in mice via repressing miRNA-10b. Circ Res 111:967–981
Williamson G, Clifford MN (2010) Colonic metabolites of berry polyphenols: the missing link to biological activity? Br J Nutr 104(Suppl 3):S48–S66
Woodward GM, Needs PW, Kay CD (2011) Anthocyanin-derived phenolic acids form glucuronides following simulated gastrointestinal digestion and microsomal glucuronidation. Mol Nutr Food Res 55:378–386
Xu J, Gordon JI (2003) Honor thy symbionts. Proc Natl Acad Sci USA 100:10452–10459
Yin MC, Lin CC, Wu HC et al (2009) Apoptotic effects of protocatechuic acid in human breast, lung, liver, cervix, and prostate cancer cells: potential mechanisms of action. J Agric Food Chem 57:6468–6473
Yokoyama S, Oshima K, Nomura I et al (2011) Complete genomic sequence of the equol-producing bacterium Eggerthella sp. strain YY7918, isolated from adult human intestine. J Bacteriol 193:5570–5571
Yuan JP, Wang JH, Liu X (2007) Metabolism of dietary soy isoflavones to equol by human intestinal microflora–implications for health. Mol Nutr Food Res 51:765–781
Acknowledgments
The authors are in part supported by National Institutes of Health Grant No. R01AT009168.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Stevens, J.F., Maier, C.S. The chemistry of gut microbial metabolism of polyphenols. Phytochem Rev 15, 425–444 (2016). https://doi.org/10.1007/s11101-016-9459-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11101-016-9459-z