
Abstract
Multidrug resistance (MDR) represents one of the major problems in pharmacotherapy of important diseases (e.g., cancer, epilepsy). Although many factors may contribute to the development of MDR phenotype, the increased expression and/or functional activity of P-glycoprotein (P-gp; active drug efflux transporter) across the cell membrane has been recognized as the main one. Therefore, a great attention has been given to the search of P-gp inhibitors as therapeutic agents to reverse the MDR mediated by P-gp. Since the chemical entities identified over the last three decades as potential P-gp inhibitors did not show suitable pharmacological properties, more recently herbal components, such as flavonoid compounds, have gained a great interest as safe P-gp inhibitors. The interest in flavonoids as P-gp inhibitors is increasing due to their potential favourable characteristics, including selectivity and non-cytotoxic effects. Flavonoids integrate the third-generation non-pharmaceutical category of P-gp inhibitors, and some of them exhibited effects comparable to those of the classic P-gp inhibitors. In fact, some flavonoids found in foods and beverages of herbal origin appear to be quite promising to inhibit the P-gp–mediated drug efflux, indicating their potential value to enhance the systemic/cellular bioavailability of P-gp drug substrates when administrated in co-therapy. This review paper summarizes the current evidence of P-gp inhibitory effects produced by flavonoids, taking into account studies performed in cell-based in vitro models, in vivo animal models and clinical trials.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The multidrug resistance (MDR) was described for the first time in 1968 by Kessel and collaborators (Borska et al. 2010). MDR refers to the phenomenon of simultaneous resistance to various structurally and functionally unrelated drugs, and it may be developed in response to a specific drug or drug combination (Ling 1997; van der Kolk et al. 2000; Deeley et al. 2006). Moreover, it often encompasses agents to which individuals have not been previously exposed, and which may or may not share targets and mechanisms of action with those that elicited the development of drug resistance (Deeley et al. 2006). Actually, the resistance to drug treatment still represents one of the major problems in the pharmacotherapy of several diseases, including cancer, infectious diseases and brain disorders such as epilepsy (Brandt et al. 2006).
Up to date, one of the most important mechanisms implicated in MDR is the over-expression and/or increased activity of drug efflux transporters belonging to the adenosine triphosphate (ATP)-binding cassette (ABC) transporter superfamily (Girardin 2006; Chang 2007; Borska et al. 2010; Giacomini et al. 2010). Many of the ABC drug efflux transporters are primarily located in the cell membrane where they can extrude a variety of structurally diverse drugs, drug conjugates and metabolites, and other compounds from the inside of cells (Schinkel and Jonker 2012). In all mammalian species, these multidomain integral membrane proteins use energy of ATP hydrolysis to the translocation of solutes across cellular membranes against the concentration gradient (Löscher and Potschka 2005), reducing, consequently, the intracellular concentration of the substrates (Dantzig et al. 2003; Löscher and Potschka 2005; Schinkel and Jonker 2012). Indeed, drugs ability to cross cellular barriers and reach their therapeutic targets in appropriate concentrations is critical for their efficacy (Chan et al. 2004), and it may be considerably determined through the active efflux mediated by membrane proteins (Pérez-Tomás 2006). Thus, transmembrane efflux proteins can be major determinants of the pharmacokinetics, safety and efficacy profiles of drugs (Giacomini et al. 2010). These membrane transporters are expressed in different cells and tissues such as lymphocytes, intestine, liver, kidney, testis, placenta, and central nervous system (CNS) and, therefore, they are critical to the disposition of many xenobiotics (Wang et al. 2002). In fact, the pharmacological properties of many drugs in current use are affected by these membrane transporters, which may determine the rate and extent of the absorption, distribution and elimination processes. Furthermore, the ABC transporters can extensively limit the drug penetration into specific cells and tissue compartments, restricting the access of drugs into important pharmacological sanctuaries such as brain, testis and fetus. Therefore, the clinical efficacy and toxicity of many drugs may be determined on a large extent by drug interactions with ABC transporters (Schinkel and Jonker 2012).
Although the family of mammalian ABC transporters is extensive and functionally highly diverse, the P-glycoprotein (P-gp), the MDR-associated proteins (MRPs) 1–5 (MRP1, MRP2, MRP3, MRP4 and MRP5), and the breast cancer resistance protein (BCRP) have demonstrated a clinical significant role in the transport of relevant drugs (Schinkel and Jonker 2012).
P-glycoprotein
P-glycoprotein is certainly the most studied drug efflux membrane transporter of the ABC transporter superfamily (Schinkel and Jonker 2012). P-gp was originally discovered in 1976 by Juliano and Ling as a surface glycoprotein, which was over-expressed in drug resistant ovary cells mutants from Chinese hamster (Juliano and Ling 1976; Linnet and Ejsing 2008; Miller et al. 2009; Zhang et al. 2012). The discovery of P-gp, more than 35 years ago, demonstrated that a single protein may confer resistance to a relatively large number of structurally diverse drugs with different mechanisms of action (Deeley et al. 2006). Consequently, the resistance conferred by P-gp efflux transporter is the best characterized process among the various putative mechanisms of MDR, and it is of considerable clinical importance (Wang et al. 2002; Löscher and Potschka 2005; Bansal et al. 2009). This transmembrane active efflux system has been found in numerous species (insects, fish, amphibians, reptiles, birds and mammalians), although there are differences among them (Löscher and Potschka 2002a; Fortuna et al. 2011).
In humans, P-gp is encoded by two MDR genes, MDR1 (ABCB1) and MDR3 (ABCB4), being the latter also designated MDR2; on the other hand, mice express mdr1a (abcb1a), mdr1b (abcb1b) and mdr2 (abcb4) (Tandon et al. 2006; Hennessy and Spiers 2007; del Amo et al. 2009; Zhang et al. 2012). The human gene encoding for MDR associated to P-gp is MDR1 (ABCB1), while in mice are mdr1a (abcb1a) and mdr1b (abcb1b) (Fortuna et al. 2011). The combined tissue distribution of mdr1a and mdr1b transcripts in rodents roughly coincides with that of the MDR1 mRNA transcripts in humans (Kwan and Brodie 2005; Linnet and Ejsing 2008; del Amo et al. 2009). Hence, it is believed that those two genes in rodents perform the same functions of the human MDR1 (Löscher and Potschka 2002a; Chan et al. 2004; Kwan and Brodie 2005; Linnet and Ejsing 2008; Zhang et al. 2012). There is 82 % homology between human MDR1 and mouse mdr1a, and the corresponding value between human MDR1 and mouse mdr1b is 79 % (del Amo et al. 2009). The denomination P-gp usually refers to the protein encoded by MDR1, mdr1a and mdr1b and, therefore, it will be herein followed.
The human P-gp is predominantly and physiologically expressed at the apical/luminal membrane of polarized cells in several normal tissues with secretory (small intestine, liver, kidney, adrenal gland) and barrier functions (small intestine, blood–brain barrier, blood–testis barrier, blood–ovarian barrier and placenta) (Volk et al. 2004; Fromm 2004; Giacomini et al. 2010; Marquez and Van Bambeke 2011). Indeed, P-gp forms a functional barrier that protects the body by actively limiting the absorption and systemic distribution of xenobiotic compounds, and/or increasing their elimination together with xenobiotic-metabolizing enzymes (Löscher and Potschka 2002b; Dantzig et al. 2003; Volk et al. 2004; Fromm 2004; Kwan and Brodie 2005; del Amo et al. 2009). Thus, many xenobiotics are metabolized and/or excreted into bile, urine, and intestinal lumen, preventing their accumulation in body tissues (Fromm 2004; Tandon et al. 2006). In fact, the strategic physiological expression of P-gp in organs that play key roles in the processes of drug absorption and disposition suggest that P-gp has a relevant impact on the pharmacokinetics of its substrate molecules, including many clinically important therapeutic agents (Tandon et al. 2006; del Amo et al. 2009; Fortuna et al. 2011). Moreover, P-gp has also been associated to other physiological functions such as the transport of steroid hormones, transport of ions, and secretion of cytokines (del Amo et al. 2009). In addition, emerging evidence shows that P-gp expression is also changed at the level of blood–brain barrier during pathophysiology and/or therapy of CNS diseases such as epilepsy and stroke (Löscher and Potschka 2002b; Seegers et al. 2002; Kubota et al. 2006; Miller et al. 2009), as well as in multidrug resistant tumour cells (Zhang et al. 2012).
The human P-gp contains two homologous and symmetrical distinct halves, called NH2- and COOH-terminal halves (Fig. 1) (Yang et al. 2008; del Amo et al. 2009; Schinkel and Jonker 2012; Breier et al. 2013). Each half comprises a transmembrane domain (TMD) and a cytosolic nucleotide-binding domain (NBD) or ABC unit (de Wet et al. 2001; Barron et al. 2003; Linnet and Ejsing 2008; del Amo et al. 2009; Zhang et al. 2012; Breier et al. 2013). The two halves are joined by a 60 amino acid linker region (Ramakrishnan 2003) and each TDM is composed by six transmembrane α-helix segments (TMs) (Di Pietro et al. 2002; Breier et al. 2013). The TMDs mediate the binding and efflux transport of P-gp substrates using energy derived from the hydrolysis of ATP at the NBD (Sheu et al. 2010). The NBDs are peripherally located at the cytoplasmic face of the cell membrane and they bind ATP and couple ATP hydrolysis with the drug transport process (Chan et al. 2009). The twelve TMs fold together to form a barrel-like structure that crosses the plasma membrane (Ramakrishnan 2003; Linnet and Ejsing 2008). The substrate specificity for P-gp is determined by the TM5, TM6, TM11 and TM12, which seem to be integrated to form a large drug binding pocket (Bellamy 1996; Breier et al. 2013). The drug recognition by P-gp, followed by ATP-binding and subsequent hydrolysis are essential to actively export relatively lipophilic substrates out of the lipid bilayer of the cell membrane to the extracellular medium (Tandon et al. 2006; Bansal et al. 2009; del Amo et al. 2009).

P-glycoprotein-mediated drug transport
Several models have been proposed to explain the mechanism of P-gp-mediated drug transport. The pore, the flippase and the hydrophobic vacuum cleaner are the most popular models of P-gp function (Fig. 2) (Krishna and Mayer 2000; Ramakrishnan 2003; Varma 2003; Hennessy and Spiers 2007; Bansal et al. 2009; del Amo et al. 2009).

Models proposed to explain the P-glycoprotein function: the pore (a), the flippase (b) and the hydrophobic vacuum cleaner model (c). ADP Adenosine diphosphate; ATP Adenosine triphosphate; R Substrate (Varma 2003)
According to the pore model, in the absence of the nucleotide, both TMDs form a single barrel with a central pore that opens to the extracellular surface and spans much of the membrane depth. On the contrary, upon binding of nucleotides, the TDMs reorganize into three compact domains that open the central pore along its length, allowing the direct access of hydrophobic drugs from the lipid bilayer to the central pore of the transporter (Fig. 2a) (Varma 2003; Bansal et al. 2009). P-gp has also been suggested to function as a flippase that flips the substrate from the inner leaflet of the lipid bilayer to the outer leaflet of the plasma membrane, or directly to the extracellular environment against the concentration gradient (Fig. 2b) (Ramakrishnan 2003; Hennessy and Spiers 2007; Bansal et al. 2009; del Amo et al. 2009). The flippase model is based on the idea that after interaction of the substrate with the membrane, the substrate can access to the core of TMDs from the lipid bilayer and, then, P-gp flips the drug from the inner to the outer leaflet in an ATP hydrolysis-dependent manner (Ramakrishnan 2003). The assumptions of the substrates partition into the lipid phase prior to interacting with P-gp and the location of a P-gp binding pocket in the internal part of the plasma membrane are considered in the both models (Hennessy and Spiers 2007; del Amo et al. 2009). These characteristics may explain the unusually broad substrate specificity of P-gp since the primary determinant for substrate specificity seems to be its ability to intercalate into the lipid bilayer appropriately, being the subsequent interaction with the substrate-binding site of secondary importance (Hennessy and Spiers 2007). Finally, the hydrophobic vacuum cleaner (Fig. 2c) combines the features of pore and flippase models (Varma 2003) and the designation of hydrophobic is due to the fact that the majority of P-gp substrates have been found to be hydrophobic compounds with a planar ring system, and often carrying a positive charge at physiological pH (Bansal et al. 2009). This model suggests that P-gp binds directly to the hydrophobic compounds embedded in the inner leaf of the plasma membrane and transports the substrate from the lipid bilayer to the extracellular medium through a protein channel (Krishna and Mayer 2000; Ramakrishnan 2003; Hennessy and Spiers 2007; Bansal et al. 2009; del Amo et al. 2009). Structural data indicate that the substrates could access to their binding sites through gates formed between TMs 5/8 and 2/11 (Hennessy and Spiers 2007). However, other mechanisms are also suggested particularly because uncharged hydrophilic molecules with planar ring systems (e.g. colchicine) are also effluxed out by P-gp. The change in electric potential and pH across membrane seems to contribute to that process (Bansal et al. 2009) as well as the conformational changes that P-gp undergoes when nucleotide binds to the intracellular NBDs (Varma 2003; Bansal et al. 2009).
