
Abstract
Amaryllidaceae alkaloids are an example of the vast diversity of secondary metabolites with great therapeutic promise. The identification of novel compounds in this group with over 300 known structures continues to be an area of active study. The recent identification of norbelladine 4′-O-methyltransferase (N4OMT), an Amaryllidaceae alkaloid biosynthetic enzyme, and the assembly of transcriptomes for Narcissus sp. aff. pseudonarcissus and Lycoris aurea highlight the potential for discovery of Amaryllidaceae alkaloid biosynthetic genes with new technologies. Recent technical advances of interest include those in enzymology, next generation sequencing, genetic modification, nuclear magnetic resonance spectroscopy, and mass spectrometry.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Amaryllidaceae alkaloids
Amaryllidaceae alkaloid introduction
The Amaryllidaceae alkaloids are largely restricted to the family Amaryllidaceae, specifically the subfamily Amaryllidoideae (Chase et al. 2009). Some noteworthy exceptions are the collection of alkaloids that have been found in the genus Hosta that is in the order Asparagales along with Amaryllidaceae (Chase et al. 2009; Li et al. 2012), the potential but un-replicated isolation of acetylcaranine and lycorine from Urginea altissima which is also a member of the order Aspergales (Miyakado et al. 1975; Pohl et al. 2001), and the isolation of crinamine from Dioscorea dregeana (Mulholland et al. 2002). New Amaryllidaceae alkaloid structures and the biosynthesis of these alkaloids have recently been reviewed (Kornienko and Evidente 2008; Bastida et al. 2011; Jin 2013; Takos and Rook 2013). Galanthamine is a prime example of the Amaryllidaceae alkaloids. It is one of the three primary drugs used for the treatment of Alzheimer’s disease. Another Amaryllidaceae alkaloid called lycorine has been shown to arrest the cell cycle and induce apoptosis in the cancer cell line HL-60 (Liu et al. 2004). In addition, the Amaryllidaceae alkaloids haemanthamine and haemanthidine have been shown to have anticancer activities (Havelek et al. 2014; Doskočil et al. 2015). The Amaryllidaceae alkaloids and their derivatives have been a source of novel acetylcholine esterase inhibitors, anti-cancer compounds, anti-viral compounds, and antibacterial compounds. The last 10 years of progress in the identification of these bioactive compounds has recently been reviewed (He et al. 2015). There are still new alkaloids being discovered in this group and even novel carbon skeletons with great potential to contribute to the list of known biologically active compounds. A diversity of carbon skeletons are known for this group of alkaloids including hostasinine, belladine, galanthamine, crinine, lycorine, galanthindole, homolycorine, galasine, montanine, cripowellin, cherylline, buflavine, plicamine, tazettine, graciline, augustamine, pancratistatin, and gracilamine (Jin 2009) (Figs. 1, 2, 3, 4, 5, 6). Many of these compounds are of potential pharmacological significance and their production through biological means is an area of great interest.

Primary Amaryllidaceae skeleton biosynthetic pathways with elaboration into the alkaloids haemanthamine, crinine, lycorine, and galanthamine

Carbon skeleton rearrangements in narciclasine, tazettine, and plicamine biosynthesis

Para-para ‘phenol–phenol’ coupling-derived skeletons found in the Amaryllidaceae and Hostasinine A from Hosta spp

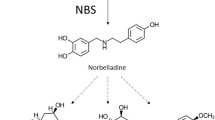
Core biosynthetic pathway for Amaryllidaceae alkaloids. Phenylalanine is converted to trans-cinnamic acid by phenylalanine ammonia lyase (PAL) and then to 4-hydroxycinnamic acid by CYP73A1. 4-hydroxycinnamic acid is potentially converted to 3,4-dihydroxycinnamic acid by CYP98A3 or to 4-hydroxybenzaldehyde and then to 3,4-dihydroxybenzaldehyde potentially by a VpVAN paralogue. Tyrosine is converted to tyramine by tyrosine decarboxylase. 3,4-dihyroxybenzaldehyde and tyramine are condensed to form a Schiff-base that is reduced by an unknown reductase into norbelladine. Norbelladine is methylated by norbelladine 4′-O-methyltransferase (N4OMT) into 4′-O-methylnorbelladine

Formation of the homolycorine carbon skeleton from norpluviine. The order of reactions between norpluviine and homolycorine are not determined, but are diagramed step by step for illustration of the enzyme types that may be involved

The pathway for cherylline and the structures of galasine, buflavine, and apogalanthamine. R indicates an undetermined methyl or hydrogen group
Prevalent biosynthetic gene superfamilies
To further the biological production of these compounds, an understanding of their biosynthetic genes is requisite and, although only one gene is known, the majority of the reactions are reaction types that are typically catalyzed by a collection of characterized enzyme families. The knowledge of these families can help inform efforts through homology searches to identify candidate genes. When studying secondary metabolism, in particular Amaryllidaceae alkaloid biosynthesis, several reaction types appear frequently, including methylation, reduction, oxidation, condensation, hydroxylation, phenol-phenol’ coupling, and oxide bridge formation (see Figs. 1, 2, 4, 5, 6 for examples). Examples of reductions found in the Amaryllidaceae include reduction of ketones, aldehydes, carbon–carbon double bonds, and imines. Two reductase superfamilies noted for their tendency to reduce aldehydes, ketones, carbon–carbon double bonds, and imines include aldo–keto reductases (AKRs) and short-chain dehydrogenase/reductases (SDRs) (Jörnvall et al. 1995; Sengupta et al. 2015). The SDR superfamily consists of three families including short-chain dehydrogenase/reductases, medium-chain dehydrogenase/reductases (MDRs) also known as alcohol dehydrogenases (ADH), and long-chain dehydrogenases/reductases (LDRs) (Kavanagh et al. 2008). The common feature of the SDR superfamily is a “Rossmann-fold” which is involved in the binding of dinucleotide cofactors including NADPH or NADH (Kavanagh et al. 2008). Oxidation reactions creating these various double bonds could be catalyzed by AKRs and SDRs as well because of the potential of these enzyme families to drive oxidations (Porté et al. 2013). 2-Oxoglutarate dependent dioxygenases and cytochrome P450 enzymes are well known for their ability to hydroxylate substrates thus making them good candidate gene families for the various hydroxylases in the biosynthesis of the Amaryllidaceae alkaloids (Lester et al. 1997; Nelson and Werck-Reichhart 2011) (Figs. 1, 2, 5, 6). The formation of an oxide bridge from a methoxy and hydroxyl group, as noted in haemanthamine and lycorine biosynthesis, is probably catalyzed by a cytochrome P450 because CYP81Q1, CYP719A1, CYP719A13, and CYP719A14 are enzymes shown to catalyze this type of reaction (Ikezawa et al. 2003; Ono et al. 2006; Díaz Chávez et al. 2011) (Fig. 1). Phenol-phenol’ coupling reactions are also likely catalyzed by cytochrome P450 enzymes. The P450s CYP81Q1, CYP719A1, CYP719A13, CYP719A14, DesC, and KtnC have been shown to catalyze phenol–phenol coupling reactions and are proposed to act by a diradical mechanism (Ikezawa et al. 2003; Ono et al. 2006; Díaz Chávez et al. 2011; Mazzaferro et al. 2015). Other enzyme groups noted for their ability to perform phenol-phenol’ coupling reactions are laccases and peroxidases (Schlauer et al. 1998; Constantin et al. 2012). In the Amaryllidaceae, two forms of methylation are common: O-methylation and N-methylation. O-Methyltransferases are divided into class I and class II methyltransferases (Ibdah et al. 2003) (see Figs. 1, 2, 5, 6 for examples). It has been shown that the class I O-methyltransferase, N4OMT is responsible for the methylation of norbelladine to 4′-O-methylnorbelladine in Amaryllidaceae alkaloid biosynthesis (Kilgore et al. 2014). Other O-methylation reactions in the biosynthesis of these compounds could be catalyzed by homologues to N4OMT or other known O-methyltransferases including reticuline 7-O-methyltransferase, (R,S)-norcoclaurine 6-O-methyltransferase, columbamine O-methyltransferase, chavicol O-methyltransferase, and eugenol O-methyltransferase (Gang et al. 2002; Morishige et al. 2002; Ounaroon et al. 2003). Examples of N-methyltransferases that could share homology with N-methyltransferases involved in several Amaryllidaceae alkaloid biosynthetic pathways include coclaurine N-methyltransferase and caffeine synthase (Kato et al. 2000; Choi et al. 2002). Homologues of O-methyltransferases would be of potential interest when looking for an N-methyltransferase as well because of the close homology that exists between the O- and N- methyltransferases (Raman and Rathinasabapathi 2003).
Amaryllidaceae alkaloid biosynthetic pathways have several reaction types with little enzymatic information in the existing literature. Examples include a potential retro-Prins reaction and a carbon–carbon bond severing reaction. During the biosynthesis of narciclasine from the para-para’ phenol-phenol’ coupling derivative 11-hydroxyvittatine a series of reactions including a retro-Prins reaction are proposed (Fuganti 1973). The retro-Prins reaction would result in the carbon-2 hydroxyl, cleavage of the 10b-11 bond, and migration of the 1-2 carbon double bond to 10b-1 (see Fig. 2 for proposed narciclasine pathway). An enzyme proposed to use the retro-Prins reaction is germacradienol/germacrene D synthase from Streptomyces coelicolor (Jiang et al. 2006). Another reaction of special mechanistic interest occurs during the biosynthesis of cripowellin. The cripowellin skeleton was discovered in 1998 in Crinum powellii and resembles a highly oxidized version of the haemanthamine skeleton that has had the 10b-4a carbon–carbon bond severed and replaced with a ketone on the 4a position (Velten et al. 1998) (see Fig. 3 for structure). If this is the pathway for generating this alkaloid, it is possible that carbon bond cleavage leading to the formation of a ketone is catalyzed by a cytochrome P450 similar to secologanin synthase that converts loganin into secologanin in an analogous manner with a ketone product (Irmler et al. 2000).