Structurally, human P-gp is a 170 kDa transmembrane glycoprotein composed by 1,280 amino acids (Bellamy 1996; Schinkel 1999; Ramakrishnan 2003; Kwan and Brodie 2005; Linnet and Ejsing 2008). This well-known drug efflux transporter contains more than one substrate binding domain and extrudes a wide range of structurally and functionally unrelated compounds. P-gp is unique in its ability to recognize and transport a plethora of diverse substrates with variable chemical properties and pharmacological actions. Indeed, P-gp substrates include several clinically important drugs from different pharmacotherapeutic or pharmacological classes (Table 1) (Hennessy and Spiers 2007; Miller et al. 2009; Borska et al. 2010; Fortuna et al. 2011). However, the ability of P-gp to transport such enormous variety of chemical compounds is still poorly understood (Raub 2006). Moreover, it has been shown that drug–drug interactions involving P-gp may decrease the therapeutic efficacy and/or potentiate toxic effects. This problem results from the large number of compounds that may interact with P-gp either as a substrate, inhibitor or inducer (Fortuna et al. 2011). Therefore, one of the key points that are increasingly considered in programs of drug discovery and development consists on testing new chemical entities as P-gp substrates. Indeed, drug candidates that are substrates of P-gp may be “victims” of drug–drug interactions where this transporter is inhibited or induced (Lin and Yamazaki 2003; Doran et al. 2005; Chang et al. 2006; Sugimoto et al. 2011; Palmeira et al. 2011). Hence, this type of studies are recommended and may be useful in the decision-making processes for optimization and selection of the most promissory drug candidates (Mizuno et al. 2003; Balayssac et al. 2005; Guidance for Industry, FDA 2006; Giacomini et al. 2010).
Furthermore, the understanding of the physiological role of P-gp as a natural detoxification mechanism in various human tissues (Lin 2003; Balayssac et al. 2005; Zhang et al. 2006; Chang et al. 2006) and the recognition of the involvement of P-gp in MDR (Matheny et al. 2001) have motivated the search of P-gp inducers and inhibitors with potential interest respectively in cases of poisoning and therapeutic inefficacy (Huisman et al. 2003; Dinis-Oliveira et al. 2006).
P-glycoprotein inhibitors: an overview
The recognition that P-gp-mediated MDR is clinically important in several diseases has been followed by concerted efforts to search for therapeutically useful P-gp inhibitors in order to overcome that functional barrier and allow drug penetration into the target tissue (Tandon et al. 2006; Hennessy and Spiers 2007). The pharmacokinetics, efficacy and safety of P-gp substrates can be affected by the level of expression and functionality of P-gp, which can be modulated by inhibition and induction (Giacomini et al. 2010). If P-gp inhibitors or reversal agents become available, it might be possible, for example, to administer drugs targeted to the brain concomitantly with reversal agents, which would overcome the extrusion of P-gp substrates from the brain, increasing its concentration at the biophase and, consequently, the intended therapeutic effects (Ramakrishnan 2003).
Although initial thoughts on the clinical application of P-gp inhibitors were focused on reversing MDR in chemo-resistant tumour cells that express significant amounts of P-gp, more recent insights indicated that such inhibitors might also be useful to modulate the general pharmacokinetic behaviour of drugs in the body (Schinkel and Jonker 2012). Therefore, P-gp inhibitors are currently in development not only to increase drug penetration into malignant tissues in order to optimize chemotherapy, but also to increase brain penetration of CNS-active drugs to circumvent the pharmacoresistance associated with several CNS diseases (e.g. epilepsy) (Kemper et al. 2003; Miller et al. 2009; Bansal et al. 2009; Coley 2010; Thomas and Coley 2013).
A P-gp inhibitor should have a high propensity for binding to P-gp without inhibiting other ABC transporters or interacting with cytochrome P450 (CYP) isoenzymes. Furthermore, it should be selectively targeted to P-gp in non-physiological tissues with a high affinity and individually adapted to the recipient’s genotype. Thus, a non-toxic compound which does not have any pharmacological activity of its own is considered the ideal P-gp inhibitor (Bansal et al. 2009). P-gp inhibitors, even though structurally divergent, are responsible for decreased drug extrusion and reversal of cellular MDR (de Wet et al. 2001). Considering their physicochemical properties, P-gp inhibitors apparently are hydrophobic molecules, with a molecular weight ranging from 200 to almost 1900 Da (Chan et al. 2004; Fromm 2004; Schinkel and Jonker 2012), with a planar ring system and weak positive charge at physiologic pH (Schinkel 1999; Kwan and Brodie 2005; Miller et al. 2009; Giacomini et al. 2010; Zhang et al. 2012). Some investigators suggest a general pharmacophore with two or three hydrogen bond acceptor (or electron donor) groups in a fixed spatial separation (Sun et al. 2003; Chan et al. 2004; Kwan and Brodie 2005). The general features of a substrate/inhibitor include also a planar aromatic domain, as well as the existence of at least one tertiary basic nitrogen atom on the drug molecule in deprotonated state at neutral pH (Sun et al. 2003; Breier et al. 2013). Thus, based on their chronological development, specificity and affinity to the efflux transporter, the P-gp inhibitors are now categorized into three generations (Dantzig et al. 2003; Varma 2003; Bansal et al. 2009).
The first-generation of P-gp inhibitors are pharmacologically active and they are in clinical use for other indications beyond the inhibition of P-gp (Dantzig et al. 2003; Varma 2003; Hennessy and Spiers 2007; Yang et al. 2008; Bansal et al. 2009; Potschka 2012). It includes calcium channel blockers such as verapamil (Fig. 3), anti-hypertensives like reserpine, quinidine and yohimbine, the immunosuppressant cyclosporin A (Fig. 3), anti-estrogens like tamoxifen and toremifene, and antiarrhytmic agents such as quinidine (Dantzig et al. 2003; Varma 2003; Hennessy and Spiers 2007; Bansal et al. 2009; Potschka 2012). However, these compounds have low potency and lack of selectivity for P-gp inhibition. Indeed, the clinical use of these agents at the concentrations necessary to inhibit P-gp is associated with severe toxicity (Krishna and Mayer 2000; Dantzig et al. 2003; Varma 2003; Chung et al. 2005; Hennessy and Spiers 2007; Bansal et al. 2009; Potschka 2012). For instance, the use of verapamil to inhibit P-gp leads to cardiotoxicity and the use of cyclosporin A induces an increase of hepatic, renal, myeloid, and neurological toxicity (Ramakrishnan 2003).

Chemical structure of some P-glycoprotein inhibitors belonging to the first-, second- and third-generations (PubChem Coumpound Database)
The second-generation of P-gp inhibitors was developed as derivatives of the first-generation compounds in order to retain the ability of the lead compound to inhibit P-gp, but decreasing the pharmacological activity and, consequently, the toxicity (Krishna and Mayer 2000; Dantzig et al. 2003; Varma 2003; Hennessy and Spiers 2007; Bansal et al. 2009; Potschka 2012). For example, the P-gp modulator valspodar lacks the immunosuppressive effect of cyclosporin A, although it is a close structural analogue of this drug (Fig. 3). On the other hand, dexverapamil (R-isomer of verapamil; Fig. 3) retains the ability to inhibit P-gp, but lacks the cardiovascular effects of its precursor (verapamil) (Dantzig et al. 2003; Hennessy and Spiers 2007). Although these compounds have been developed to be less toxic, some of their characteristics still limit their clinical usefulness. Indeed, the affinity of these compounds towards P-gp was shown to be too low to produce significant P-gp inhibition in vivo at tolerable doses (Bansal et al. 2009). Additionally, first- and second-generation P-gp inhibitors also lack specificity as they inhibit two or more ABC transporters, which leads to complicated drug–drug interactions (Varma 2003; Hennessy and Spiers 2007). For instance, valspodar inhibits not only P-gp but also MRP2 and the bile acid export protein (ABCB11) (Hennessy and Spiers 2007). Moreover, P-gp also has overlapping substrate specificity with CYP3A. Therefore, numerous studies have demonstrated clinically important drug–drug interactions when a P-gp inhibitor is co-administered with another drug metabolised by CYP3A4; as example, valspodar interferes with CYP enzyme activity thereby affecting clearance and systemic exposure of co-administered compounds (Hennessy and Spiers 2007; Potschka 2012). Thus, in spite of the early expected favourable contributions of the first- and second-generations of P-gp inhibitors, compounds have failed to yield the desired clinical benefit (Dantzig et al. 2003). Besides that, many first- and second-generation P-gp inhibitors are just competitive inhibitors, competing as substrates for the P-gp (Yang et al. 2008).
In contrast to first- and second-generation compounds, the third-generation of P-gp modulators has been designed to have an increased potency, inhibiting P-gp at concentrations in the nanomolar ranges, and to exhibit an increased specificity being simultaneously devoid of additional effects on drug metabolic enzymes. These include compounds as annamycin, elacridar, laniquidar, mitotane, OC 144-093, tariquidar and zosuquidar (Fig. 3) (Krishna and Mayer 2000; Dantzig et al. 2003; Hennessy and Spiers 2007; Potschka 2012). Bansal et al. (2009) presented a list of P-gp modulators belonging to different classes, which includes compounds that are inducers and inhibitors of P-gp. In the third-generation of P-gp inhibitors are included not only synthetic compounds, but also pharmaceutical excipients like polysorbates (Tween 20 or 80), herbs and fruits like curcumin or orange, and herbal constituents like flavonoids (Bansal et al. 2009). Among the herbal compounds, several new diterpene compounds isolated from Euphorbia spp. also showed to be potent MDR modulators acting specifically on P-gp transporter (Corea et al. 2009).
Competitive inhibitors, which are P-gp substrates themselves, and non-competitive inhibitors, are included in the three-generation of P-gp modulators. Non-competitive inhibitors can interfere with the activation and transport cycle of P-gp and induce alterations in the conformation of the transporter molecule which affect transport efficacy (Potschka 2012). Different mechanisms by which these inhibitors might interact with the transport proteins have been suggested (Brand et al. 2006). In general, P-gp can be inhibited by (a) blocking drug binding site either competitively, non-competitive or allosterically; (b) interfering with ATP hydrolysis; or (c) changing integrity of cell membrane lipids (Varma 2003; Brand et al. 2006). However, most of the drugs inhibits P-gp function by blocking drug binding sites (Varma 2003). Furthermore, many of the inhibitors have their potential use reduced in clinical settings probably because they seem to be relatively nonspecific and participate in unwanted drug–drug interactions or interference with other physiological systems (Brand et al. 2006). Thereby, it is urgent the emergence of new agents with potential to reverse the MDR mediated by P-gp. These drug candidates should be potent, non-cytotoxic, and have minimal adverse effects (Hadjeri et al. 2003).
Over the last years, phytochemicals are gaining a great attention as P-gp modulators (Bansal et al. 2009), because, as potentially non-toxic compounds, they present characteristics of ideal P-gp inhibitors (Bansal et al. 2008, 2009). Herbal products are taken by about 10 % or more of the general population, and by 30–70 % of individuals with specific diseases (Chung et al. 2005). Hence, it has also been reported that some phytochemicals found in vegetables, fruits and some plants, which have, among others, anticancer, antiviral, and antioxidant properties, may modulate or inhibit P-gp activity and may have potential to be developed as MDR reversing agents (Chung et al. 2005). Accordingly, herbal constituents are considered good candidates for increasing the bioavailability and tissue-penetration (e.g. overpass the blood–brain barrier) or decreasing biliary excretion of P-gp substrates, reversing thus the MDR; although there are also safety concerns related to the phytochemical compounds, their continuous and long history of use in large amounts as a part of normal daily diet constitutes a favourable feature (Bansal et al. 2009).
Flavonoids
For a long time, flavonoids have been consumed in order to improve health status of humans (Bansal et al. 2009); indeed, the medicinal use of natural products, such as extracts of plants, is now a common practice worldwide (Aszalos 2008). Since several flavonoids share some of the optimal properties of an ideal P-gp inhibitor, the interest on testing whether they inhibit P-gp has significantly increased during the search for selective and non-cytotoxic P-gp inhibitors (Brand et al. 2006; Bansal et al. 2009). Currently, they are considered good drug candidates (or lead compounds) as modulators of MDR due to their inhibitory activities on P-gp function and because of their physiological safety (Kitagawa et al. 2005).
Flavonoids consist on a large group of polyphenolic herbal constituents, which integrate the third-generation, non-pharmaceutical category of P-gp inhibitors (Ross and Kasum 2002; Brand et al. 2006; Bansal et al. 2009). Some of these compounds produced effects that are found to be comparable to those of the classic P-gp inhibitors, verapamil and cyclosporin A (Fig. 3) (Bansal et al. 2009). Actually, flavonoids and/or their metabolites are considered interesting modulators or substrates of intestinal membrane bound transport proteins, including P-gp (Brand et al. 2006; Chan et al. 2009).