Core biosynthetic pathway
Intermediate discovery
The core biosynthetic pathway of the Amaryllidaceae alkaloids consists of the reactions required to produce 3,4-dihydroxybenzaldehyde and tyramine, the condensation and reduction of these precursors to norbelladine, and the subsequent methylation of norbelladine to 4′-O-methylnorbelladine (Fig. 4). Phenylalanine and tyrosine were shown to be precursors for haemanthamine by incorporation of [3-14C]phenylalanine and [3-14C]tyramine into haemanthamine in Nerine bowdenii (Wildman et al. 1962b). Degradation experiments of haemanthamine generated from radiolabeled tyramine were used to demonstrate the placement of the labeled carbons on positions 11 and 12 in experiments with [2-14C]tyrosine in Sprekelia formosissima and [1-14C]tyrosine in Narcissus ‘Twink’ daffodil (Battersby et al. 1961a; Wildman et al. 1962a). [3-14C]Tyramine has also been documented to incorporate into haemanthamine, haemanthidine, and 6-hydroxycrinamine in Haemanthus natalensis bulbs (Jeffs 1962). Lycorine and norpluviine have been shown to incorporate [2-14C]tyramine and [1-14C]tyramine in Narcissus “Twink” (Battersby and Binks 1960; Battersby et al. 1961b). [14C]Phenylalanine and [3H]3,4-dihydroxybenzaldehyde were both shown to be precursors to the aromatic half of haemanthamine and lycorine (Suhadolnik et al. 1962, 1963a). The pathway from phenylalanine to the intermediate 3,4-dihydroxybenzaldehyde was determined by feeding to Narcissus pseudonarcissus [3-14C]trans-cinnamic acid, [3-14C]4-hydroxycinnamic acid, [7-14C]benzaldehyde, [7-14C]4-hydroxybenzaldehyde, [3H]3,4-dihydroxybenzaldehyde and [3H]threo-DL-phenylserine and monitoring production of haemanthamine. The precursors [3-14C]trans-cinnamic acid, [3-14C]4-hydroxycinnamic acid, [3H]3,4-dihydroxybenzaldehyde and [7-14C]4-hydroxybenzaldehyde showed incorporation into haemanthamine. This led to the conclusion that the pathway for conversion of phenylalanine to 3,4-dihydroxybenzaldehyde is in the following sequence: phenylalanine, trans-cinnamic acid, 4-hydroxycinnamic acid, 3,4-dihydroxycinnamic acid or 4-hydroxybenzaldehyde, and 3,4-dihydroxybenzaldehyde (Suhadolnik et al. 1963b). 3,4-Dihydroxybenzaldehyde has been documented in Hydnophytum formicarum and other plants outside the Amaryllidaceae (Prachayasittikul et al. 2008). It is possible that the 3,4-dihydroxybenzaldehyde pathway is more phylogenetically spread than the latter steps or convergent evolution of product formation has occurred. Carbon fourteen-labeled norbelladine has been shown to incorporate into the alkaloids lycorine, crinamine, belladine, haemanthamine, and norpluviine (Battersby et al. 1961a, b; Wildman et al. 1962c). 4′-O-methylnorbelladine has been shown to be a precursor of all the primary alkaloid skeletons including crinine (crinine), haemanthamine (vittatine, 11-hydroxyvittatine), galanthamine (galanthamine, N-demethylgalanthamine, and N-demethylnarwedine), and lycorine (lycorine, norpluviine, and galanthine) (Kirby and Tiwari 1966; Bruce and Kirby 1968; Fuganti and Mazza 1972a, b; Fuganti 1973; Eichhorn et al. 1998). 4′-O-methylnorbelladine has long been considered the direct substrate for creation of the para-para’ and ortho-para’ carbon skeletons. 4′-O-methylnorbelladine has recently been established as the direct precursor of the para-ortho’ skeleton as well (Eichhorn et al. 1998). This universal requirement in all phenol-phenol coupling branches for 4′-O-methylnorbelladine makes it the last common intermediate before a three way split in the Amaryllidaceae biosynthetic pathway.
The three common divisions at 4′-O-methylnorbelladine are the para-para’ coupling that leads to the crinine and vittatine enantiomeric series, the ortho-para’ phenol coupling that is elaborated into the classic alkaloid lycorine, and the para-ortho’ coupling that is elaborated into the most widely used Amaryllidaceae alkaloid galanthamine (Fig. 1). Most other Amaryllidaceae alkaloid carbon skeletons are thought to be derivatives of these four skeletons. Examples include the pancratistatin and tazettine carbon skeletons derived from the haemanthamine skeleton and the homolycorine skeleton derived from the lycorine skeleton (Figs. 2, 3, 5). The belladine-type alkaloids are thought to originate by the simple methylation of norbelladine, though the order of methylations is not determined. The cherylline skeleton is thought to originate from hydroxylation at the 11-position of the norbelladine skeleton and subsequent cyclization with the dioxygenated phenol group (Chan 1973) (Fig. 6).
Enzymology
The biosynthesis of 3,4-dihydroxybenzaldehyde from phenylalanine likely involves the early phenylpropanoid biosynthetic pathway through to caffeic acid (3,4-dihydroxycinnamic acid). Assuming the involvement of the phenylpropanoid pathway, 3,4-dihyroxycinnamic acid is a more likely intermediate in the biosynthesis than 4-hydroxybenzaldehyde. This is in agreement with the relatively low incorporation of 4-hydroxybenzaldehyde in radiolabeling experiments (Suhadolnik et al. 1963b). The deamination of phenylalanine to trans-cinnamic acid is done by phenylalanine ammonia-lyase (PAL) (Tanaka et al. 1989). LrPAL1 and LrPAL2 have been cloned from the Amaryllidaceae plant Lycoris radiata demonstrating the presence of this enzyme in the Amaryllidaceae (Jiang et al. 2011, 2013b). The hydroxylation of trans-cinnamic acid to 4-hydroxycinnamic acid is done by cinnamate 4-hydroxylase (CYP73A1) (Fahrendorf and Dixon 1993; Teutsch et al. 1993). CYP98A3 has been documented to hydroxylate the 3-position of free 4-hydroxycinnamic acid (Franke et al. 2002). However, CYP98A3 has been shown to prefer the shikimic acid or quinic acid esters over free 4-hydroxycinnamic acid (Schoch et al. 2001; Franke et al. 2002). For this reason, it is possible a detour is required through shikimic acid, quinic acid, or acyl-CoA esters to obtain hydroxylated 3,4-dihydroxycinnamic acid. The conversion of 3,4-dihydroxycinnamic acid to 3,4-dihydroxybenzaldehyde appears very similar to the conversion of ferulic acid to vanillin by vanillin synthase (VpVAN), a hydratase/lyase (Gallage et al. 2014). The only difference is that the 3-hydroxyl is methylated in vanillin biosynthesis. Because of the substrate and reaction similarity, it is possible that this reaction is catalyzed by an enzyme related to VpVAN. Interestingly, there has been debate regarding VpVAN’s preference for ferulic acid or 4-hydroxycinnamic acid (Havkin-Frenkel et al. 2003). If a similar enzyme in 3,4-dihydroxybenzaldehyde biosynthesis shares the ability to perform this reaction on substrates that have or have not been hydroxylated at the 3-position, it would explain some of the ambiguity observed in earlier radiolabeling experiments. The conversion of tyrosine to tyramine is likely done by a homologue to the enzyme responsible for this reaction in other systems, tyrosine decarboxylase (Lehmann and Pollmann 2009). This homologue in Narcissus sp. aff. pseudonarcissus, KT378599, has been cloned and confirmed to have tyrosine decarboxylase activity.
The formation of the predicted Schiff-base intermediate from tyramine and 3,4-dihydroxybenzaldehyde is possibly a spontaneous reaction occurring in solution, an enzymatically catalyzed condensation, or both. This Schiff-base exists as three interchanging isomeric structures. This condensation is followed by a reduction of the imine double bond to make norbelladine. The reductase catalyzing this reaction could be an AKR or SDR and could also facilitate the formation of the Schiff-base by binding the tyramine and 3,4-dihydroxybenzaldehyde and causing an increase in local concentration for condensation. An SDR in the ADH family, tetrahydroalstonine synthase, from Catharanthus roseus has been shown to reduce the imine bond on strictosidine to form tetrahydroalstonine (Stavrinides et al. 2015). Several more NADPH dependent imine reductases have been characterized in bacteria (Wetzl et al. 2015). After this reduction, norbelladine has been shown to be methylated by the class I methyltransferase N4OMT in Narcissus sp. aff. pseudonarcissus (Kilgore et al. 2014). The three common phenol-phenol’ coupling reactions that follow require the same biochemistry to operate and are likely catalyzed by cytochrome P450 enzymes, laccases, or peroxidases (Schlauer et al. 1998; Ikezawa et al. 2003; Ono et al. 2006; Díaz Chávez et al. 2011; Constantin et al. 2012). The extensive work studying the biosynthesis of the derivatives generated from phenol–phenol’ coupling products has been reviewed recently and is beyond the scope of this review (Kornienko and Evidente 2008; Bastida et al. 2011; Jin 2013; Takos and Rook 2013).