The flavonoids are herbal secondary metabolites (Kitagawa 2006; Shohai et al. 2011; Thilakarathna and Rupasinghe 2013; Romano et al. 2013). According to Kitagawa (2006) and Sheu et al. (2010), flavonoids comprise more than 4,000 known compounds, resulting from the various combinations of multiple hydroxyl and methoxyl group substituents of the basic flavonoid skeleton (Fig. 4). Moon et al. (2006) reported more than 8000 compounds with a flavonoid structure. Although occasionally they are found, in nature, as aglycones, most of the flavonoids are attached to sugars (glycosides) (Ross and Kasum 2002; Morris and Zhang 2006). Many of the flavonoids have a very limited bioavailability because of high intestinal metabolism or poor absorption in vivo (Tsuji et al. 2013). In humans and animals, these compounds are absorbed from the gastrointestinal tract and are excreted in the urine and faeces, either unchanged or as flavonoid metabolites (Cook and Samman 1996; Morris and Zhang 2006; Brand et al. 2006). In the small intestine and liver, dietary flavonoids are substrates for phase I and phase II metabolizing enzymes (Wasowski and Marder 2012). Upon ingestion, and before oral absorption, flavonoid glycosides are deglycosylated, by lactase phlorizin hydrolase or cytosolic β-glucosidase. The compounds that are not absorbed in the intestine will reach the colon, where the enzymes of the gut microflora degrade flavonoids to simple phenolic acids, that may undergo absorption and further hepatic metabolism (Spencer 2007; Vauzour et al. 2008; Thilakarathna and Rupasinghe 2013). The absorbed aglycone is metabolized into glucuronide-, sulphate- and methoxylated conjugates (Brand et al. 2006; Spencer 2007; Vauzour et al. 2008; Bansal et al. 2009; Jäger and Saaby 2011; Wasowski and Marder 2012).

Chemistry
The term flavonoid is used to describe herbal pigments mostly derived from benzo-γ-pyrone (rings A and C; Fig. 4), with an assortment of basic structures (Hendrich 2006). These compounds are found in all vascular plants and have low molecular weights (Wasowski and Marder 2012; Romano et al. 2013). The basic structure of flavonoids consists of two benzene moieties (rings A and B, Fig. 4), which are linked in the middle through a heterocyclic pyran or pyrone with a double bond (ring C; Fig. 4) (Di Pietro et al. 2002; Ross and Kasum 2002; Shohai et al. 2011; Wasowski and Marder 2012; Tsuji et al. 2013). Hence, the flavonoids can be divided into several classes depending on their structure (Kitagawa 2006; Jäger and Saaby 2011), namely the oxidation status of ring C, the hydroxylation pattern of the ring structure and the substituent located on the 3rd position (Morris and Zhang 2006; Spencer 2007; Bansal et al. 2009; Alvarez et al. 2010; Tsuji et al. 2013). Taking into account the chemical nature of each molecule and the positions of moieties substituting rings A, B, and C, the flavonoids are divided into six major subclasses that are particularly well known and characterized and they correspond to the main dietary groups of flavonoids (Ross and Kasum 2002; Spencer 2007). These subclasses include the anthocyanidines (Fig. 5); the flavanols or catechins, in which the 2,3-bond is reduced, contain an additional hydroxyl substituent at position 3 of the ring C, and the carbonyl at position 4 is deleted (Fig. 6); the flavanones, where the 2,3-bond is reduced, losing electron conjugation and ring planarity (Fig. 7); the flavones (Fig. 8); the flavonols, which contain an additional hydroxyl substituent at the position 3 of the ring C and where the 4′,5′-bond is reduced (Fig. 9); and the isoflavones, where the ring B is branched at position 3 of the ring C (Fig. 10) (Ross and Kasum 2002; Deferme and Augustijns 2003; Hendrich 2006; Kitagawa 2006; Moon et al. 2006; Spencer 2007; Crozier et al. 2009; Sheu et al. 2010; Thilakarathna and Rupasinghe 2013). Indeed, flavonoids and isoflavonoids (e.g. isoflavones) are distinguished by the position at which the ring B is attached into the benzo-γ-pyrone core of the molecule. Thus, in flavonoids the ring B is bound at position 2 of ring C, whereas in isoflavonoids the ring B is bound at position 3 of ring C (Figs. 5, 6, 7, 8, 9, 10) (Hendrich 2006). Minor dietary flavonoids include dihydroflavonols, flavan-3,4-diols, coumarins, chalcones, dihydrochalcones, and aurones (Crozier et al. 2009). Although a modified number system is used for chalcones and isoflavones derivatives, the individual carbon atoms are identified with ordinary numerals for the ring A and the ring C, and “primed numerals” for the ring B (Fig. 4) (Tapas et al. 2008; Wasowski and Marder 2012).



Chemical structures of the compounds belonging to the flavanones subgroup of flavonoids and their glycosides (PubChem Coumpound Database; Horowitz and Gentili 1960; Wagner et al. 1969; Sacco and Maffei 1997; Rakwal et al. 2000; Ley et al. 2005; Ogawa et al. 2006; Miyake et al. 2007; Chokchaisiri et al. 2009; Sheu et al. 2010; Shohai et al. 2011; Jäger and Saaby 2011; Tran et al. 2011; Wesołowska 2011)

Chemical structures of the compounds belonging to the flavones subgroup of flavonoids and their glycosides (PubChem Coumpound Database; Cook and Samman 1996; Kim et al. 2004; Tapas et al. 2008; Crozier et al. 2009; Kim et al. 2010; Shohai et al. 2011; Rao et al. 2011; Tran et al. 2011; Wasowski and Marder 2012; Moreno Escobosa et al. 2012; Lam et al. 2012; Kumar et al. 2013)

Chemical structures of the compounds belonging to the flavonols subgroup of flavonoids and their glycosides (PubChem Coumpound Database; Harborne 1962; Cook and Samman 1996; Raj 2001; Hasegawa and Shirato 2002; Boumendjel et al. 2002; Tapas et al. 2008; Ali et al. 2008; Matsui et al. 2009; Zhou et al. 2009; Crozier et al. 2009; Sheu et al. 2010; Shohai et al. 2011; Jäger and Saaby 2011; Tran et al. 2011; Wasowski and Marder 2012; Alonso-Castro et al. 2013; Yang et al. 2013; Makino et al. 2013)

Chemical structures of the compounds belonging to the isoflavones subgroup of flavonoids and their glycosides (PubChem Coumpound Database; Keung and Vallee 1993; Khan et al. 2000; Tapas et al. 2008; Chokchaisiri et al. 2009; Sheu et al. 2010; Tran et al. 2011; Wesołowska 2011; Zhang et al. 2011; Zou et al. 2012; Shajib et al. 2012)
Structure–activity relationship
Specifically, there are structural requirements necessary for the inhibitory effects of flavonoids on P-gp function (Kitagawa et al. 2005). The structure–activity relationships for flavonoid–P-gp interaction have been extensively studied mainly evaluating the binding affinity of different flavonoids with mouse NBD2 (Morris and Zhang 2006). The presence of the 5-hydroxyl group, the 3-hydroxyl group, and the 2,3-double bond appears to be important for potent flavonoid-NBD2 interaction and are required for the P-gp inhibitory activity (Figs. 1, 11) (Conseil et al. 1998; Boumendjel et al. 2002; Hendrich 2006; Morris and Zhang 2006; Bansal et al. 2009). Isoflavonoids have lower P-gp interaction activity due to the different position where the ring B is branched (Boumendjel et al. 2002; Morris and Zhang 2006). Boumendjel et al. (2002) evaluate the binding affinity toward the NBD2 of P-gp of a set of naturally occurring flavones, isoflavones, flavonols, flavanones and chalcones. The structure–activity relationship analysis of flavonoids–P-gp concluded that flavonols, chalcones and flavones are the most active and the binding affinities are lower for flavanones and isoflavones (Boumendjel et al. 2002; Sheu et al. 2010). On the other hand, Sheu et al. (2010) conducted a study aiming to establish the qualitative and quantitative structure–activity relationships of flavonoids modulation effects on P-gp, using the human colorectal adenocarcinoma cells (HCT15) as an in vitro model and fexofenadine as a P-gp substrate probe. Accordingly, the flavonoid compounds with hydroxyl groups attached at the 5- and 7-positions of the ring A and with the total number of hydroxyl groups, at an optimal of three, possess a high inhibitory effect on P-gp activity (Fig. 11). However, flavonoids with four hydroxyl groups such as kaempferol (3,5,7,4′-tetrahydroxyflavone; Fig. 9) and fisetin (3,7,3′,4′-tetrahydroxyflavone; Fig. 9) have not shown this high inhibitory effect on P-gp activity, whereas flavonoids with a greater number of hydroxyl groups such as quercetin (3,5,7,3′,4-pentahydroxyflavone; Fig. 9) and taxifolin (3,5,7,3´,4′-pentahydroxyflavanone; Fig. 7), were able to enhance P-gp activity (Sheu et al. 2010). Wesołowska (2011) besides stating that flavonoids should possess hydroxyl groups at positions 3 and 5 of ring A and the 2,3-double bond to exhibit high affinity for NBD2 of P-gp, also described the importance of the carbonyl group at position 4 and a hydrophobic motif on either ring A or B (Fig. 11) (Wesołowska 2011). The planar structure of flavonoids also seems to be important for their interaction with P-gp (Fig. 11). As a result, for flavanones, which lack the double-bond between the 2- and 3-position in ring C, the stereoscopic relationship of the ring B with other rings (A and C) is different from that of flavones (with the double-bond in that position) (Figs. 7, 8). This double bond confers a special structure on flavonoids molecules that are largely planar so that they may readily intercalate between the hydrophobic amino acid residues of P-gp (Kitagawa et al. 2005).

Over the years, flavonoids have been subjected to various chemical modifications in order to obtain improved chemical entities as P-gp inhibitors. In general, it was found that chemical modifications that increased hydrophobicity of the flavonoid molecules, such as prenylation, geranylation, or a linear sequence of two isoprenyl residues, significantly increased the modulatory activity of P-gp (Wesołowska 2011). Therefore, in some cases, prenylated flavonoids bind with high affinity to P-gp and show an increased inhibitory potency (Di Pietro et al. 2002; Hendrich 2006). In this context, it was demonstrated that the introduction of alkoxy goups at position 4 of chalcones resulted in compounds much more actives, being recognised that the binding affinity increases as a function of the chain length (Bois et al. 1999); on the other side, it was reported that halogenated chalcones exhibited high-affinity binding to P-gp and that the increase in binding affinity is related to the increase in hydrophobicity of the halogenate substituent (Bois et al. 1998). The intracyclic oxygen may be also required, since, when it was replaced by a nitrogen atom, the P-gp inhibitory activity dramatically reduced (Fig. 11) (Kitagawa et al. 2005). The hydroxyl groups in polyphenols, such as those in the gallic acid moiety of tea catechins and alkyl gallates may be important in polar interactions with P-gp, possibly at the ATP-binding site. Accordingly, the hydrophobic moiety of polyphenols may be important for interaction at the steroid-interacting hydrophobic sequence of P-gp (Kitagawa 2006). Thus, the hydrophobicity of flavonoid compounds is a critical parameter for binding affinity towards NBD2 and consequently for their inhibitory effects on P-gp-mediated efflux of substrates (Di Pietro et al. 2002; Kitagawa et al. 2005; Kitagawa 2006). Thereby, the P-gp inhibitory activity of flavonoids increases as their molecular hydrophobicity increases (Kitagawa et al. 2005). Additionally, Wesołowska (2011) underlined the role of the hydrophobicity of ring substituents because the compounds containing geranyl groups are more active than those substituted by halogen atoms, which are followed by flavonoids with methoxy and hydroxyl groups. On the other hand, the results of a virtual screening of flavonoids as P-gp inhibitory agents indicated that glutamine-47, tyrosine-53, serine-83, isoleucine-87, glycine-100 and arginine-154 of the P-gp molecule are determinant residues for binding interactions as they establish strong hydrogen bonding with flavonoid molecules (Udaya Kumar et al. 2011). Udaya Kumar et al. (2011) also referred that for the stability of the complex an important role is played by hydrogen binding interactions.
Although detailed studies have been performed on the interaction of flavonoids with NBD2, a similar binding site certainly also exists on NBD1 since flavonoids compete with ATP. Indeed, Trompier et al. (2003) reported that a series of flavonoids bind to both NBDs (1 and 2) of the human MRP1, being stated that isoprenylation of flavonoid molecules leads to a preferential binding to NBD1. Thus, it is expected that additional binding(s) is(are) also found within the TMDs’ drug-binding sites, as described for MRP1 (Trompier et al. 2003).
Sources of flavonoids
The flavonoids are among the most ubiquitous groups of polyphenolic compounds found in foods and beverages of herbal origin (Boumendjel et al. 2002; Kitagawa et al. 2005; Badhan and Penny 2006; Brand et al. 2006; Wesołowska et al. 2009; Chan et al. 2009). These compounds, diverse in chemical structure and characteristics, are found in high levels in fruits, vegetables, nuts, stems, flowers, red wine, soybeans and tea (Cook and Samman 1996; Deferme and Augustijns 2003; Badhan and Penny 2006; Brand et al. 2006; Aszalos 2008; Chan et al. 2009). The anthocyanidines are mainly found in berries, cherries, grapes, red cabbage and red wine; the flavanols or catechins in apples, black tea, chocolate, bilberry, cocoa, grapes, hawthorn, motherwort, red wine and tea; the flavanones in tomato and citrus fruits and juices; the flavones in celery, honey, parsley, peppers, propolis, sweet red and thyme; the flavonols in apples, berries, broccoli, cherries, kale, leeks, lettuce, onions red wine, tea and tomato; and, finally, the isoflavones can be found in legumes like peas, red clover and soy (Moon et al. 2006; Alvarez et al. 2010; Shohai et al. 2011; Thilakarathna and Rupasinghe 2013; Toh et al. 2013).