Methods of interest to pathway elucidation
Introduction
The themes of miniaturization and increased throughput in methods supporting secondary metabolism research promise to accelerate discovery of biosynthetic enzymes in these systems. These trends are particularly relevant because they enable studies in non-model systems with increased efficiency. How these methods and their associated computational tools relate to metabolomics has recently been reviewed (Misra and van der Hooft 2015; Sumner et al. 2015). In this section, advances in methods and theory of potential use to secondary metabolism research in non-model species are examined, including gene discovery, next generation sequencing, gene editing, NMR, and MS.
Gene clusters and co-regulation of biosynthetic pathways
Gene clusters have been observed in the secondary metabolism of Zea mays, Avena spp., Oryza sativa, Arabidopsis thaliana, Lotus japonicus, Sorghum bicolor, Manihot esculenta, Papaver somniferum, and Solanum spp. as reviewed recently (Boycheva et al. 2014; Chae et al. 2014). Current theory for gene cluster formation postulates that gene clusters form when a particular set of genes or alleles of genes are favored in one environment but disfavored in another and the alleles interact positively together or negatively apart (Takos and Rook 2012). In secondary metabolism, intermediates lacking modifications, for example glycosylation in the case of cyanogenic glucosides, are often toxic (Takos et al. 2011). In this scenario, the presence of the entire pathway generates a beneficial compound, but an incomplete pathway may lead to a loss in fitness. When looking at Amaryllidaceae alkaloid biosynthesis, several intermediates are catechols, which could form reactive oxygen species, form DNA adducts, form protein adducts, or cause protein–protein cross-linking (Schweigert et al. 2001). Also, Amaryllidaceae alkaloids are thought to function primarily as herbivore deterrents, but the drain on the plant’s nitrogen supply would have a fitness cost. Under a low-nitrogen, low-herbivore pressure, the pathway would be unfavorable. Corroborating this perspective, Narcissus rupicola, one of the only Narcissus spp. without Amaryllidaceae alkaloids, grows on rocky soil where nutrients such as nitrogen may be limiting (Berkov et al. 2014). Variability in the composition of alkaloids between Galanthus elwesii populations has been observed; this could indicate environment-specific benefits for selected alkaloids (Berkov et al. 2004). Considering that Amaryllidaceae alkaloids are favorable in particular environments, but perhaps not in others, and that intermediates possess reactive functional groups, they meet all the criteria that favor the generation of gene clusters (Fisher 1930; Takos and Rook 2012). Genome assemblies can be used to discover genes surrounding known biosynthetic genes and assuming a gene cluster organization these genes could be tested for involvement in the biosynthetic pathway (Itkin et al. 2013). Members of gene families with the same enzymatic mechanism for a proposed reaction are prime candidates because evolutionary changes in substrate preference are more likely than changes in the underlying chemistry (Furnham et al. 2012). This clustering information could be combined with co-expression analysis to filter and support candidate gene lists (Itkin et al. 2013). Changes in chromatin structural proteins have been noted to change expression in genes from gene clusters of Arabidopsis thaliana and this is thought to contribute to co-expression of genes in clusters (Nützmann and Osbourn 2015). This could be one of the mechanisms for the co-expression observed in many biosynthetic genes in secondary metabolism pathways. Another potential mechanism is a shared transcription factor as noted for the transcription factor OsTGAP1 in momilactone biosynthesis (Okada et al. 2009). One approach to elucidating biosynthetic pathways is to address correlation between metabolites or known biosynthetic genes and potential biosynthetic genes with regard to expression patterns, presence, absence, or pseudo gene status either within a species or between species. If, for example, genes that co-express with a known biosynthetic gene in multiple species can be found, it is more likely to be related to the function of this known biosynthetic gene than a gene co-expressing in only one species. However, when looking for a biosynthetic gene in this way, the possibility of non-homologous genes doing the same reaction should be considered and misannotation of the known gene should be guarded against. This is possible when considering the history of convergent evolution in secondary metabolism (Pichersky and Lewinsohn 2011; Takos et al. 2011). This may be the case for the more derivatized Amaryllidaceae alkaloids, since there is not a clear correlation between phylogeny and the presence or absence of particular alkaloids (Rønsted et al. 2012). Due to the limitations of detection methods, however, the absence of these compounds in intervening lineages cannot be asserted with any certainty. The wide distribution of Amaryllidaceae alkaloids within the Amaryllidaceae would indicate a conserved core biosynthetic pathway and therefore a lack of convergent evolution. Examination of alkaloid composition in species closely related to the species in question may be helpful. If the biosynthetic pathway seems to be present in most common lineages as indicated by the presence of the end product, for example the ubiquitous Amaryllidaceae alkaloid lycorine, then there is no evidence for convergent evolution and common origin can be assumed in gene discovery workflows.
Sequencing technologies
Advances in sequencing technologies show great promise to improve de novo genome and transcriptome assemblies in non-model systems. These improved datasets will facilitate identification of gene clusters, co-expression analysis, and cloning of candidate genes. Second generation sequencing has improved the efficiency with which genomic and transcriptomic sequence information can be obtained. It is of particular value when studying systems without previous sequencing information (Kilgore et al. 2014). Early platforms for high throughput sequencing are Roche 454 sequencing by Life sciences corporation; second generation MiSeq, NextSeq, HiSeq, and HiSeq X Illumina platforms; and SOLiD from life technologies. De novo genome assemblies can be made with second generation sequencing data using programs such as ALLPATHS, Velvet, ABySS, and SOAPdenovo (Zerbino and Birney 2008; Simpson et al. 2009; Li et al. 2010; Gnerre et al. 2011). These genomes can provide information for the majority of the genome including genes, intergenic space, promoters, and introns. If the actual transcripts or the proteins they encode are the primary interest of the study, then de novo transcriptome assemblies will be more practical because of the reduced level of information required for sequence coverage and ability to focus only on transcripts that are being transcribed in samples of interest. The ability to focus on expressed material is of particular value for Amaryllidaceae species with Narcissus spp. genome sizes in the C2 range of 14–38 pg (Zonneveld 2008). De novo transcriptome assembly can provide a combination of sequence information, alternative splicing, and expression information in one experiment (Wang et al. 2009; Liu et al. 2014). Several prominent de novo transcriptome assemblers include Trinity, Oases, and Trans-ABySS (Robertson et al. 2010; Schulz et al. 2012; Haas et al. 2013). Programs commonly used to align reads back to the transcriptomes and obtain expression estimates in the form of read counts include Bowtie and BWA (Li and Durbin 2009; Langmead 2010). This combination of information allows workflows for candidate gene selection based on homology and co-expression to be carried out with a very manageable initial investment and no prior sequence information (Giddings et al. 2011; Yeo et al. 2013; Kilgore et al. 2014). Many transcriptomes have been assembled using second generation sequencing, thereby providing information on a genetic level to previously uncharacterized systems such as Camptotheca acuminata, Catharanthus roseus, Rauvolfia serpentina, Valeriana officinalis, and Veratrum californicum (Góngora-Castillo et al. 2012; Yeo et al. 2013; Augustin et al. 2015). Transcriptomes for the Amaryllidaceae species Narcissus sp. aff. pseudonarcissus and Lycoris aurea have been reported (Wang et al. 2013; Kilgore et al. 2014). In addition, transcriptomes are available for Galanthus elwesii and Galanthus sp. in the MedPlant RNA Seq Database, http://www.medplantrnaseq.org. The Alliums are close relatives of the Amaryllidaceae and transcriptomes have been reported for Allium sativum, Allium cepa, Allium fistulosum, and Allium tuberosum (Kamenetsky et al. 2015; Rajkumar et al. 2015; Tsukazaki et al. 2015; Zhou et al. 2015). Given the agricultural importance of alliums (garlic and onion) they will likely be examined more extensively on a molecular level in the near future and could provide a valuable point of comparison when examining genes in the closely related Amaryllidaceae. As more organisms are sequenced, homology-based comparisons become more meaningful because these sequences can be used to prepare phylogenies. When these phylogenies are combined with biochemical validation data for proteins contained in the phylogenies, annotations will be more accurate using programs such as SIFTER (Engelhardt et al. 2009).
Second generation sequencing generates short reads that create fragmented genomes in non-model and model systems alike. The inability to differentiate reads from highly similar transcripts can make de novo transcriptome and genome assemblies prone to collapsing these similar sequences into one contig. Sophisticated analysis workflows have been developed to resolve this problem in second generation sequencing (Spannagl et al. 2013). In addition, third and fourth generation sequencing technologies that provide longer sequencing reads are a promising new tool. PacBio from pacific biosciences is an instrument that monitors the incorporation of fluorescently labeled bases into a DNA strand by a polymerase tethered to a pore on a sequencing cell. The system is able to routinely generate sequences ~ 5 kb long with up to 50 kb possible. The down side is a ~80 % error rate (Lee et al. 2014). Using circular libraries, the polymerase used for sequencing can read the same sequence multiple times and this data can be processed for error reduction. This results in a tradeoff between read length and accuracy determined by the number of times the polymerase can make a cycle around a loop. This system has been used for the sequencing and distinguishing of members of the highly similar vomeronasal receptor class 1 gene family in the non-model lemur Microcebus murinus (Larsen et al. 2014). Using PacBio single-molecule sequencing an assembly of Oropetium thomaeum was constructed by VanBuren et al. 99 % of this 245 Mb genome was assembled into 625 contigs and highly repetitive regions including a ~25 kb inverted repeat in the chloroplast genome were completely assembled (VanBuren et al. 2015). One advantage of the PacBio system is its ability to simultaneously detect DNA modifications including the modified bases N6-methyladenosine, 5-methylcytosine, and 5-hydroxymethylcytosine. This information could be of use when using genomic DNA as a template and searching for regulatory modifications (Flusberg et al. 2010). A fourth generation system for DNA sequencing that commercializes nanopore technology is the MinION system that measures changes in electrical conductance as a DNA strand passes through a protein pore. This instrument is produced by Oxford Nanopore Technologies. One of the advantages of this technology is its size, measuring only 4 inches long and the ability to be powered by a standard USB 3.0 port making it the first highly portable sequencer. The consistency of this system will need to be improved and the fail rate reduced (Camilla et al. 2015). The MinION has been shown to have an average read size of ~5 kb with reads reaching 10 kb. The low accuracy of MinION has improved from ~65 to ~85% since its first appearance, but still makes the technology impractical when used alone (Mikheyev and Tin 2014; Loman and Watson 2015). A combination of Illumina sequencing and MinION to make accurate Nanopore Synthetic-long reads prior to assembly has been used, however, to generate an Acinetobacter baylyi assembly with 99.99 % accuracy (Madoui et al. 2015). A third approach to the generation of long reads is Illumina TruSeq which is a variant of second generation sequencing. This technique shears genomes into 10 kb sections and then performs a short read sequencing and assembly workflow on these 10 kb sections. This has been shown to be effective in the placement of the highly related transposable elements within the Drosophila melanogaster genome (McCoy et al. 2014). The longer sequence reads from PacBio, MinION, and Illumina TruSeq will help improve genome assemblies and connect contigs separated by highly repetitive regions. PacBio and MinION should also be able to provide start to end sequencing information of transcripts enabling splice variant analysis. The current downside to PacBio and MinION technologies is the high error rate associated with the raw read data. The Illumina TruSeq technology has low error rates ~0.03 % because its reads are built with short reads. As for MinION, the high error rate can be corrected in PacBio with very high coverage with circular libraries or by combining the second generation short reads with third generation long reads as the programs LSC, proovread, and LoRDEC are designed to do (Au et al. 2012; Hackl et al. 2014; Salmela and Rivals 2014).