Apart from food components, flavonoids are also present in medicinal herbs or dietary supplements including silibin, which is extracted from milk thistle, Silybum marianum, Alpina officinarum, Soy Isoflavones, Ginkgo Biloba or Hypericum perforatum (Hendrich 2006; Moon et al. 2006; Sheu et al. 2010). Although in the usual human diet, the intake level of these compounds can be up to several hundred milligrams a day (Wang et al. 2005; Badhan and Penny 2006; Brand et al. 2006), in people consuming over-the-counter botanical and dietary supplements the intake may be considerably higher (Lohner et al. 2007).
Biological properties and flavonoid–P-glycoprotein interactions
Flavonoids are water-soluble pigments and play diverse functions in the plant kingdom including defence, growth, development, photosensitization, ultraviolet protection, auxin transport inhibition, allelopathy and flower colouring (Buer et al. 2010; Shohai et al. 2011; Jäger and Saaby 2011). In 1936, Rusznyak and Szent-Gyorgyi reported the first observation regarding the biological activities of flavonoids (Ross and Kasum 2002). As integral constituents of the diet, they may exert a wide range of beneficial effects on human health in a multitude of disease states including cancer, cardiovascular diseases, neurodegenerative disorders and osteoporosis (Boumendjel et al. 2002; Badhan and Penny 2006; Morris and Zhang 2006; Brand et al. 2006; Wesołowska et al. 2009; Chan et al. 2009; Wasowski and Marder 2012). Moreover, there are recent evidence of an inverse relationship between dietary flavonoid intake and cardiovascular disease mortality and its risk factors (Toh et al. 2013). These broad spectrum of biological activities also include anti-inflammatory, antimicrobial, antiviral, anti-proliferative, pro-apoptotic, free-radical scavenging, antioxidant and hormonal effects (Boumendjel et al. 2002; Deferme and Augustijns 2003; Hendrich 2006; Badhan and Penny 2006; Brand et al. 2006; Wesołowska et al. 2009; Chan et al. 2009; Buer et al. 2010; Wasowski and Marder 2012). However, Choi et al. (2011) also referred that the protective defence mechanism against oxidants can be overcome by the inadvertent overproduction of reactive oxygen species. Interactions with key enzymes, signalling cascades involving cytokines and transcription factors, or antioxidant systems, may originate the health-promoting effects of flavonoids (Buer et al. 2010; Wasowski and Marder 2012). Flavonoids have also attracted interest as new chemical entities with activity on the CNS. These actions can result from their binding to the benzodiazepine site on the γ-aminobutyric acid receptor type A (GABAA receptor) resulting in sedation, anxiolytic or anti-convulsive effects, or from their action as inhibitors of monoamine oxidase A or B, working as anti-depressants or antiparkinson agents (Romano et al. 2013). However, although some of these activities have been demonstrated by Fernández et al. (2005), the depressant effects were not observed in rodents.
Flavonoids produce their modulating effects by a number of possible mechanisms (Chan et al. 2009). Some of them are good inhibitors of a variety of ATP binding proteins including protein kinases such as cyclic adenosine monophosphate dependent protein kinase, protein kinase C, serine/threonine kinases, tyrosine kinase, topoisomerase II, and myosin. They also inhibit various membrane ATPases, such as mitochondrial H+-ATPase, Na+/K+-ATPase, Ca2+-ATPase, and H+/K+-ATPase (Di Pietro et al. 2002; Boumendjel et al. 2002; Chan et al. 2009). Moreover, the activities of many other enzymes are also inhibited by flavonoids, for instance β-glucuronidase, lipoxygenase, cyclooxygenase, inducible nitric oxide synthase, monooxygenase, thyroid peroxidase, xanthine oxidase, mitochondrial succinoxidase and nicotinamide adenine dinucleotide-oxidase, phosphodiesterase and phospholipase A2 (Moon et al. 2006).
Although the molecular mechanisms particularly underlying flavonoid–P-gp interactions are not clear, several hypotheses have been proposed (Chan et al. 2009; Go et al. 2009; Cassidy and Setzer 2010). Among the proposed mechanisms are the following: (a) flavonoids modulate P-gp by interacting with the vicinal ATP-binding site and the steroid binding site; (b) flavonoids act as substrate and may interact with P-gp directly either by competitive binding to the substrate-binding site or by binding to other drug-binding sites and changing the P-gp conformation; and (c) flavonoids may bind to the allosteric site or other binding sites (Morris and Zhang 2006; Lohner et al. 2007; Go et al. 2009; Alvarez et al. 2010). Thus, due to the complexity of interactions between P-gp and its modulators, and also because the interactions of flavonoids with P-gp may be different for specific flavonoids, the elucidation of the exact interaction mechanism between these polyphenolic compounds and P-gp is difficult (Zhang and Morris 2003a; Go et al. 2009). Therefore, it has been shown that flavonoids, such as flavonols (e.g., quercetin; Fig. 9), flavanones (e.g., naringenin; Fig. 7), isoflavones (e.g., genistein; Fig. 10) and glycosylated flavonol derivatives (e.g., rutin; Fig. 9), directly interact with the ATP-binding site and the vicinal steroid binding site, leading to the inhibition of P-gp function (Morris and Zhang 2006; Lohner et al. 2007; Chen et al. 2010). Others flavonoids compounds, like epicatechin (Fig. 6) from green tea, activate P-gp by a heterotrophic allosteric mechanism (Lohner et al. 2007). In addition, Morris and Zhang (2006) reported that flavonoids may also directly bind to the C-terminal NBD2 of the purified recombinant mouse P-gp; for instance chrysin (Fig. 8), quercetin (Fig. 9) and kaempferol (Fig. 9) directly bound to the NBD2 cytosolic domain of mouse P-gp (Chan et al. 2009). According to Badhan and Penny (2006) flavonoids have also shown to interact with NBDs of ATPase transporters. The inhibition of the ATPase activity and direct binding of flavonoids to the C-terminal NBD2 of murine Abcb1 has been demonstrated using saturation transfer difference-nuclear magnetic resonance spectroscopy (Badhan and Penny 2006).
Thus, the use of flavonoids as potential modulators of P-gp-mediated drug efflux is highly attractive and NBDs represent potential targets for therapeutic intervention (Badhan and Penny 2006). Flavonoids bind to NBD of P-gp by overlapping simultaneously both binding sites of the cytosolic domains (Trompier et al. 2003; Kitagawa 2006). As a result, these compounds have been suggested to be modulators with bifunctional interactions at vicinal ATP-binding sites and steroid-interacting regions of the P-gp, which are expected to be in close proximity to the ATP-binding site, within a cytosolic domain of P-gp (Conseil et al. 1998; Kitagawa et al. 2005; Kitagawa 2006; Morris and Zhang 2006; Tran et al. 2011). In such situation, the hydrophobic ring B would bind to the steroid-binding region, whereas rings A and C would bind to the ATP pocket (Kitagawa et al. 2005). The results presented by Sheu et al. (2010) confirm that flavonoids have bifunctional interactions in their rings A and C at the ATP-binding site, and the hydrophobic ring B at the steroid-interacting hydrophobic sequence of P-gp. Thus, flavonoids may induce their binding affinity towards NBD2 of P-gp through their ability to mimic the adenine moiety of ATP (Di Pietro et al. 2002; Boumendjel et al. 2002; Kitagawa 2006). However, it is also postulated that these compounds may also interact with P-gp by other mechanisms once opposite effects on P-gp ATPase activity have been observed for different flavonoids(Morris and Zhang 2006).
Although chalcones, flavones and flavonols have demonstrated to possess MDR reversing activity through their high affinity to bind P-gp, the isoflavones have been reported as inactive as modulators of the P-gp functioning (Hadjeri et al. 2003). Overall, the concentrations of flavonoids required to produce a significant modulation of P-gp activity seem to be 10 μM or higher. These concentrations appear to be easily achievable in the intestine after food ingestion and, especially, in the course of dietary supplementation (Morris and Zhang 2006; Bansal et al. 2009).
Indeed, many of flavonoid glycosides may not potently interact with P-gp. However, the aglycones released from these glycosides could be present in intestine at high enough concentrations to inhibit intestinal P-gp, resulting in potential drug interactions (Morris and Zhang 2006; Bansal et al. 2009). Nevertheless, except for the more potent flavonoids, the concentration of flavonoid aglycones in the systemic circulation may not be high enough for the occurrence of significant P-gp interactions in other tissues (Morris and Zhang 2006). Moreover, because the main metabolites of flavonoids (glucuronides and sulphate conjugates) are organic anions, these may not interact with P-gp. Hence, even after regular supplementation the systemic inhibition of P-gp by flavonoids or their metabolites may be, in general, insignificant. However, a significant interaction could occur after administration of an extremely high dose, especially by intravenous (IV) injection (Morris and Zhang 2006; Bansal et al. 2009). In that way, flavonoid modulators also have limitations besides their moderate activity as P-gp inhibitors. Thus, they have a broad spectrum of biological activities including anti-estrogenic activity and the inhibition of other ATPases (Chan et al. 2009); they can also inhibit the absorption of some nutrients, change the pharmacokinetics of certain drugs, and affect neurobehavioral development (Wang et al. 2007).
Interactions of flavonoids with P-glycoprotein
Currently, with the resurgence of the use of medicinal herbs in the western world, the combined use of modern and traditional therapies is increasingly becoming more common (Pathak and Udupa 2010). Although many ingredients in the dietary supplements are not thoroughly studied, a significant number of people regularly consume them in the hope of improving their health and prevent or treat specific diseases (Peng et al. 2006). However, either harmful or beneficial food–drug interactions can occur, particularly due to changes in drug bioavailability. Therefore, the interactions of dietary supplements with drugs may be exploited as a way to improve the pharmacokinetic properties of the co-administered drug (Peng et al. 2006; Bansal et al. 2008; Pathak and Udupa 2010). In the process of drug discovery and development, several drug candidates are found to be potent, safe, selective and affordable, but often do not have a favourable pharmacokinetic profile. Thus, the co-administration of another drug or molecule may improve the drug’s pharmacokinetic properties. In this way a promising approach to overcome the P-gp-mediated drug resistance can be the co-administration of safe dietary phytochemicals that are inhibitors of known drug transporters or metabolizing enzymes which are determinants for drug biodisposition (Peng et al. 2006). This strategy can result in a more cost-effective therapy, by saving additional amounts of drugs due to the optimization of drug delivery (Amin 2013).
The resistance against a variety of chemically unrelated drugs may be due to the overexpression of P-gp in the plasma membrane (Breier et al. 2013). The dietary flavonoids have gained a great attention in respect of the discovery of effective P-gp inhibitors owing to their various health promoting effects and favourable safety profiles. Indeed, because some of these compounds appear to inhibit P-gp-mediated drug efflux, they have potential value to enhance the cellular availability of diverse drugs (Go et al. 2009). Hence, through an effect on the ATPase activity, the flavonoids can modulate ABC transporters by inhibition (e.g., genistein and quercetin) or induction (e.g., glabridin) (Alvarez et al. 2010).
Many prototypic inhibitors and inducers affect both CYP3A4 and P-gp and thus many drug interactions caused by these inhibitors and inducers involve these two systems. Nevertheless, although the P-gp participates in drug efflux phenomena affecting absorption, distribution and elimination of numerous drugs, the CYP enzymes are only involved in drug metabolism (Lin 2003). Due to their beneficial pharmacological activities and their additional ability to modulate both CYP3A4 and P-gp, flavonoids have attracted much attention in recent years (Hsiu et al. 2002; Fang et al. 2005; Choi and Li 2005; Rao et al. 2007; Cho et al. 2011; Li et al. 2011). To eliminate foreign compounds, P-gp co-localizes with CYP3A4 in the polarized epithelial cells of excretory organs, such as the liver, kidney and intestine (Li et al. 2011). Since CYP3A4 and P-gp share a substantial overlap in substrate specificity, their function can modulate the bioavailability of many drugs, affecting their pre-systemic metabolism (Choi and Burm 2006; Choi and Kang 2008; Li et al. 2009, 2011) and promoting the circulation of P-gp substrates between the lumen and epithelial cells. These facts prolong the drug exposure to CYP3A4, reducing its systemic absorption (Choi and Li 2005; Choi and Burm 2006; Choi and Kang 2008). Thus, it seems important to understand the actions of flavonoids not only on the function of P-gp, but also at the P-gp expression levels (Lohner et al. 2007).
Actually, the effects of flavonoids compounds on the level of P-gp expression and/or P-gp activity in cells and tissues may globally change the pharmacokinetic parameters of many drugs. Obviously, this can lead to modifications in drug bioavailability, as well as in biodisposition and toxicity (Chieli et al. 2009). However, many phytochemicals have poor bioavailability and show limited efficacy in vivo (Go et al. 2009). With the great interest in herbal components as alternative medicines, more preclinical and clinical investigation of interactions between herbal constituents and drugs needs to be performed to prevent potential adverse reactions, or to utilize those interactions for therapeutic benefit (Piao and Choi 2008a; Shin et al. 2009; Pathak and Udupa 2010; Singh et al. 2012). However, there is far less information on the pharmacokinetic interactions between herbal products and drugs (Shin et al. 2009; Pathak and Udupa 2010; Singh et al. 2012) and several efforts are currently being expended to identify natural phytocompounds that modulate P-gp and metabolic enzymes (Shin et al. 2009; Cho et al. 2011).