Genetic modification
Recent advances in genome editing have enabled the testing of candidate genes in non-model organisms through the generation of knockout mutations with substantially reduced effort. Prior to genome editing techniques, random mutagenesis techniques such as EMS mutagenesis and transposon-based methods were used for the generation of knockout mutations in a gene of interest (Page and Grossniklaus 2002; Kim et al. 2006). Several techniques have been applied to induce targeted mutations through double stranded breaks including meganucleases, zinc-finger nucleases (ZFNs), transcription activator-like effector nucleases (TALENs), and clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR associated (Cas) protein. These double stranded breaks are repaired through non-homologous end joining resulting in error prone repair with deletions, additions, and substitutions or with homologous recombination using a provided template sequence to add a desired insertion, substitution, or deletion.
Meganucleases, ZFN, and TALENs, though extremely sequence specific, are limited by the technical difficulties associated with their use in the lab. The first enzymes used for targeted mutagenesis were meganucleases. These are large restriction enzymes with long motifs 14–40 nt long. They are specific, but engineering new sequence specificity is a complicated endeavor (Smith et al. 2006). ZFNs are modular with the ability to place motifs for 3 nt sequences in tandem and build sequence specificity with an added FokI domain for target DNA cleavage. The applications of ZFNs are limited by availability of compatible sequences, potential for incompatibility between specific modules, and difficult cloning requirements as reviewed previously (Carroll 2011). TALENs are easier to use with one motif per nucleotide and no restrictions on the sequence targeted except technical limitations that come with the cloning of repeats and the recommendation that the target sequence should start with a T (Gaj et al. 2013). TALENs also are very sequence specific and generate a low level of background mutations (Guilinger et al. 2014). However, TALENs require extensive cloning and, as a result, applications are limited by cloning efforts.
Due to the simplicity of its application, the CRISPR/Cas system has been used in a diversity of ways since its first application for targeted genetic modification in human and mouse cell lines (Jinek et al. 2012; Cong et al. 2013; Mali et al. 2013). In this system, the Cas9 protein binds to a guide RNA (gRNA) and the gRNA directs Cas9 to a specific sequence of DNA for cleavage. CRISPR/Cas is very simple to implement with a compatible sequence; given the Cas9 gene, only a gRNA sequence is required, which can easily be synthesized. This makes CRISPR/Cas a far easier system to apply to non-model species than its predecessors (Gilles and Averof 2014). The CRSPR/Cas system has some sequence constraints. The gRNA used to designate the target sequence contains ~20 nt of sequence complementary to the sequence of interest. Typically, the gRNA is transcribed by RNA polymerase III using a U6 promoter and as a result the preferred first base of the template, and therefore the gRNA target, is G. However, if the gRNA is placed between tRNAs in a tRNA array and the tRNA generating machinery is used to make the gRNA, this restraint on the 5′ nucleotide is lifted (Xie et al. 2015). Upstream from the 3′ end on the ~20 nt targeted sequence, a Cas9 protein defined protospacer adjacent motif (PAM), typically NGG, is required on the target sequence. The most extensively used Cas9 systems have NGG motifs, although Cas9 proteins with altered PAM motifs have been generated (Kleinstiver et al. 2015). Strong variability in mutation rate exists between constructs and room for improvement exists (Johnson et al. 2015; Mikami et al. 2015). Potential ways to improve the system are to use geminivirus- or tobacco rattle virus-based delivery systems that allow the spread of construct and the desired mutation through a plant by systemic infection (Ali et al. 2015; Yin et al. 2015). The CRISPR/Cas system also generates a variable level of off target mutations and several approaches have been applied to reduce these mutations. These include using FokI dimerization dependent cleavage domains with nuclease deficient Cas9 for sequence targeting. Dimerization allows two Cas9 proteins to target neighboring sequences doubling the sequence used for targeting from 20 to 40 nt (Wyvekens et al. 2015). Another approach is to shorten the 20 nt complementary sequence on the gRNA to 19–17 nt. The shortened sequence decreases the tolerance of mismatches typically observed towards the 5′ end. Large decreases in mutagenesis efficiency are not observed with 19 nt, but are observed with 18 or 17 nt (Wyvekens et al. 2015). Another approach to reduce off target mutations and broaden PAM specificity is to make a fusion protein for Cas9 and a TALEN or ZFN construct (Bolukbasi et al. 2015). In some cases, applications of CRISPR/Cas have resulted in the successful mutation of both copies of the gene (Oriza sativa and Populus tomentosa), mutations in close paralogues, or multiplexing of mutations for multiple genes (Zhang et al. 2014; Ma et al. 2015; Xie et al. 2015). The ability to target paralogues and potentially get homozygous mutations in one mutagenesis without subsequent breeding would be of particular interest when working on the Amaryllidaceae because some commercial cultivars are polypoid, sterile, and/or have a 3–7 year seed-to-seed generation time (Zonneveld 2010). The CRISPR/Cas system has been applied to Nicotiana benthamiana, Oryza sativa, Arabidopsis thaliana, Sorghum bicolor, Citrus sinensis var. Valencia, Solanum lycopersicum, Nicotiana tabacum, Triticum aestivum, Zea maize (in press accepted), Glycine max, Populus tomentosa, and Marchantia polymorpha showing the versatility of this technique in plants (Feng et al. 2015; Jiang et al. 2013a; Shan et al. 2013; Brooks et al. 2014; Jia and Wang 2014; Ron et al. 2014; Sugano et al. 2014; Fan et al. 2015; Gao et al. 2015; Johnson et al. 2015; Sun et al. 2015). To apply any of these targeted gene-editing systems to the Amaryllidaceae and obtain knockouts for candidate biosynthetic genes, the ability to make stable transformants is desirable and has been demonstrated in Narcissus tazzeta (Lu et al. 2007). The application of virus-induced gene silencing (VIGS) for the down regulation of genes of interest is another alternative for the examination genes in any species where infiltration, transient expression, and appropriate interaction with viral components can be achieved (Senthil-Kumar and Mysore 2014).
Nuclear magnetic resonance spectroscopy
NMR techniques have become more practical for the identification of the small quantities of compound observed in metabolomics surveys and generated during many enzyme assays. This will allow more complete catalogs of compounds in plant species to determine the presence or absence of compounds and their associated metabolic pathways. It will also facilitate identification of unknown products observed in enzyme assays. The usage of SPE to concentrate metabolite fractions coming out of a separation technique like HPLC and the subsequent release of metabolite for NMR analysis has been of great utility. This workflow enables the structural elucidation of compounds from complex mixtures. When used in parallel with 2D NMR techniques for deconvolution of co-eluting compounds chromatography issues can be avoided (Mahrous and Farag 2015). Another improvement of NMR technique is the invention of microprobes and miniaturized coils with current volumes of 10 µl and the potential of nanoliter sample sizes (Fratila and Velders 2011). The low volumes required allow for the elucidation of structures with microgram quantities of compound (Aramini et al. 2007). In addition, algorithms for combining NMR data with MS data during compound identification for greater accuracy have been developed (Bingol et al. 2015; Bingol and Brüschweiler 2015). Without these innovations for deconvolution and miniaturization of NMR experiments, the workflows that utilize NMR in high throughput systems with LC–MS outlined in the following section would not be practical.
Mass spectrometry
MS has improved in mass accuracy with the development of fourier transform mass spectrometers (FTMS). The increased mass accuracy of FTMS such as the Orbitrap can be of immense value in metabolomics by providing the accuracy needed to infer the molecular formula of compounds (Krauss et al. 2010). Combined with MS/MS data, the compounds can be searched against databases for the inference of structure. This is of great value in metabolomic studies and in enzyme product identification when the product is a characterized structure for which standard is lacking. Several workflows apply MSn and HPLC with SPE and NMR for the systematic structure elucidation of components in complex mixtures of metabolites (Castro et al. 2010; Sumner et al. 2015). An additional separation technique that can be used is ion mobility spectrometry, which uses a gas phase to slow compounds of different shapes. Compounds that interact with the gas phase more arrive at the detector later than compounds that interact with the gas phase less. This technique can be modified to separate chiral compounds by adding a chiral modifier to the gas phase (Dwivedi et al. 2006). The ability to use this technique on small molecules and to use it in parallel with LC make this another useful tool for the identification of compounds in complex mixtures (Budimir et al. 2007). To obtain quantification of compounds, evaporative light scattering detection (ELSD) can be used in combination with LC–MS (LC–ELSD–MS) (Cremin and Zeng 2002). Another example of a combination workflow utilizes LC–ELSD–MS detection for initial characterization of complex plant extracts, followed by structural elucidation of select compounds by NMR. A significant effect in a high throughput screen for a biological activity of interest, such as a greater than 32 % reduction in growth of the cancer cell lines MCF7, NCI-H460, or SF-26, is used to prioritize compounds for structural elucidation with NMR (Eldridge et al. 2002). These systematic catalogs of structures are very useful to secondary metabolism research because the associated MSn and NMR data become available in databases for future analysis of enzyme products or identification of metabolites from other complex mixtures in workflows lacking MSn or NMR components. In GC–MS data analysis, the use of libraries such as just described is relatively simple because of the standard ionization settings and resulting reproducibility of spectra. In LC–MSn, the fragmentation of compounds can vary greatly depending on the instrument and settings (Hopley et al. 2008). To deal with this problem, there are prediction algorithms that utilize LC–MSn data with fragmentation rules for ion trees and algorithms for relating MSn data to databases with different instruments and settings; an example is the Mass Frontier software from Thermo Scientific. Quantification and identification of small metabolites is complemented by the use of proteomic methods to acquire information on expressed proteins. This is illustrated by the use of proteomics on Lycoris aurea to identify proteins responsive to nitrogen treatment and the quantification of galanthamine changes during nitrogen treatment. This study demonstrated that galanthamine levels correlated on nitrogen treatment with the change observed in phenylalanine ammonia-lyase protein, an early enzyme in galanthamine biosynthesis (Ru et al. 2013) (Fig. 4).
Substrate considerations
Advances in MS and NMR promise to lower the quantity of product of enzyme assays necessary for structure elucidation. This also lowers the quantity of substrate required, which can also be a limiting factor when performing enzyme assays. When trying to acquire substrates for enzyme assays the substrates can be bought, synthesized, or isolated from the source. Purchasing the substrate is frequently not an option for highly specialized pathway intermediates, so the latter two alternatives become necessary. In the case of Amaryllidaceae alkaloids, there is a large diversity of specialized synthesis methods that have regularly been reviewed by Zhong Jin and can be used for production of various pathway intermediates (Jin 2013). As biosynthetic genes are discovered, functional expression of these genes in a heterologous system will facilitate chemo-enzymatic syntheses (Augustin et al. 2015). It is possible that in the future reactions for which the native enzyme is not known could have an alternate enzyme engineered to perform the desired reaction (Arnold 2015). Methods for isolation are provided in the publication of a compound’s discovery, but the availability of plant material can be a major constraint, particularly for endangered species.
Conclusion
In conclusion, the Amaryllidaceae alkaloids are a diverse group of alkaloids with many biosynthetic enzymes yet to be discovered. Advances in sequencing will facilitate genomic and transcriptomic analyses of these plants to identify candidate biosynthetic genes. Several sequencing projects have already generated transcriptomes for Narcissus sp. aff. pseudonarcissus and Lycoris aurea (Wang et al. 2013; Kilgore et al. 2014). The combination of sequencing with other methods such as proteomics can be a powerful approach for identification candidate genes. This combination was implemented during the discovery of VpVAN in Vanilla planifolia by selectively looking for transcripts and proteins highly concentrated in the biosynthetic tissue for vanillin, the inner bean pod (Gallage et al. 2014). Transcriptomic and genomic sequencing can also provide complementary information during candidate gene selection by allowing the combination of co-expression analysis and gene cluster searches. This combination of transcriptomic and genomic resources have become available in Catharanthus roseus through next generation sequencing (Kellner et al. 2015). Testing the candidate enzymes that will be identified through combinations of omics approaches, including transcriptomics, proteomics, and genomics, will be facilitated by sensitive detection and structural elucidation of substrates and products by MS and NMR techniques, either through plant-to-plant comparisons or direct assay. The discovery of N4OMT is an example: a de novo transcriptome assembly of Narcissus sp. aff. pseudonarcissus provided both the sequence and expression information needed to identify a candidate methyltransferase. Once the methyltransferase was shown to make a methylated product from norbelladine with MS/MS, small volume NMR technologies were utilized to identify the product 4′-O-methylnorbelladine (Kilgore et al. 2014). Another example of combining transcriptomics with sensitive techniques such as small volume NMR is the discovery of the first 6 steps of cyclopamine biosynthesis from cholesterol up to the intermediate verazine. In this study, a de novo transcriptome assembly of Veratrum californicum is used to find candidates through co-expression analysis. The candidates are expressed in insect cells and the various products are identified by a combination of MS/MS and NMR (Augustin et al. 2015). Existing and new transcriptomic data can be used in combination for the discovery of biosynthetic enzymes. This is the case for the discovery of the 6 missing biosynthetic genes in mayapple for etoposide aglycone which is a precursor for etoposide, a topoisomerase inhibitor used in chemotherapy. The study uses the Nicotiana benthamiana transient expression system to build the pathway step by step in planta while supplementing with the early intermediate matairesinol. The creation of biosynthetic intermediates in planta avoided the need to poses every intermediate during enzyme testing. Untargeted metabolomics enabled the observation of the biosynthetic intermediates generated during transient expression of different candidate biosynthetic gene combinations (Lau and Sattely 2015). These examples of enzyme discovery in different species with different compound classes show these improvements are generally applicable to secondary metabolism in non-model systems when looking for novel enzymes and will be of use when pursuing biosynthetic genes in the Amaryllidaceae.
Abbreviations
- ADH:
-
Alcohol dehydrogenases
- AKR:
-
Aldo-keto reductase
- CRISPR:
-
Clustered regularly interspaced short palindromic repeats
- Cas:
-
CRISPR associated
- ELSD:
-
Evaporative light scattering detection
- FTMS:
-
Fourier transform mass spectrometers
- gRNA:
-
Guide RNA
- LDR:
-
Long-chain dehydrogenases/reductase
- MS:
-
Mass spectrometry
- MDR:
-
Medium-chain dehydrogenase/reductase
- N4OMT :
-
Norbelladine 4′-O-methyltransferase
- NMR:
-
Nuclear magnetic resonance spectroscopy
- PAL:
-
Phenylalanine ammonia-lyase
- PAM:
-
Protospacer adjacent motif
- SDR:
-
Short-chain dehydrogenase/reductase
- SPE:
-
Solid phase extraction
- TALENs:
-
Transcription activator-like effector nucleases
- VpVAN:
-
Vanillin synthase
- VIGS:
-
Virus induced gene silencing
- ZFNs:
-
Zinc-finger nucleases
References
Ali Z, Abul-faraj A, Li L et al (2015) Efficient virus-mediated genome editing in plants using the CRISPR/Cas9 system. Mol Plant 8(8):1288–1291