Due to the popular use of flavonoid-containing dietary supplements, flavonoid–drug interactions have been increasingly reported (Singh et al. 2012). Therefore, the discovery of promising compounds for P-gp inhibition among flavonoids still continuous (Go et al. 2009). The literature cites various examples of flavonoids as inhibitors of P-gp transport, which affect the bioavailability and uptake of many drugs. These include in vitro studies on the effect of flavonoids on intracellular accumulation, efflux or transport of P-gp substrates using cells over-expressing P-gp, or a variety of animal and clinical studies (Bansal et al. 2009). Then, several in vitro, in vivo non-clinical and clinical assays performed with different flavonoids are presented below, constituting a summary of the literature data and a starting point for new studies.
Cell-based in vitro models
Over the years, a great diversity of flavonoids compounds has been tested in in vitro conditions, particularly in cell-based models, aiming to assess their effects on the expression levels and functional activity of P-gp (Table 2). In many studies, molecular biology assays prior to functional tests were performed either as confirmatory tests to effectively verify the presence of P-gp in the cells, or as quantization assays to determine the effects of the flavonoids on the expression levels of P-gp.
In order to evaluate the potential effect that a flavonoid may have in the P-gp activity (as inhibitor or inducer), at least one P-gp probe substrate must be used in the in vitro functional assays which include accumulation/efflux and/or transport assays. However, the selection of the adequate P-gp probe substrate is not easy, especially because they may interact with other transport proteins and metabolizing enzymes. Thus, according to the Food and Drug Administration draft guidance for drug interaction studies, an in vitro P-gp probe substrate should be selective for the P-gp transporter, exhibit low to moderate passive membrane permeability through the cell monolayer, not suffer significant metabolism, be commercially available, and be suitable to be also used as an in vivo P-gp probe substrate (Guidance for Industry, FDA, 2006).
Analyzing Table 2, there are flavonoids that stand out as being simultaneously inducers and inhibitors of P-gp. For example, the flavanol epigallocatechin gallate (Fig. 6) appeared to be a strong inducer of P-gp expression in Caco-2 cells when used at a concentration of 10 μM (Lohner et al. 2007); however, its uses at concentrations levels in the order of 100 μM inhibited P-gp function in the same cell model (Jodoin et al. 2002). An and Morris (2010) and Mitsunaga et al. (2000) also reported a biphasic effect of biochanin A (Fig. 10) and quercetin (Fig. 9) in the P-gp function. Mitsunaga et al. (2000) concluded that low concentrations of quercetin indirectly activate the efflux transport of [3H]vincristine by enhancing the phosphorylation, and thus the activity of P-gp, whereas high concentrations of quercetin lead to P-gp inhibition. Thus, the uptake of [3H]vincristine by MBEC4 cells in the steady-state was decreased by 10 μM quercetin, but increased by 50 mM quercetin (Mitsunaga et al. 2000). Likewise, Scambia et al. (1994) reported that 10 μM of quercetin reduces the expression of the immunoreactive P-gp in MCF-7/ADR-resistant cells as evaluated by flow cytofluorimetry. In the study performed by An and Morris (2010), the intracellular fluorescence of mitoxantrone, a P-gp substrate, decreased when co-incubated with 10 μM of biochanin A while there was no significant difference in mitoxantrone cell accumulation in the presence of 50 μM biochanin A. These results indicate that biochanin A may induce the P-gp activity at lower concentrations, and, hence, decreasing the intracellular concentration of mitoxantrone. Actually, several studies summarized in Table 2 revealed that the inhibitory effects of various flavonoids on P-gp function were concentration-dependent (e.g., diosmin, fisetin, naringin, silymarin, tangeritin) (Romiti et al. 2004; Kitagawa et al. 2005; Castro et al. 2007; Mertens-Talcott et al. 2007; Yoo et al. 2007). The increase in P-gp expression levels caused by some flavonoids such as catechin (Fig. 6), naringenin (Fig. 7) or chrysin (Fig. 8) in in vitro intestinal epithelial cells may represent an adaptation or defence mechanism limiting the entry of lipophilic xenobiotics into the organism (Lohner et al. 2007). In this context, through intracellular drug accumulation studies, it was demonstrated that the C-isoprenylation of chrysin enhances the binding affinity toward P-gp. Actually, the 8-(3,3-dimethylallyl)chrysin was described to be more efficient than cyclosporin A in the inhibition of P-gp in leukemic cells (Comte et al. 2001).
The in vitro results shown by Ofer et al. (2005) also demonstrated that the flavonoids hesperetin, naringin (Fig. 7), isoquercetin, kaempferol, quercetin and spiraeoside (Fig. 9) bear the ability to interfere with processes of secretory intestinal transport. These effects may occur due to the interaction of flavonoids with P-gp, but apparently not via competition at the talinolol (P-gp probe substrate) binding site of P-gp as demonstrated by the radioligand binding assay, where none of the tested flavonoids showed to induce the displacement of talinolol. Another mode of interaction may be the inhibition of members of the organic cation transporters-family, which are located at the basolateral membrane of intestinal epithelial cells. These organic cation transporters are mainly located in the basolateral membranes of kidney and liver and usually mediate the uptake of compounds with a transiently or permanently positive net charge into the cells of these excretory organs (Ofer et al. 2005).
In some cases, the use of distinct in vitro systems and/or assays yielded slightly different results. Therefore, it is important to test flavonoid compounds in different cell lines and using different assays in order to increase the confidence in their classification as inhibitors or inducers of P-gp. The inhibition of P-gp activity by many flavonoids and the increased cell accumulation of many drugs (P-gp substrates) suggests the possibility of important changes in absorption/bioavailability of such drugs by the co-administration of these compounds (Zhang and Morris 2003b). In this context, despite the valuable evidences often obtained from these in vitro studies on the modulation of P-gp by flavonoids, more conclusive results are only achieved through interaction studies conducted in in vivo conditions (Choi et al. 2010, 2011a).
In vivo animal models
Table 3 summarizes the main features regarding the in vivo studies conducted to assess the effects of the oral administration of flavonoids in the bioavailability of different drugs (P-gp substrates) administered orally and/or intravenously. Indeed, it has been reported that pharmacokinetic interaction between herbal constituents and drugs may be mediated by CYP450 metabolizing enzymes and also by drug transporters such as P-gp (Cho et al. 2009). Based on the broad overlap in their substrate specificities as well as their co-localization in the small intestine (the primary site of absorption for orally administered drugs), CYP3A4 and P-gp have been recognized as a concerted barrier to drug intestinal absorption (Shin et al. 2009). Since P-gp is co-localized with CYP3A4 in the small intestine, P-gp and CYP3A4 may act synergistically in pre-systemic metabolism, limiting the absorption of drugs which are substrates of both proteins (Piao et al. 2008; Cho et al. 2009; Li et al. 2011). Therefore, dual inhibitors against CYP3A4 and P-gp can have a great impact on the bioavailability of many drugs (Shin et al. 2009). Bearing this in mind, several studies using flavonoids have been performed in order to develop P-gp inhibitors that reduce the drug efflux in the intestine and, hence, enhance oral bioavailability of P-gp substrate drugs (Park et al. 2012). The dosage of drug should be also concerned in case of the increased drug toxicity following the combination with flavonoids (Li et al. 2009). In most studies included in the Table 3, the pre-treatment of the animals with a flavonoid (10–30 min before drug administration) is sufficient to significantly increase the oral drug bioavailability. For instance, the oral bioavailability of diltiazem was increased in male Sprague-Dawley rats submitted to a pre-treatment with the flavanone hesperidin (Fig. 7). Li et al. (2011) also describe the higher bioavailability of tamoxifen in male Sprague-Dawley rats pre-treatment with the flavone baicalein (Fig. 8). According to the authors, this increase might be mainly due to the flavonoid-mediated inhibition of CYP3A-mediated metabolism and P-gp efflux (Choi et al. 2004a, 2006; Yeum and Choi 2006; Li et al. 2007, 2009; Shin et al. 2008; Piao and Choi 2008a; Li and Choi 2008; Choi and Kang 2008; Cho et al. 2009, 2011; Shin et al. 2009; Lee and Choi 2010)
In addition, some studies also compared the effect of the flavonoid pre-treatment on the bioavailability of a drug administered either orally or intravenously; as expected, in the majority of these studies the impact of the flavonoid pre-treatment on pharmacokinetics of the drug administered intravenously was less significant than after oral administration (Choi et al. 2006; Li et al. 2007,2009; Piao and Choi 2008a; Bansal et al. 2008; Shin et al. 2009; Pathak and Udupa 2010; Lee and Choi 2010; Cho et al. 2011). Accordingly, the enhanced oral drug exposure in the presence of the flavonoid together with insignificant changes on drug pharmacokinetics after IV administration, could be mainly due to the increased intestinal absorption via flavonoid-mediated P-gp inhibition rather than the reduced elimination of the drug (Choi et al. 2006; Lee and Choi 2010). As shown in Table 3, the study performed by Li and Choi (2007), in which genistein (Fig. 10) was pre-administered at a dose of 10 mg kg−1, the area under the concentration–time curve (AUC) significantly increased and the total body clearance (CLt) was reduced for IV paclitaxel. Hence, since genistein is not only subjects to CYP2C8 and CYP3A4 mediated metabolism, but also provides inhibition of P-gp, BCRP and MRP2 efflux function, the decreased CLt and increased AUC of etoposide suggests that the metabolism and excretion of paclitaxel in the liver and kidney might be inhibited. This study is consistent with the results reported by Lim and Choi (2006) where a dose of 10 mg kg−1 of naringin (Fig. 7), a dual inhibitor of CYP3A and P-gp, significantly increased the AUC and reduced the CLt of paclitaxel administered by IV route to rats. According to the authors, the significantly greater value of AUC could be due mainly to an inhibition of metabolism of paclitaxel via CYP3A1/2 by oral naringin, but the inhibition of hepatic P-gp by oral naringin could also contribute (Lim and Choi 2006).
In other studies, the animals were not pre-treated with the flavonoid before the administration of drug, but a simultaneous co-administration was performed. The results obtained after oral and/or IV administration of the drug are similar to the aforementioned (Choi and Han 2005; Shin et al. 2006; Piao and Choi 2008b; de Castro et al. 2008; Li and Choi 2009; Go et al. 2009). In other words, the oral drug bioavailability significantly increased after co-administration of the flavonoid, but the same was not true when the drug was administered intravenously. However, the results reported by Choi and Burm (2006) and Choi and Li (2005) disagree with this theory. Indeed, in male New Zealand white rabbits pre-treated with the flavanone morin (Fig. 7), the oral bioavailability of nimodipine was significantly increased, but the same did not occur when the drug was simultaneously co-administered with the flavonoid. Accordingly, the morin can interact with nimodipine in the gastrointestinal lumen forming a flavonoid-drug complex that is not absorbed, or the absorption of morin in the gastrointestinal mucosa is early enough to inhibit nimodipine-CYP3A4 metabolizing enzyme and P-gp efflux pump by a pre-treatment period of 30 min before nimodipine administration (Choi and Burm 2006). The same results were obtained by Choi and Li (2005) wherein the bioavailability of diltiazem in rabbits pre-treated with quercetin (Fig. 9) is significantly increased, but not in the rabbits co-administered with quercetin. The co-administration of naringenin (Fig. 7) with talinolol in male Sprague-Dawley rats also did not result in a significant increase of the oral drug bioavailability (de Castro et al. 2008).
As biochanin A (Fig. 10) is an inhibitor of CYP3A and P-gp and tamoxifen a well-known substrate of CYP3A4/P-gp, after their co-administration an increase of the oral bioavailability of tamoxifen was expected; however this was not observed, and a significant decrease in the relative bioavailability of tamoxifen after the co-administration of the flavonoid was reported by Singh et al. (2012). This isoflavone also caused a significant decrease in oral bioavailability of fexodenadine which is not metabolized by CYP450 isoenzymes at a large extent (Peng et al. 2006). Similarly, Hsiu et al. (2002) reported the decrease in the oral bioavailability of cyclosporin when co-administered with quercetin (Fig. 9) in male Sprague-Dawley rats and in male Yorkshire pigs. Finally, Park et al. (2012) studied the effect of the co-administration of silymarin [a flavonoid composed mainly by silibinin (90 %) and small amounts of silibinin stereoisomers as isosilybin A, silydianin, and silychristin (Fig. 12)] on oral bioavailability of paclitaxel, and the results obtained demonstrated an increased oral bioavailability of the drug (a P-gp substrate). Actually, all the previously reported results suggest that the concurrent use of flavonoids or flavonoids-containing dietary supplement or herbs with medications whose absorption and metabolism are mediated by P-gp and/or CYP3A4 should be given special (Hsiu et al. 2002).