Aramini JM, Rossi P, Anklin C et al (2007) Microgram-scale protein structure determination by NMR. Nat Methods 4(6):491–493
Arnold FH (2015) The nature of chemical innovation: new enzymes by evolution. Q Rev Biophys 48(4):404–410
Au KF, Underwood JG, Lee L et al (2012) Improving PacBio long read accuracy by short read alignment. PLoS One 7(10):e46679
Augustin MM, Ruzicka DR, Shukla AK et al (2015) Elucidating steroid alkaloid biosynthesis in Veratrum californicum: production of verazine in Sf9 cells. Plant J 82(6):991–1003
Bastida J, Berkov S, Torras L et al (2011) Chemical and biological aspects of Amaryllidaceae alkaloids. Transworld Research Network, Kerala, India
Battersby AR, Binks R (1960) Biosynthesis of lycorine. In: Proceedings of the chemical society of London, pp 410–411
Battersby AR, Fales HM, Wildman WC (1961a) Biosynthesis in the Amaryllidaceae. Tyrosine and norbelladine as precursors of haemanthamine. J Am Chem Soc 83(19):4098–4099
Battersby AR, Bink R, Breuer SW (1961b) Biosynthesis in the Amaryllidaceae: incorporation of norbelladine into lycorine and norpluvine. In: Proceedings of the chemical society of London, pp 243
Berkov S, Sidjimova B, Evstatieva L et al (2004) Intraspecific variability in the alkaloid metabolism of Galanthus elwesii. Phytochemistry 65(5):579–586
Berkov S, Martínez-Francés V, Bastida J et al (2014) Evolution of alkaloid biosynthesis in the genus Narcissus. Phytochemistry 99:95–106
Bingol K, Brüschweiler R (2015) Two elephants in the room: new hybrid nuclear magnetic resonance and mass spectrometry approaches for metabolomics. Curr Opin Clin Nutr Metab Care 18(5):471–477
Bingol K, Bruschweiler-Li L, Yu C et al (2015) Metabolomics beyond spectroscopic databases: a combined MS/NMR strategy for the rapid identification of new metabolites in complex mixtures. Anal Chem 87(7):3864–3870
Bolukbasi MF, Gupta A, Oikemus S et al (2015) DNA-binding-domain fusions enhance the targeting range and precision of Cas9. Nat Method 12(12):1150–1156
Boycheva S, Daviet L, Wolfender JL et al (2014) The rise of operon-like gene clusters in plants. Trends Plant Sci 19(7):447–459
Brooks C, Nekrasov V, Lippman ZB et al (2014) Efficient gene editing in tomato in the first generation using the clustered regularly interspaced short palindromic repeats/CRISPR-associated9 system. Plant Physiol 166(3):1292–1297
Bruce IT, Kirby GW (1968) Stereochemistry of protonation and hydroxylation in the biosynthesis of norpluviine and lycorine. Chem Commun (London) 4:207–208
Budimir N, Weston DJ, Creaser CS (2007) Analysis of pharmaceutical formulations using atmospheric pressure ion mobility spectrometry combined with liquid chromatography and nano-electrospray ionisation. Analyst 132(1):34–40
Camilla LC, Loose M, Tyson JR et al (2015) MinION analysis and reference consortium: phase 1 data release and analysis [version 1; referees: 1 approved]. F1000Research 4:1075
Carroll D (2011) Genome engineering with zinc-finger nucleases. Genetics 188(4):773–782
Castro A, Moco S, Coll J et al (2010) LC-MS-SPE-NMR for the isolation and characterization of neo-clerodane diterpenoids from Teucrium luteum subsp. flavovirens (perpendicular). J Nat Prod 73(5):962–965
Chae L, Kim T, Nilo-Poyanco R et al (2014) Genomic signatures of specialized metabolism in plants. Science 344(6183):510–513
Chan JLA (1973) Biosynthesis of cherylline using doubly-labled norbelladine-type precursors. Chemistry, Doctor of Philosophy. Retrospective Theses and Dissertations: Iowa State University, pp 116
Chase MW, Reveal JL, Michael FF (2009) A subfamilial classification for the expanded asparagalean families Amaryllidaceae, Asparagaceae and Xanthorrhoeaceae. Bot J Linn Soc 161(2):132–136
Choi KB, Morishige T, Shitan N et al (2002) Molecular cloning and characterization of coclaurine N-methyltransferase from cultured cells of Coptis japonica. J Biol Chem 277(1):830–835
Cong L, Ran FA, Cox D et al (2013) Multiplex genome engineering using CRISPR/Cas systems. Science 339(6121):819–823
Constantin MA, Conrad J, Beifuss U (2012) Laccase-catalyzed oxidative phenolic coupling of vanillidene derivatives. Green Chem 14:2375–2379
Cremin PA, Zeng L (2002) High-throughput analysis of natural product compound libraries by parallel LC–MS evaporative light scattering detection. Anal Chem 74(21):5492–5500
Díaz Chávez ML, Rolf M, Gesell A et al (2011) Characterization of two methylenedioxy bridge-forming cytochrome P450-dependent enzymes of alkaloid formation in the Mexican prickly poppy Argemone mexicana. Arch Biochem Biophys 507(1):186–193
Doskočil I, Hošťálková A, Šafratová M et al (2015) Cytotoxic activities of Amaryllidaceae alkaloids against gastrointestinal cancer cells. Phytochem Lett 13:394–398
Dwivedi P, Wu C, Matz LM et al (2006) Gas-phase chiral separations by ion mobility spectrometry. Anal Chem 78(24):8200–8206
Eichhorn J, Takada T, Kita Y et al (1998) Biosynthesis of the Amaryllidaceae alkaloid galanthamine. Phytochemistry 49(4):1037–1047
Eldridge GR, Vervoort HC, Lee CM et al (2002) High-throughput method for the production and analysis of large natural product libraries for drug discovery. Anal Chem 74(16):3963–3971
Engelhardt BE, Jordan MI, Repo ST et al (2009) Phylogenetic molecular function annotation. J Phys Conf Ser 180(1):12024
Fahrendorf T, Dixon RA (1993) Stress responses in alfalfa (Medicago sativa L.). XVIII: molecular cloning and expression of the elicitor-inducible cinnamic acid 4-hydroxylase cytochrome P450. Arch Biochem Biophys 305(2):509–515
Fan D, Liu T, Li C et al (2015) Efficient CRISPR/Cas9-mediated targeted mutagenesis in Populus in the first generation. Sci Rep 5:12217
Feng C, Yuan J, Wang R et al (2015) Efficient targeted genome modification in maize using CRISPR/Cas9 system. J Genet Genom. doi:10.1016/j.jgg.2015.10.002
Fisher RA (1930) The genetical theory of natural selection. Oxford University Press, Oxford
Flusberg BA, Webster DR, Lee JH et al (2010) Direct detection of DNA methylation during single-molecule, real-time sequencing. Nat Methods 7(6):461–465
Franke R, Humphreys JM, Hemm MR et al (2002) The Arabidopsis REF8 gene encodes the 3-hydroxylase of phenylpropanoid metabolism. Plant J 30(1):33–45
Fratila RM, Velders AH (2011) Small-volume nuclear magnetic resonance spectroscopy. Annu Rev Anal Chem (Palo Alto Calif) 4:227–249
Fuganti C (1973) Evidence for the intermediacy of 11-hydroxyvittatine in the biosynthesis of narciclasine. Gazz Chim Ital 103:1255–1258
Fuganti C, Mazza M (1972a) The absolute configuration of narciclasine: a biosynthetic approach. J Chem Soc Chem Commun 4:239
Fuganti C, Mazza M (1972b) Stereochemistry of hydorxylation in the biosynthesis of lycorine in Clivia miniata regel. J Chem Soc Chem Commun 16:936–937
Furnham N, Sillitoe I, Holliday GL et al (2012) Exploring the evolution of novel enzyme functions within structurally defined protein superfamilies. PLoS Comput Biol 8(3):e1002403
Gaj T, Gersbach CA, Barbas CF III (2013) ZFN, TALEN, and CRISPR/Cas-based methods for genome engineering. Trends Biotechnol 31(7):397–405
Gallage NJ, Hansen EH, Kannangara R et al (2014) Vanillin formation from ferulic acid in Vanilla planifolia is catalysed by a single enzyme. Nat Commun 5:4037
Gang DR, Lavid N, Zubieta C et al (2002) Characterization of phenylpropene O-methyltransferases from sweet basil: facile change of substrate specificity and convergent evolution within a plant O-methyltransferase family. Plant Cell 14(2):505–519
Gao J, Wang G, Ma S et al (2015) CRISPR/Cas9-mediated targeted mutagenesis in Nicotiana tabacum. Plant Mol Biol 87(1–2):99–110
Giddings LA, Liscombe DK, Hamilton JP et al (2011) A stereoselective hydroxylation step of alkaloid biosynthesis by a unique cytochrome P450 in Catharanthus roseus. J Biol Chem 286(19):16751–16757
Gilles AF, Averof M (2014) Functional genetics for all: engineered nucleases, CRISPR and the gene editing revolution. Evodevo 5:43
Gnerre S, Maccallum I, Przybylski D et al (2011) High-quality draft assemblies of mammalian genomes from massively parallel sequence data. Proc Natl Acad Sci USA 108(4):1513–1518
Góngora-Castillo E, Childs KL, Fedewa G et al (2012) Development of transcriptomic resources for interrogating the biosynthesis of monoterpene indole alkaloids in medicinal plant species. PLoS One 7(12):e52506
Guilinger JP, Pattanayak V, Reyon D et al (2014) Broad specificity profiling of TALENs results in engineered nucleases with improved DNA cleavage specificity. Nat Method 11(4):429–435
Haas BJ, Papanicolaou A, Yassour M et al (2013) De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat Protoc 8(8):1494–1512
Hackl T, Hedrich R, Schultz J et al (2014) proovread: large-scale high-accuracy PacBio correction through iterative short read consensus. Bioinformatics 30(21):3004–3011
Havelek R, Seifrtova M, Kralovec K et al (2014) The effect of Amaryllidaceae alkaloids haemanthamine and haemanthidine on cell cycle progression and apoptosis in p53-negative human leukemic Jurkat cells. Phytomedicine 21(4):479–490
Havkin-Frenkel D, Podstolski A, Dixon R (2003) Vanillin biosynthetic pathway enzyme from Vanilla planifolia. US Patent 10/087,714, 9 Apr 2003
He M, Qu C, Gao O et al (2015) Biological and pharmacological activities of Amaryllidaceae alkaloids. RSC Adv 5:16562–16574
Hopley C, Bristow T, Lubben A et al (2008) Towards a universal product ion mass spectral library—reproducibility of product ion spectra across eleven different mass spectrometers. Rapid Commun Mass Spectrom 22(12):1779–1786
Ibdah M, Zhang XH, Schmidt J et al (2003) A novel Mg(2+)-dependent O-methyltransferase in the phenylpropanoid metabolism of Mesembryanthemum crystallinum. J Biol Chem 278(45):43961–43972
Ikezawa N, Tanaka M, Nagayoshi M et al (2003) Molecular cloning and characterization of CYP719, a methylenedioxy bridge-forming enzyme that belongs to a novel P450 family, from cultured Coptis japonica cells. J Biol Chem 278(40):38557–38565
Irmler S, Schröder G, St-Pierre B et al (2000) Indole alkaloid biosynthesis in Catharanthus roseus: new enzyme activities and identification of cytochrome P450 CYP72A1 as secologanin synthase. Plant J 24(6):797–804
Itkin M, Heinig U, Tzfadia O et al (2013) Biosynthesis of antinutritional alkaloids in solanaceous crops is mediated by clustered genes. Science 341(6142):175–179
Jeffs P (1962) The Alkaloids of the Amaryllidaceae. Part X. Biosynthesis of haemanthamine. In: Proceedings of the chemical society of London, pp 80–81
Jia H, Wang N (2014) Targeted genome editing of sweet orange using Cas9/sgRNA. PLoS One 9(4):e93806
Jiang J, He X, Cane DE (2006) Geosmin biosynthesis. Streptomyces coelicolor germacradienol/germacrene D synthase converts farnesyl diphosphate to geosmin. J Am Chem Soc 128(25):8128–8129
Jiang Y, Xia N, Li X et al (2011) Molecular cloning and characterization of a phenylalanine ammonia-lyase gene (LrPAL) from Lycoris radiata. Mol Biol Rep 38(3):1935–1940
Jiang W, Zhou H, Bi H et al (2013a) Demonstration of CRISPR/Cas9/sgRNA-mediated targeted gene modification in Arabidopsis, tobacco, sorghum and rice. Nucleic Acids Res 41(20):e188
Jiang Y, Xia B, Liang L et al (2013b) Molecular and analysis of a phenylalanine ammonia-lyase gene (LrPAL2) from Lycoris radiata. Mol Biol Rep 40(3):2293–2300
Jin Z (2009) Amaryllidaceae and Sceletium alkaloids. Nat Prod Rep 26(3):363–381
Jin Z (2013) Amaryllidaceae and Sceletium alkaloids. Nat Prod Rep 30(6):849–868
Jinek M, Chylinski K, Fonfara I et al (2012) A programmable dual-RNA–guided DNA endonuclease in adaptive bacterial immunity. Science 337(6096):816–821
Johnson RA, Gurevich V, Filler S et al (2015) Comparative assessments of CRISPR-Cas nucleases’ cleavage efficiency in planta. Plant Mol Biol 87(1–2):143–156
Jörnvall H, Persson B, Krook M et al (1995) Short-chain dehydrogenases/reductases (SDR). Biochemistry 34(18):6003–6013
Kamenetsky R, Faigenboim A, Shemesh Mayer E et al (2015) Integrated transcriptome catalogue and organ-specific profiling of gene expression in fertile garlic (Allium sativum L.). BMC Genom 16(1):12
Kato M, Mizuno K, Crozier A et al (2000) Caffeine synthase gene from tea leaves. Nature 406(6799):956–957
Kavanagh KL, Jörnvall H, Persson B et al (2008) Medium- and short-chain dehydrogenase/reductase gene and protein families: the SDR superfamily: functional and structural diversity within a family of metabolic and regulatory enzymes. Cell Mol Life Sci 65(24):3895–3906
Kellner F, Kim J, Clavijo BJ et al (2015) Genome-guided investigation of plant natural product biosynthesis. Plant J 82(4):680–692
Kilgore MB, Augustin MM, Starks CM et al (2014) Cloning and characterization of a norbelladine 4′-O-methyltransferase involved in the biosynthesis of the Alzheimer’s drug galanthamine in Narcissus sp. aff. pseudonarcissus. PLoS One 9(7):e103223
Kim Y, Schumaker KS, Zhu JK (2006) EMS mutagenesis of Arabidopsis. Methods Mol Biol 323:101–103
Kirby GW, Tiwari HP (1966) Phenol oxidation and biosynthesis. Part IX. The biosynthesis of norpluviine and galanthine. J Chem Soc (January):676–682
Kleinstiver BP, Prew MS, Tsai SQ et al (2015) Engineered CRISPR-Cas9 nucleases with altered PAM specificities. Nature 523(7561):481–485
Kornienko A, Evidente A (2008) Chemistry, biology, and medicinal potential of narciclasine and its congeners. Chem Rev 108(6):1982–2014
Krauss M, Singer H, Hollender J (2010) LC-high resolution MS in environmental analysis: from target screening to the identification of unknowns. Anal Bioanal Chem 397(3):943–951
Langmead B (2010) Aligning short sequencing reads with Bowtie. Curr Protoc Bioinform Chapter: 11, Unit: 11–7. doi:10.1002/0471250953.bi1107s32
Larsen PA, Heilman AM, Yoder AD (2014) The utility of PacBio circular consensus sequencing for characterizing complex gene families in non-model organisms. BMC Genom 15:720
Lau W, Sattely ES (2015) Six enzymes from mayapple that complete the biosynthetic pathway to the etoposide aglycone. Science 349(6253):1224–1228
Lee H, Gurtowski J, Yoo S et al (2014) Error correction and assembly complexity of single molecule sequencing reads. bioRxiv. doi:10.1101/006395
Lehmann T, Pollmann S (2009) Gene expression and characterization of a stress-induced tyrosine decarboxylase from Arabidopsis thaliana. FEBS Lett 583(12):1895–1900
Lester DR, Ross JJ, Davies PJ et al (1997) Mendel’s stem length gene (Le) encodes a gibberellin 3 beta-hydroxylase. Plant Cell 9(8):1435–1443
Li H, Durbin R (2009) Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25(14):1754–1760
Li R, Zhu H, Ruan J et al (2010) De novo assembly of human genomes with massively parallel short read sequencing. Genome Res 20(2):265–272
Li R, Wang MY, Li XB (2012) Chemical constituents and biological activities of genus Hosta (Liliaceae). J Med Plant Res 6(14):2704–2713
Liu J, Hu WX, He LF et al (2004) Effects of lycorine on HL-60 cells via arresting cell cycle and inducing apoptosis. FEBS Lett 578(3):245–250
Liu R, Loraine AE, Dickerson JA (2014) Comparisons of computational methods for differential alternative splicing detection using RNA-seq in plant systems. BMC Bioinformatics 15:364
Loman NJ, Watson M (2015) Successful test launch for nanopore sequencing. Nat Methods 12(4):303–304
Lu G, Zou Q, Guo D et al (2007) Agrobacterium tumefaciens-mediated transformation of Narcissus tazzeta var. chinensis. Plant Cell Rep 26(9):1585–1593
Ma X, Zhang Q, Zhu Q et al (2015) A robust CRISPR/Cas9 system for convenient, high-efficiency multiplex genome editing in monocot and dicot plants. Mol Plant 8(8):1274–1284
Madoui MA, Engelen S, Cruaud C et al (2015) Genome assembly using Nanopore-guided long and error-free DNA reads. BMC Genom 16(1):327
Mahrous EA, Farag MA (2015) Two dimensional NMR spectroscopic approaches for exploring plant metabolome: a review. J Adv Res 6(1):3–15
Mali P, Yang L, Esvelt KM et al (2013) RNA-guided human genome engineering via Cas9. Science 339(6121):823–826
Mazzaferro LS, Hüttel W, Fries A et al (2015) Cytochrome P450-catalyzed regio- and stereoselective phenol coupling of fungal natural products. J Am Chem Soc 137(38):12289–12295
McCoy RC, Taylor RW, Blauwkamp TA et al (2014) Illumina TruSeq synthetic long-reads empower de novo assembly and resolve complex, highly-repetitive transposable elements. PLoS ONE 9(9):e106689
Mikami M, Toki S, Endo M (2015) Comparison of CRISPR/Cas9 expression constructs for efficient targeted mutagenesis in rice. Plant Mol Biol 88(6):561–572
Mikheyev AS, Tin MM (2014) A first look at the Oxford Nanopore MinION sequencer. Mol Ecol Resour 14(6):1097–1102
Misra BB, van der Hooft JJJ (2015) Updates in metabolomics tools and resources: 2014–2015. Electrophoresis. doi:10.1002/elps.201500417
Miyakado M, Kato T, Ohno N et al (1975) Alkaloids of Urginea altissima and their antimicrobial activity against Phytophthora capsici. Phytochemistry 14(12):2717
Morishige T, Dubouzet E, Choi KB et al (2002) Molecular cloning of columbamine O-methyltransferase from cultured Coptis japonica cells. Eur J Biochem 269(22):5659–5667
Mulholland DA, Crouch N, Decker B et al (2002) The isolation of the Amaryllidaceae alkaloid crinamine from Dioscorea dregeana (Dioscoreaceae). Biochem Syst Ecol 30(2):183–185
Nelson D, Werck-Reichhart D (2011) A P450-centric view of plant evolution. Plant J 66(1):194–211
Nützmann HW, Osbourn A (2015) Regulation of metabolic gene clusters in Arabidopsis thaliana. New Phytol 205(2):503–510
Okada A, Okada K, Miyamoto K et al (2009) OsTGAP1, a bZIP transcription factor, coordinately regulates the inductive production of diterpenoid phytoalexins in rice. J Biol Chem 284(39):26510–26518
Ono E, Nakai M, Fukui Y et al (2006) Formation of two methylenedioxy bridges by a Sesamum CYP81Q protein yielding a furofuran lignan, (+)-sesamin. Proc Natl Acad Sci USA 103(26):10116–10121
Ounaroon A, Decker G, Schmidt J et al (2003) (R, S)-Reticuline 7-O-methyltransferase and (R, S)-norcoclaurine 6-O-methyltransferase of Papaver somniferum—cDNA cloning and characterization of methyl transfer enzymes of alkaloid biosynthesis in opium poppy. Plant J 36(6):808–819
Page DR, Grossniklaus U (2002) The art and design of genetic screens: arabidopsis thaliana. Nat Rev Genet 3(2):124–136
Pichersky E, Lewinsohn E (2011) Convergent evolution in plant specialized metabolism. Annu Rev Plant Biol 62:549–566
Pohl T, Koorbanally C, Crouch NR et al (2001) Bufadienolides from Drimia robusta and Urginea altissima (Hyacinthaceae). Phytochemistry 58(4):557–561