Clinical studies
Up to date there are few clinical studies published in literature documenting the effect of a particular flavonoid on the pharmacokinetics of a given drug. Two of those studies were developed by Rao et al. (2007) and Kim et al. (2009) testing the flavonoids silymarin (Fig. 7) and quercetin (Fig. 5) respectively. Rao et al. (2007) investigated the effect of silymarin pre-treatment on the pharmacokinetics of ranitidine in twelve healthy male human volunteers (age range 19–26 years, weight 50–68 kg). The study design involved the administration of 150 mg ranitidine to the volunteers, either alone, or after a 7 days pre-treatment period with thrice-daily dose of 140 mg silymarin, after an overnight fast. The washout period between each ranitidine treatment was 7 days and the serum levels of the drug were determined by high-performance liquid chromatography with ultraviolet detection. The results showed that the pharmacokinetics of ranitidine was not influenced by silymarin. The plasma peak concentration (C max), the plasma AUC from zero to 12 h (AUC0–12), the plasma AUC from zero to infinity (AUC0–∞), elimination rate constant (Kel), terminal elimination half-life (T1/2el) and time to reach C max (T max) were not significantly altered after pre-treatment with silymarin. However, the area under the first moment curve (AUMC) and the mean residence time (MRT) showed a significant increase after the silymarin pre-treatment. Indeed, despite ranitidine is a substrate of CYP3A4 and P-gp, and silymarin a flavonoid compound that has potential to influence drug biodisposition by decreasing the metabolic activity of CYP3A4 and by inhibiting P-gp function, important changes were not evidenced in the majority of the pharmacokinetic parameters of ranitidine. Rao et al. (2007) suggested that silymarin was so poorly absorbed after oral administration that its blood concentrations were too low to cause an appreciable effect on CYP3A4 or P-gp.
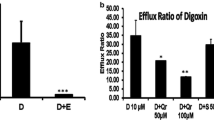
In turn, Kim et al. (2009) developed a study aimed at evaluating whether quercetin exhibited any inhibitory effect on P-gp-mediated drug disposition in humans using fexofenadine as a P-gp probe substrate. In this study, twelve healthy male subjects (age range 24–31 years, weight 67–82 kg) were treated for 7 days with 500 mg quercetin or placebo three times daily. Subsequently, a single dose of 60 mg fexofenadine was administered orally on day 7. Afterwards, the fexofenadine drug concentrations in plasma and urine samples were quantified using high-performance liquid chromatography coupled to fluorescence detection. After a pharmacokinetic analysis of the concentration–time profiles, a significant increase in the mean plasma concentrations of fexofenadine after quercetin treatment was registered; more specifically, the AUC of plasma fexofenadine was increased by 55 % and the C max was elevated by 68 % during the quercetin treatment. Nevertheless, no differences were observed in the renal clearance or terminal elimination half-life (t1/2) of fexofenadine between placebo and quercetin phases, but the oral clearance of the drug was significantly decreased (37 %) under quercetin treatment. The results of this clinical study showed that the short-term use of quercetin elevated the plasma concentrations of fexofenadine and it may occur due to the inhibition of P-gp-mediated drug efflux (Kim et al. 2009). Hence, more clinical trials are needed to reliably assess the effects of flavonoids in modulating P-gp function in humans.
Conclusion
Over the last years the strategies for drug discovery and development have changed considerably and new features are taken into account by pharmaceutical industry in making decision process (Mizuno et al. 2003). The therapeutic failure due to the development of MDR is a growing concern in the clinical practice. As a result, due to the recognition that P-gp-mediated drug efflux is one of the major factors contributing for MDR in several diseases (Wang et al. 2002; Löscher and Potschka 2005; Hennessy and Spiers 2007; Bansal et al. 2009), the investigation of the role of this drug transporter on the biodisposition of new drug candidates has been emphasized as an integral component of the processes of drug discovery, drug development and clinical therapy (Sun et al. 2004). Moreover, the advances in molecular biology have allowed to increase the knowledge on the structure, localization and functional role of P-gp, as well the subjacent drug efflux mechanisms (Varma 2003).
Because of the broad substrate specificity and the ubiquitous distribution of P-gp through tissues frequently involved in absorption, distribution and elimination, the modulation of the activity of this transporter may be responsible by substantial changes in the drug disposition of P-gp substrates (Matheny et al. 2001; Chang et al. 2006). The realization of the importance of drug efflux transporters in the course of disease processes and their treatments, led to the rapid identification of P-gp substrates and/or inhibitors (Chang et al. 2006), and promoted the search of selective P-gp inhibitors as add-on therapy in order to overcome the MDR phenotype (Dantzig et al. 2003). However, the selective P-gp inhibition is a complex and difficult task and much work still remaining to optimize this strategy. Nevertheless, the growing understanding of P-gp efflux mechanisms, the development of improved screening methods, and the increasing number of structure–activity relationship studies will provide new opportunities not only to enhance the bioavailability of life saving drugs, but also to improve their pharmacokinetics, tissue distribution and cell uptake (Varma 2003). Even though, there are particular criteria that should be satisfied in order to find clinically promising P-gp inhibitors (e.g. specificity for P-gp, lack of interference with CYP450 enzymes) (Dantzig et al. 2003).
The herbal products are increasingly used worldwide for medicinal purposes and there has been a growing interest in assessing the modulating effects of dietary constituents on P-gp function (Sun et al. 2004). Among these, flavonoids appear to have optimal properties as potential inhibitors of the P-gp multidrug transporter (Ross and Kasum 2002; Brand et al. 2006; Schinkel and Jonker 2012). This evidence is supported by many in vitro and in vivo assays, as well as by a study in humans. Accordingly, it is essential to perform more and better studies, in order to recognize and select which flavonoids are the best candidates to enhance the cellular availability of diverse drugs and, thus, their pharmacological potential. This therapeutic approach can be extremely important for the treatment of important diseases, such as epilepsy and cancer, for which the existing treatments are often ineffective, particularly due to the MDR phenomena (Dantzig et al. 2003; Varma 2003; Brandt et al. 2006; Lazarowski et al. 2007; Zhang et al. 2012).
Abbreviations
- ABC:
-
Adenosine triphosphate (ATP)-binding cassette
- AUC:
-
Area under the concentration–time curve
- BCRP:
-
Breast cancer resistance protein
- Clt :
-
Total body clearance
- C max :
-
Peak concentration
- CNS:
-
Central nervous system
- CYP:
-
Cytochrome P450
- IV:
-
Intravenous
- MDR:
-
Multidrug resistance
- MRP:
-
Multidrug resistance-associated protein
- NBD:
-
Nucleotide-binding domain
- P-gp:
-
P-glycoprotein
- TMs:
-
Transmembrane α-helix segments
- TMD:
-
Transmembrane domain
References
Ali H-A, Chowdhury AKA, Rahman AKM et al (2008) Pachypodol, a flavonol from the leaves of Calycopteris floribunda, inhibits the growth of CaCo-2 colon cancer cell line in vitro. Phytother Res 22:1684–1687
Alonso-Castro AJ, Ortiz-Sánchez E, García-Regalado A et al (2013) Kaempferitrin induces apoptosis via intrinsic pathway in HeLa cells and exerts antitumor effects. J Ethnopharmacol 145:476–489
Alvarez AI, Real R, Pérez M et al (2010) Modulation of the activity of ABC transporters (P-Glycoprotein, MRP2, BCRP) by flavonoids and drug response. J Pharm Sci 99:598–617
Amin ML (2013) P-glycoprotein inhibition for optimal drug delivery. Drug Target Insights 7:27–34
An G, Morris ME (2010) Effects of the isoflavonoid biochanin A on the transport of mitoxantrone in vitro and in vivo. Biopharm Drug Dispos 31:340–350
Aszalos A (2008) Role of ATP-binding cassette (ABC) transporters in interactions between natural products and drugs. Curr Drug Metab 9:1010–1018
Badhan R, Penny J (2006) In silico modelling of the interaction of flavonoids with human P-glycoprotein nucleotide-binding domain. Eur J Med Chem 41:285–295
Balayssac D, Authier N, Cayre A, Coudore F (2005) Does inhibition of P-glycoprotein lead to drug–drug interactions? Toxicol Lett 156:319–329
Bansal T, Awasthi A, Jaggi M et al (2008) Pre-clinical evidence for altered absorption and biliary excretion of irinotecan (CPT-11) in combination with quercetin: possible contribution of P-glycoprotein. Life Sci 83:250–259
Bansal T, Jaggi M, Khar RK, Talegaonkar S (2009) Emerging significance of flavonoids as P-glycoprotein inhibitors in cancer chemotherapy. J Pharm Pharm Sci 12:46–78
Barron D, Di Pietro A, Dumontet C, Mcintosh DB (2003) Isoprenoid flavonoids are new leads in the modulation of chemoresistance. Phytochem Rev 1:325–332
Bellamy WT (1996) P-glycoproteins and multidrug resistance. Annu Rev Pharmacol Toxicol 36:161–183
Bois F, Beney C, Boumendjel A et al (1998) Halogenated chalcones with high-affinity binding to P-glycoprotein: potential modulators of multidrug resistance. J Med Chem 41:4161–4164
Bois F, Boumendjel A, Mariotte AM et al (1999) Synthesis and biological activity of 4-alkoxy chalcones: potential hydrophobic modulators of P-glycoprotein-mediated multidrug resistance. Bioorg Med Chem 7:2691–2695
Borska S, Sopel M, Chmielewska M et al (2010) Quercetin as a potential modulator of P-glycoprotein expression and function in cells of human pancreatic carcinoma line resistant to daunorubicin. Molecules 15:857–870
Boumendjel A, Di Pietro A, Dumontet C, Barron D (2002) Recent advances in the discovery of flavonoids and analogs with high-affinity binding to P-glycoprotein responsible for cancer cell multidrug resistance. Med Res Rev 22:512–529
Brand W, Schutte ME, Williamson G et al (2006) Flavonoid-mediated inhibition of intestinal ABC transporters may affect the oral bioavailability of drugs, food-borne toxic compounds and bioactive ingredients. Biomed Pharmacother 60:508–519
Brandt C, Bethmann K, Gastens AM, Löscher W (2006) The multidrug transporter hypothesis of drug resistance in epilepsy: proof-of-principle in a rat model of temporal lobe epilepsy. Neurobiol Dis 24:202–211
Breier A, Gibalova L, Seres M et al (2013) New insight into p-glycoprotein as a drug target. Anticancer Agents Med Chem 13:159–170
Buer CS, Imin N, Djordjevic MA (2010) Flavonoids: new roles for old molecules. J Integr Plant Biol 52:98–111
Cassidy CE, Setzer WN (2010) Cancer-relevant biochemical targets of cytotoxic Lonchocarpus flavonoids: a molecular docking analysis. J Mol Model 16:311–326
Castro AF, Altenberg GA (1997) Inhibition of drug transport by genistein in multidrug-resistant cells expressing P-glycoprotein. Biochem Pharmacol 53:89–93
Castro WVDE, Mertens-Talcott S, Derendorf H (2007) Grapefruit juice-drug interactions: grapefruit juice and its components inhibit P-glycoprotein (ABCB1) mediated transport of talinolol in Caco-2 cells. J Pharm Sci 96:2808–2817
Chan LMS, Lowes S, Hirst BH (2004) The ABCs of drug transport in intestine and liver: efflux proteins limiting drug absorption and bioavailability. Eur J Pharm Sci 21:25–51
Chan K-F, Zhao Y, Chow TWS et al (2009) Flavonoid dimers as bivalent modulators for p-glycoprotein-based multidrug resistance: structure-activity relationships. ChemMedChem 4:594–614
Chang X (2007) A molecular understanding of ATP-dependent solute transport by multidrug resistance-associated protein MRP1. Cancer Metastasis Rev 26:15–37
Chang C, Bahadduri PM, Polli JE et al (2006) Rapid identification of P-glycoprotein substrates and inhibitors. Drug Metab Dispos 34:1976–1984
Chen C, Zhou J, Ji C (2010) Quercetin: a potential drug to reverse multidrug resistance. Life Sci 87:333–338
Chieli E, Romiti N, Rodeiro I, Garrido G (2009) In vitro effects of Mangifera indica and polyphenols derived on ABCB1/P-glycoprotein activity. Food Chem Toxicol 47:2703–2710
Cho Y-A, Choi D-H, Choi J-S (2009) Effect of hesperidin on the oral pharmacokinetics of diltiazem and its main metabolite, desacetyldiltiazem, in rats. J Pharm Pharmacol 61:825–829
Cho Y-A, Choi J-S, Burm J-P (2011) Effects of the antioxidant baicalein on the pharmacokinetics of nimodipine in rats: a possible role of P-glycoprotein and CYP3A4 inhibition by baicalein. Pharmacol Rep 63:1066–1073
Choi J-S, Burm JP (2006) Enhanced nimodipine bioavailability after oral administration of nimodipine with morin, a flavonoid, in rabbits. Arch Pharm Res 29:333–338
Choi J-S, Han H-K (2005) Pharmacokinetic interaction between diltiazem and morin, a flavonoid, in rats. Pharmacol Res 52:386–391
Choi J-S, Kang KW (2008) Enhanced tamoxifen bioavailability after oral administration of tamoxifen in rats pretreated with naringin. Arch Pharm Res 31:1631–1636
Choi J-S, Li X (2005) Enhanced diltiazem bioavailability after oral administration of diltiazem with quercetin to rabbits. Int J Pharm 297:1–8
Choi J-S, Choi H-K, Shin S-C (2004a) Enhanced bioavailability of paclitaxel after oral coadministration with flavone in rats. Int J Pharm 275:165–170
Choi J-S, Jo B-W, Kim Y-C (2004b) Enhanced paclitaxel bioavailability after oral administration of paclitaxel or prodrug to rats pretreated with quercetin. Eur J Pharm Biopharm 57:313–318
Choi B-C, Choi J-S, Han H-K (2006) Altered pharmacokinetics of paclitaxel by the concomitant use of morin in rats. Int J Pharm 323:81–85
Choi D-H, Li C, Choi J-S (2010) Effects of myricetin, an antioxidant, on the pharmacokinetics of losartan and its active metabolite, EXP-3174, in rats: possible role of cytochrome P450 3A4, cytochrome P450 2C9 and P-glycoprotein inhibition by myricetin. J Pharm Pharmacol 62:908–914
Choi J-S, Piao Y-J, Kang KW (2011a) Effects of quercetin on the bioavailability of doxorubicin in rats: role of CYP3A4 and P-gp inhibition by quercetin. Arch Pharm Res 34:607–613
Choi S-J, Shin S-C, Choi J-S (2011b) Effects of myricetin on the bioavailability of doxorubicin for oral drug delivery in rats: possible role of CYP3A4 and P-glycoprotein inhibition by myricetin. Arch Pharm Res 34:309–315
Chokchaisiri R, Suaisom C, Sriphota S et al (2009) Bioactive flavonoids of the flowers of Butea monosperma. Chem Pharm Bull (Tokyo) 57:428–432
Chung SY, Sung MK, Kim NH et al (2005) Inhibition of P-glycoprotein by natural products in human breast cancer cells. Arch Pharm Res 28:823–828
Chung SY, Jang DS, Han A et al (2007) Modulation of P-glycoprotein-mediated resistance by kaempferol derivatives isolated from Zingiber zerumbet. Phytother Res 21:565–569
Coley HM (2010) Overcoming multidrug resistance in cancer: clinical studies of p-glycoprotein inhibitors. Methods Mol Biol 596:341–358
Comte G, Daskiewicz JB, Bayet C et al (2001) C-Isoprenylation of flavonoids enhances binding affinity toward P-glycoprotein and modulation of cancer cell chemoresistance. J Med Chem 44:763–768
Conseil G, Baubichon-Cortay H, Dayan G et al (1998) Flavonoids: a class of modulators with bifunctional interactions at vicinal ATP- and steroid-binding sites on mouse P-glycoprotein. Proc Natl Acad Sci USA 95:9831–9836
Cook N, Samman S (1996) Flavonoids: chemistry, metabolism, cardioprotective effects, and dietary sources. J Nutr Biochem 7:66–76
Corea G, Di Pietro A, Dumontet C et al (2009) Jatrophane diterpenes from Euphorbia spp. as modulators of multidrug resistance in cancer therapy. Phytochem Rev 8:431–447
Crozier A, Jaganath IB, Clifford MN (2009) Dietary phenolics: chemistry, bioavailability and effects on health. Nat Prod Rep 26:1001–1043
Dantzig AH, de Alwis DP, Burgess M (2003) Considerations in the design and development of transport inhibitors as adjuncts to drug therapy. Adv Drug Deliv Rev 55:133–150
De Castro WV, Mertens-Talcott S, Derendorf H, Butterweck V (2008) Effect of grapefruit juice, naringin, naringenin, and bergamottin on the intestinal carrier-mediated transport of talinolol in rats. J Agric Food Chem 56:4840–4845
De Wet H, McIntosh DB, Conseil G et al (2001) Sequence requirements of the ATP-binding site within the C-terminal nucleotide-binding domain of mouse P-glycoprotein: structure-activity relationships for flavonoid binding. Biochemistry 40:10382–10391
Deeley RG, Westlake C, Cole SPC (2006) Transmembrane transport of endo- and xenobiotics by mammalian ATP-binding cassette multidrug resistance proteins. Physiol Rev 86:849–899
Deferme S, Augustijns P (2003) The effect of food components on the absorption of P-gp substrates: a review. J Pharm Pharmacol 55:153–162
Del Amo EM, Heikkinen AT, Mönkkönen J (2009) In vitro-in vivo correlation in P-glycoprotein mediated transport in intestinal absorption. Eur J Pharm Sci 36:200–211
Di Pietro A, Conseil G, Pérez-victoria JM et al (2002) Cellular and molecular life sciences modulation by flavonoids of cell multidrug resistance mediated by P-glycoprotein and related ABC transporters. Cell Mol Life Sci 59:307–322
Dinis-Oliveira RJ, Remião F, Duarte J et al (2006) P-glycoprotein induction: an antidotal pathway for paraquat-induced lung toxicity. Free Radic Biol Med 41:1213–1224
Doran A, Obach RS, Smith BJ et al (2005) The impact of P-glycoprotein on the disposition of drugs targeted for indications of the central nervous system: evaluation using the MDR1A/1B knockout mouse model. Drug Metab Dispos 33:165–174
Du G-J, Zhang Z, Wen X-D et al (2012) Epigallocatechin Gallate (EGCG) is the most effective cancer chemopreventive polyphenol in green tea. Nutrients 4:1679–1691
Fang S-H, Hou Y-C, Chao P-DL (2005) Pharmacokinetic and pharmacodynamic interactions of morin and cyclosporin. Toxicol Appl Pharmacol 205:65–70
Fernández SP, Wasowski C, Paladini AC, Marder M (2005) Synergistic interaction between hesperidin, a natural flavonoid, and diazepam. Eur J Pharmacol 512:189–198
Fortuna A, Alves G, Falcão A (2011) In vitro and in vivo relevance of the P-glycoprotein probe substrates in drug discovery and development: focus on rhodamine 123, digoxin and talinolol. J Bioequiv Availab 01:1–23
Fromm MF (2004) Importance of P-glycoprotein at blood-tissue barriers. Trends Pharmacol Sci 25:423–429
Gazák R, Walterová D, Kren V (2007) Silybin and silymarin-new and emerging applications in medicine. Curr Med Chem 14:315–338
Giacomini KM, Huang S-M, Tweedie DJ et al (2010) Membrane transporters in drug development. Nat Rev Drug Discov 9:215–236
Girardin F (2006) Membrane transporter proteins: a challenge for CNS drug development. Dialogues Clin Neurosci 8:311–321
Go WJ, Ryu JH, Qiang F, Han H-K (2009) Evaluation of the flavonoid oroxylin A as an inhibitor of P-glycoprotein-mediated cellular efflux. J Nat Prod 72:1616–1619
Guidance for Industry (2006) Drug interaction studies: study design, data analysis, and implications for dosing and labeling. U.S. Department of Health and Human Services, FDA, CDER, CBER
Hadjeri M, Barbier M, Ronot X et al (2003) Modulation of P-glycoprotein-mediated multidrug resistance by flavonoid derivatives and analogues. J Med Chem 46:2125–2131
Harborne JB (1962) Plant polyphenols. 5: occurrence of azalein and related pigments in flowers of Plumbago and Rhododendron species. Arch Biochem Biophys 96:171–178
Hasegawa M, Shirato T (2002) Two new flavonoid glycosides from the leaves of Phellodendron amurense Ruprecht. J Am Chem Soc 75:5507–5511
Hendrich AB (2006) Flavonoid-membrane interactions: possible consequences for biological effects of some polyphenolic compounds. Acta Pharmacol Sin 27:27–40
Hennessy M, Spiers JP (2007) A primer on the mechanics of P-glycoprotein the multidrug transporter. Pharmacol Res 55:1–15
Horowitz RM, Gentili B (1960) Flavonoids of the Ponderosa lemon. Nature 185:319
Hsiu S-L, Hou Y-C, Wang Y-H et al (2002) Quercetin significantly decreased cyclosporin oral bioavailability in pigs and rats. Life Sci 72:227–235
Huisman MT, Smit JW, Wiltshire HR et al (2003) Assessing safety and efficacy of directed P-glycoprotein inhibition to improve the pharmacokinetic properties of saquinavir coadministered with ritonavir. J Pharmacol Exp Ther 304:596–602
Ikegawa T, Ushigome F, Koyabu N, Morimoto S (2000) Inhibition of P-glycoprotein by orange juice components, polymethoxyflavones in adriamycin-resistant human myelogenous leukemia (K562/ADM) cells. Cancer Lett 160:21–28
Jäger AK, Saaby L (2011) Flavonoids and the CNS. Molecules 16:1471–1485
Jeong J-M, Choi C-H (2007) Enhancement of paclitacel transport and cytotoxicity by 7,3’,4’-trimethoxyflavone, a P-glycoprotein inhibitor. J Pharm Pharm Sci 10:547–553
Jodoin J, Demeule M, Beliveau R (2002) Inhibition of the multidrug resistance P-glycoprotein activity by green tea polyphenols. Biochim Biophys Acta 1542:149–159
Juliano RL, Ling V (1976) A surface glycoprotein modulating drug permeability in Chinese hamster ovary cell mutants. Biochim Biophys Acta 455:152–162
Katiyar SK, Meleth S, Sharma SD (2008) Silymarin, a flavonoid from milk thistle (Silybum marianum L.), inhibits UV-induced oxidative stress through targeting infiltrating CD11b+ cells in mouse skin. Photochem Photobiol 84:266–271
Kellenberger E, Kuhn I, Schuber F, Muller-Steffner H (2011) Flavonoids as inhibitors of human CD38. Bioorg Med Chem Lett 21:3939–3942
Kemper EM, van Zandbergen AE, Cleypool C et al (2003) Increased penetration of paclitaxel into the brain by inhibition of P-Glycoprotein. Clin Cancer Res 9:2849–2855
Keung WM, Vallee BL (1993) Daidzin: a potent, selective inhibitor of human mitochondrial aldehyde dehydrogenase. Proc Natl Acad Sci USA 90:1247–1251
Khan IA, Avery MA, Burandt CL et al (2000) Antigiardial activity of isoflavones from Dalbergia frutescens bark. J Nat Prod 63:1414–1416
Khantamat O, Chaiwangyen W, Porn-ngarm L (2004) Screening of flavonoids for their potential inhibitory effect on p-glycoprotein activity in human cervical carcinoma KB cells. Chiang Mai Med Bull 43:45–56
Kim D-H, Na H-K, Oh TY et al (2004) Eupatilin, a pharmacologically active flavone derived from Artemisia plants, induces cell cycle arrest in ras-transformed human mammary epithelial cells. Biochem Pharmacol 68:1081–1087
Kim K-A, Park P-W, Park J-Y (2009) Short-term effect of quercetin on the pharmacokinetics of fexofenadine, a substrate of P-glycoprotein, in healthy volunteers. Eur J Clin Pharmacol 65:609–614
Kim YH, Lee YS, Choi EM (2010) Chrysoeriol isolated from Eurya cilliata leaves protects MC3T3-E1 cells against hydrogen peroxide-induced inhibition of osteoblastic differentiation. J Appl Toxicol 30:666–673
Kitagawa S (2006) Inhibitory effects of polyphenols on p-glycoprotein-mediated transport. Biol Pharm Bull 29:1–6
Kitagawa S, Nabekura T, Kamiyama S (2004) Inhibition of P-glycoprotein function by tea catechins in KB-C2 cells. J Pharm Pharmacol 56:1001–1005
Kitagawa S, Nabekura T, Takahashi T et al (2005) Structure-activity relationships of the inhibitory effects of flavonoids on P-glycoprotein-mediated transport in KB-C2 cells. Biol Pharm Bull 28:2274–2278
Krishna R, Mayer LD (2000) Multidrug resistance (MDR) in cancer: mechanisms, reversal using modulators of MDR and the role of MDR modulators in influencing the pharmacokinetics of anticancer drugs. Eur J Pharm Sci 11:265–283
Kubota H, Ishihara H, Langmann T et al (2006) Distribution and functional activity of P-glycoprotein and multidrug resistance-associated proteins in human brain microvascular endothelial cells in hippocampal sclerosis. Epilepsy Res 68:213–228
Kumar G, Karthik L, Rao KVB (2013) Phytochemical composition and in vitro antioxidant activity of aqueous extract of Aerva lanata (L.) Juss. ex Schult. Stem (Amaranthaceae). Asian Pac J Trop Med 6:180–187