Porté S, Xavier Ruiz F, Giménez J et al (2013) Aldo–keto reductases in retinoid metabolism: search for substrate specificity and inhibitor selectivity. Chem Biol Interact 202(1–3):186–194
Prachayasittikul S, Buraparuangsang P, Worachartcheewan A et al (2008) Antimicrobial and antioxidative activities of bioactive constituents from Hydnophytum formicarum Jack. Molecules 13(4):904–921
Rajkumar H, Ramagoni RK, Anchoju VC et al (2015) De novo transcriptome analysis of Allium cepa L. (onion) bulb to identify allergens and epitopes. PLoS One 10(8):e0135387
Raman SB, Rathinasabapathi B (2003) β-alanine N-methyltransferase of Limonium latifolium. cDNA cloning and functional expression of a novel N-methyltransferase implicated in the synthesis of the osmoprotectant beta-alanine betaine. Plant Physiol 132(3):1642–1651
Robertson G, Schein J, Chiu R et al (2010) De novo assembly and analysis of RNA-seq data. Nat Methods 7(11):909–912
Ron M, Kajala K, Pauluzzi G et al (2014) Hairy root transformation using Agrobacterium rhizogenes as a tool for exploring cell type-specific gene expression and function using tomato as a model. Plant Physiol 166(2):455–469
Rønsted N, Symonds MR, Birkholm T et al (2012) Can phylogeny predict chemical diversity and potential medicinal activity of plants? A case study of Amaryllidaceae. BMC Evol Biol 12:182
Ru Q, Wang X, Liu T et al (2013) Physiological and comparative proteomic analyses in response to nitrogen application in an Amaryllidaceae plant, Lycoris aurea. Acta Physiol Plant 35(1):271–282
Salmela L, Rivals E (2014) LoRDEC: accurate and efficient long read error correction. Bioinformatics 30(24):3506–3514
Schlauer J, Rückert M, Wiesen B et al (1998) Characterization of enzymes from Ancistrocladus (Ancistrocladaceae) and Triphyophyllum (Dioncophyllaceae) catalyzing oxidative coupling of naphthylisoquinoline alkaloids to michellamines. Arch Biochem Biophys 350(1):87–94
Schoch G, Goepfert S, Morant M et al (2001) CYP98A3 from Arabidopsis thaliana is a 3′-hydroxylase of phenolic esters, a missing link in the phenylpropanoid pathway. J Biol Chem 276(39):36566–36574
Schulz MH, Zerbino DR, Vingron M et al (2012) Oases: robust de novo RNA-seq assembly across the dynamic range of expression levels. Bioinformatics 28(8):1086–1092
Schweigert N, Zehnder AJ, Eggen RI (2001) Chemical properties of catechols and their molecular modes of toxic action in cells, from microorganisms to mammals. Environ Microbiol 3(2):81–91
Sengupta D, Naik D, Reddy AR (2015) Plant aldo–keto reductases (AKRs) as multi-tasking soldiers involved in diverse plant metabolic processes and stress defense: a structure-function update. J Plant Physiol 179:40–55
Senthil-Kumar M, Mysore KS (2014) Tobacco rattle virus-based virus-induced gene silencing in Nicotiana benthamiana. Nat Protoc 9(7):1549–1562
Shan Q, Wang Y, Li J et al (2013) Targeted genome modification of crop plants using a CRISPR-Cas system. Nat Biotechnol 31(8):686–688
Simpson JT, Wong K, Jackman SD et al (2009) ABySS: a parallel assembler for short read sequence data. Genome Res 19(6):1117–1123
Smith J, Grizot S, Arnould S et al (2006) A combinatorial approach to create artificial homing endonucleases cleaving chosen sequences. Nucleic Acids Res 34(22):e149–e149
Spannagl M, Martis MM, Pfeifer M et al (2013) Analysing complex Triticeae genomes—concepts and strategies. Plant Methods 9(1):35
Stavrinides A, Tatsis EC, Foureau E et al (2015) Unlocking the diversity of alkaloids in Catharanthus roseus: nuclear localization suggests metabolic channeling in secondary metabolism. Chem Biol 22(3):336–341
Sugano SS, Shirakawa M, Takagi J et al (2014) CRISPR/Cas9-mediated targeted mutagenesis in the liverwort Marchantia polymorpha L. Plant Cell Physiol 55(3):475–481
Suhadolnik RJ, Fischer AG, Zulalian J (1962) The biogenic origin of the C6–C1 unit of lycorine. J Am Chem Soc 84(22):4348–4349
Suhadolnik RJ, Fischer AG, Zulalian J (1963a) Biogenesis of the Amaryllidaceae alkaloids. Part III. Phenylalanine and protocatechuic aldehyde as C6–C1 precursors of haemanthamine and lycorine. In: Proceedings of the chemical society of London, pp 132
Suhadolnik RJ, Fischer AG, Zulalian J (1963b) Biogenesis of the Amaryllidaceae alkaloids. II. Studies with whole plants, floral primordia and cell free extracts. Biochem Biophys Res Commun 11:208–212
Sumner LW, Lei Z, Nikolau BJ et al (2015) Modern plant metabolomics: advanced natural product gene discoveries, improved technologies, and future prospects. Nat Prod Rep 32(2):212–229
Sun X, Hu Z, Chen R et al (2015) Targeted mutagenesis in soybean using the CRISPR-Cas9 system. Sci Rep 5:10342
Takos AM, Rook F (2012) Why biosynthetic genes for chemical defense compounds cluster. Trends Plant Sci 17(7):383–388
Takos AM, Rook F (2013) Towards a molecular understanding of the biosynthesis of Amaryllidaceae alkaloids in support of their expanding medical use. Int J Mol Sci 14(6):11713–11741
Takos AM, Knudsen C, Lai D et al (2011) Genomic clustering of cyanogenic glucoside biosynthetic genes aids their identification in Lotus japonicus and suggests the repeated evolution of this chemical defence pathway. Plant J 68(2):273–286
Tanaka Y, Matsuoka M, Yamanoto N et al (1989) Structure and characterization of a cDNA clone for phenylalanine ammonia-lyase from cut-injured roots of sweet potato. Plant Physiol 90(4):1403–1407
Teutsch HG, Hasenfratz MP, Lesot A et al (1993) Isolation and sequence of a cDNA encoding the Jerusalem artichoke cinnamate 4-hydroxylase, a major plant cytochrome P450 involved in the general phenylpropanoid pathway. Proc Natl Acad Sci USA 90(9):4102–4106
Tsukazaki H, Yaguchi S, Sato S et al (2015) Development of transcriptome shotgun assembly-derived markers in bunching onion (Allium fistulosum). Mol Breeding 35(1):1–11
VanBuren R, Bryant D, Edger PP et al (2015) Single-molecule sequencing of the desiccation-tolerant grass Oropetium thomaeum. Nature. doi:10.1038/nature15714
Velten R, Erdelen C, Gehling M et al (1998) Cripowellin A and B, a novel type of Amaryllidaceae alkaloid from Crinum powellii. Tetrahedron Lett 39:1737–1740
Wang Z, Gerstein M, Snyder M (2009) RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet 10(1):57–63
Wang R, Xu S, Jiang Y et al (2013) De novo sequence assembly and characterization of Lycoris aurea transcriptome using GS FLX titanium platform of 454 pyrosequencing. PLoS One 8(4):e60449
Wetzl D, Berrera M, Sandon N et al (2015) Expanding the imine reductase toolbox by exploring the bacterial protein sequence space. ChemBioChem 16(12):1749–1756
Wildman WC, Fales HM, Battersby AR (1962a) Biosynthesis in the Amaryllidaceae. The incorporation of 3-C14-tyrosine in Sprekelia formosissima. J Am Chem Soc 84(4):681–682
Wildman WC, Battersby AR, Breuer SW (1962b) Biosynthesis in the Amaryllidaceae. Incorporation of 3-C14-tyrosine and phenylalanine in Nerine bowdenii W. Wats. J Am Chem Soc 84(23):4599–4600
Wildman WC, Fales HM, Highet RJ et al (1962c) Biosynthesis in the Amaryllidaceae: Evidence for intact incorporation of norbelladine into lycorine, crinamine, and belladine. In: Proceedings of the chemical society of London, pp 180–181
Wyvekens N, Topkar VV, Khayter C et al (2015) Dimeric CRISPR RNA-guided FokI-dCas9 nucleases directed by truncated gRNAs for highly specific genome editing. Hum Gene Ther 26(7):425–431
Xie K, Minkenberg B, Yang Y (2015) Boosting CRISPR/Cas9 multiplex editing capability with the endogenous tRNA-processing system. PNAS 112(11):3570–3575
Yeo YS, Nybo SE, Chittiboyina AG et al (2013) Functional identification of valerena-1,10-diene synthase, a terpene synthase catalyzing a unique chemical cascade in the biosynthesis of biologically active sesquiterpenes in Valeriana officinalis. J Biol Chem 288(5):3163–3173
Yin K, Han T, Liu G et al (2015) A geminivirus-based guide RNA delivery system for CRISPR/Cas9 mediated plant genome editing. Sci Rep 5:14926
Zerbino DR, Birney E (2008) Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res 18(5):821–829
Zhang H, Zhang J, Wei P et al (2014) The CRISPR/Cas9 system produces specific and homozygous targeted gene editing in rice in one generation. Plant Biotechnol J 12(6):797–807
Zhou SM, Chen LM, Liu SQ et al (2015) De novo assembly and annotation of the chinese chive (Allium tuberosum Rottler ex Spr.) cranscriptome using the illumina platform. PLoS One 10(7):e0133312
Zonneveld BJM (2008) The systematic value of nuclear DNA content for all species of Narcissus L. (Amaryllidaceae). Plant Syst Evol 275(1-2):109–132
Zonneveld B (2010) The involvement of Narcissus hispanicus Gouan in the origin of Narcissus bujei and of cultivated trumpet daffodils (Amaryllidaceae). Anales del Jardín Botánico de Madrid 67(1):29–39
Acknowledgments
This review was supported with the National Institutes of Health Award Number 1RC2GM092561 (NIGMS).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Kilgore, M.B., Kutchan, T.M. The Amaryllidaceae alkaloids: biosynthesis and methods for enzyme discovery. Phytochem Rev 15, 317–337 (2016). https://doi.org/10.1007/s11101-015-9451-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11101-015-9451-z