Kwan P, Brodie MJ (2005) Potential role of drug transporters in the pathogenesis of medically intractable epilepsy. Epilepsia 46:224–235
Lam IK, Alex D, Wang Y-H et al (2012) In vitro and in vivo structure and activity relationship analysis of polymethoxylated flavonoids: identifying sinensetin as a novel antiangiogenesis agent. Mol Nutr Food Res 56:945–956
Lazarowski A, Czornyj L, Lubienieki F et al (2007) ABC transporters during epilepsy and mechanisms underlying multidrug resistance in refractory epilepsy. Epilepsia 48(Suppl 5):140–149
Lee C-K, Choi J-S (2010) Effects of silibinin, inhibitor of CYP3A4 and P-glycoprotein in vitro, on the pharmacokinetics of paclitaxel after oral and intravenous administration in rats. Pharmacology 85:350–356
Ley JP, Krammer G, Reinders G et al (2005) Evaluation of bitter masking flavanones from Herba Santa (Eriodictyon californicum (H. and A.) Torr., Hydrophyllaceae). J Agric Food Chem 53:6061–6066
Li X, Choi J-S (2007) Effect of genistein on the pharmacokinetics of paclitaxel administered orally or intravenously in rats. Int J Pharm 337:188–193
Li C, Choi JS (2008) Effects of epigallocatechin gallate on the bioavailability and pharmacokinetics of diltiazem in rats. Pharmazie 63:815–818
Li X, Choi J-S (2009) Effects of quercetin on the pharmacokinetics of Etoposide after oral or intravenous administration of etoposide in rats. Anticancer Res 29:1411–1415
Li X, Yun J-K, Choi J-S (2007) Effects of morin on the pharmacokinetics of etoposide in rats. Biopharm Drug Dispos 28:151–156
Li C, Li X, Choi J-S (2009) Enhanced bioavailability of etoposide after oral or intravenous administration of etoposide with kaempferol in rats. Arch Pharm Res 32:133–138
Li C, Kim M, Choi H, Choi J (2011) Effects of baicalein on the pharmacokinetics of tamoxifen and its main metabolite, 4-hydroxytamoxifen, in rats: possible role of cytochrome P450 3A4 and P-glycoprotein inhibition by baicalein. Arch Pharm Res 34:1965–1972
Lim S-C, Choi J-S (2006) Effects of naringin on the pharmacokinetics of intravenous paclitaxel in rats. Biopharm Drug Dispos 27:443–447
Lin JH (2003) Drug–drug interaction mediated by inhibition and induction of P-glycoprotein. Adv Drug Deliv Rev 55:53–81
Lin JH, Yamazaki M (2003) Role of P-glycoprotein in pharmacokinetics: clinical implications. Clin Pharmacokinet 42:59–98
Ling V (1997) Multidrug resistance: molecular mechanisms and clinical relevance. Cancer Chemother Pharmacol 40(Suppl):S3–S8
Linnet K, Ejsing TB (2008) A review on the impact of P-glycoprotein on the penetration of drugs into the brain: focus on psychotropic drugs. Eur Neuropsychopharmacol 18:157–169
Lohner K, Schnäbele K, Daniel H et al (2007) Flavonoids alter P-gp expression in intestinal epithelial cells in vitro and in vivo. Mol Nutr Food Res 51:293–300
Löscher W, Potschka H (2002a) Role of multidrug transporters in pharmacoresistance to antiepileptic drugs. J Pharmacol Exp Ther 301:7–14
Löscher W, Potschka H (2002b) Role of drug efflux transporters in the brain for drug disposition and treatment of brain diseases. Prog Neurobiol 76:22–76
Löscher W, Potschka H (2005) Blood-brain barrier active efflux transporters: aTP-binding cassette gene family. NeuroRx 2:86–98
Makino T, Kanemaru M, Okuyama S et al (2013) Anti-allergic effects of enzymatically modified isoquercitrin (α-oligoglucosyl quercetin 3-O-glucoside), quercetin 3-O-glucoside, α-oligoglucosyl rutin, and quercetin, when administered orally to mice. J Nat Med 67:881–886
Marquez B, Van Bambeke F (2011) ABC multidrug transporters: target for modulation of drug pharmacokinetics and drug–drug interactions. Curr Drug Targets 12:600–620
Matheny CJ, Lamb MW, Brouwer KR, Pollack GM (2001) Pharmacokinetic and pharmacodynamic implications of P-glycoprotein modulation. Pharmacotherapy 21:778–796
Matsui T, Ito C, Itoigawa M et al (2009) Effect of natsudaidain isolated from Citrus plants on TNF-alpha and cyclooxygenase-2 expression in RBL-2H3 cells. J Pharm Pharmacol 61:109–114
Mertens-Talcott SU, De Castro WV, Manthey JA et al (2007) Polymethoxylated flavones and other phenolic derivates from citrus in their inhibitory effects on P-glycoprotein-mediated transport of talinolol in Caco-2 cells. J Agric Food Chem 55:2563–2568
Miller DS, Bauer B, Hart AMS (2009) Modulation of P-glycoprotein at the blood-brain barrier: opportunities to improve CNS pharmacotherapy. Pharmacol Rev 60:196–209
Mitsunaga Y, Takanaga H, Matsuo H et al (2000) Effect of bioflavonoids on vincristine transport across blood–brain barrier. Eur J Pharmacol 395:193–201
Miyake Y, Mochizuki M, Okada M et al (2007) Isolation of antioxidative phenolic glucosides from lemon juice and their suppressive effect on the expression of blood adhesion molecules. Biosci Biotechnol Biochem 71:1911–1919
Mizuno N, Niwa T, Yotsumoto Y, Sugiyama Y (2003) Impact of drug transporter studies on drug discovery and development. Pharmacol Rev 55:425–461
Moon YJ, Wang X, Morris ME (2006) Dietary flavonoids: effects on xenobiotic and carcinogen metabolism. Toxicol Vitr 20:187–210
Moreno Escobosa MC, Cruz Granados S, Moya Quesada MC (2012) Erythema and hand edema due to flavoxate. J Investig Allergol Clin Immunol 22:390–391
Morris ME, Zhang S (2006) Flavonoid-drug interactions: effects of flavonoids on ABC transporters. Life Sci 78:2116–2130
Ofer M, Wolffram S, Koggel A et al (2005) Modulation of drug transport by selected flavonoids: involvement of P-gp and OCT? Eur J Pharm Sci 25:263–271
Ogawa Y, Oku H, Iwaoka E et al (2006) Allergy-preventive phenolic glycosides from Populus sieboldii. J Nat Prod 69:1215–1217
Palmeira A, Rodrigues F, Sousa E et al (2011) New uses for old drugs: pharmacophore-based screening for the discovery of P-glycoprotein inhibitors. Chem Biol Drug Des 78:57–72
Park JH, Park JH, Hur HJ et al (2012) Effects of silymarin and formulation on the oral bioavailability of paclitaxel in rats. Eur J Pharm Sci 45:296–301
Patanasethanont D, Nagai J, Yumoto R et al (2007) Effects of Kaempferia Parviflora extracts and their flavone constituents on P-glycoprotein function. J Pharm Sci 96:223–233
Pathak SM, Udupa N (2010) Pre-clinical evidence of enhanced oral bioavailability of the P-glycoprotein substrate talinolol in combination with morin. Biopharm Drug Dispos 31:202–214
Peng SX, Ritchie DM, Cousineau M et al (2006) Altered oral bioavailability and pharmacokinetics of P-glycoprotein substrates by coadministration of biochanin A. J Pharm Sci 95:1984–1993
Pérez-Tomás R (2006) Multidrug resistance: retrospect and prospects in anti-cancer drug treatment. Curr Med Chem 13:1859–1876
Piao Y-J, Choi J-S (2008a) Effects of morin on the pharmacokinetics of nicardipine after oral and intravenous administration of nicardipine in rats. J Pharm Pharmacol 60:625–629
Piao Y-J, Choi J-S (2008b) Enhanced bioavailability of verapamil after oral administration with hesperidin in rats. Arch Pharm Res 31:518–522
Piao Y-J, Shin S-C, Choi J-S (2008) Effects of oral kaempferol on the pharmacokinetics of tamoxifen and one of its metabolites, 4-hydroxytamoxifen, after oral administration of tamoxifen to rats. Biopharm Drug Dispos 29:245–249
Potschka H (2012) Role of CNS efflux drug transporters in antiepileptic drug delivery: overcoming CNS efflux drug transport. Adv Drug Deliv Rev 64:943–952
PubChem Coumpound Database PubChem Coumpound Database. http://www.ncbi.nlm.nih.gov/pccompound/
Raj (2001) Bioflavonoids classification, pharmacological, biochemical effects and therapeutic potential. Indian J Pharmacol 33:2
Rakwal R, Agrawal G, Yonekura M, Kodama O (2000) Naringenin 7-O-methyltransferase involved in the biosynthesis of the flavanone phytoalexin sakuranetin from rice (Oryza sativa L.). Plant Sci 155:213–221
Ramakrishnan P (2003) The role of P-glycoprotein in the blood–brain barrier. Einstein J Biol Med 19:160–165
Rao BN, Srinivas M, Kumar YS, Rao YM (2007) Effect of silymarin on the oral bioavailability of ranitidine in healthy human volunteers. Drug Metab Drug Interact 22:175–185
Rao YK, Lee M-J, Chen K et al (2011) Insulin-mimetic action of rhoifolin and cosmosiin isolated from Citrus grandis (L.) Osbeck leaves: enhanced adiponectin secretion and insulin receptor phosphorylation in 3T3-L1 Cells. Evid Based Complement Alternat Med 2011:624375
Raub TJ (2006) P-glycoprotein recognition of substrates and circumvention through rational drug design. Mol Pharm 3:3–25
Romano B, Pagano E, Montanaro V et al (2013) Novel Insights into the pharmacology of flavonoids. Phytother Res 27:1588–1596
Romiti N, Tramonti G, Donati A, Chieli E (2004) Effects of grapefruit juice on the multidrug transporter P-glycoprotein in the human proximal tubular cell line HK-2. Life Sci 76:293–302
Ross JA, Kasum CM (2002) Dietary flavonoids: bioavailability, metabolic effects, and safety. Annu Rev Nutr 22:19–34
Sacco S, Maffei M (1997) The effect of isosakuranetin (5,7-dihydroxy 4′-methoxy flavanone) on potassium uptake in wheat root segments. Phytochemistry 46:245–248
Sauna ZE, Smith MM, Marianna M, Kerr KM (2001) The mechanism of action of multidrug-resistance-linked P-glycoprotein. J Bioenerg Biomembr 33:481–491
Scambia G, Ranelletti FO, Panici PB et al (1994) Quercetin potentiates the effect of Adriamycin in a multidrug-resistant MCF-7 human breast-cancer cell line: P-glycoprotein as a possible target. Cancer Chemother Pharmacol 34:459–464
Schinkel A (1999) P-Glycoprotein, a gatekeeper in the blood–brain barrier. Adv Drug Deliv Rev 36:179–194
Schinkel AH, Jonker JW (2012) Mammalian drug efflux transporters of the ATP binding cassette (ABC) family: an overview. Adv Drug Deliv Rev 55:3–29
Seegers U, Potschka H, Löscher W (2002) Lack of effects of prolonged treatment with phenobarbital or phenytoin on the expression of P-glycoprotein in various rat brain regions. Eur J Pharmacol 451:149–155
Shajib MTI, Pedersen HA, Mortensen AG et al (2012) Phytotoxic effect, uptake, and transformation of biochanin A in selected weed species. J Agric Food Chem 60:10715–10722
Sheu M-T, Liou Y-B, Kao Y-H et al (2010) A quantitative structure-activity relationship for the modulation effects of flavonoids on p-glycoprotein-mediated transport. Chem Pharm Bull (Tokyo) 58:1187–1194
Shin S-C, Choi J-S, Li X (2006) Enhanced bioavailability of tamoxifen after oral administration of tamoxifen with quercetin in rats. Int J Pharm 313:144–149
Shin S-C, Piao Y-J, Choi J-S (2008) Effects of morin on the bioavailability of tamoxifen and its main metabolite, 4-hydroxytamoxifen, in rats. In Vivo (Brooklyn) 22:391–395
Shin SC, Li C, Choi JS (2009) Effects of baicalein, an antioxidant, on the bioavailability of doxorubicin in rats: possible role of P-glycoprotein inhibition by baicalein. Pharmazie 64:579–583
Shohai T, Shafique M, Dhanya N, Divakar MC (2011) Importance of flavonoides in therapeutics. Hygeia J D Med 3:1–18
Singh SP, Wahajuddin Raju KSR et al (2012) Reduced bioavailability of tamoxifen and its metabolite 4-hydroxytamoxifen after oral administration with biochanin A (an isoflavone) in rats. Phytother Res 26:303–307
Spencer JPE (2007) The interactions of flavonoids within neuronal signalling pathways. Genes Nutr 2:257–273
Stouch TR, Gudmundsson O (2002) Progress in understanding the structure-activity relationships of P-glycoprotein. Adv Drug Deliv Rev 54:315–328
Sugimoto H, Hirabayashi H, Kimura Y et al (2011) Quantitative investigation of the impact of P-glycoprotein inhibition on drug transport across blood–brain barrier in rats. Drug Metab Dispos 39:8–14
Sun H, Dai H, Shaik N, Elmquist WF (2003) Drug efflux transporters in the CNS. Adv Drug Deliv Rev 55:83–105
Sun J, He Z-G, Cheng G et al (2004) Multidrug resistance P-glycoprotein: crucial significance in drug disposition and interaction. Med Sci Monit 10:5–14
Takanaga H, Ohnishi A, Matsuo H, Sawada Y (1998) Inhibition of vinblastine efflux mediated by P-glycoprotein by grapefruit juice components in caco-2 cells. Biol Pharm Bull 21:1062–1066
Takanaga H, Ohnishi A, Yamada S et al (2000) Polymethoxylated flavones in orange juice are inhibitors of P-glycoprotein but not cythocrome P450 3A4. J Pharmacol Exp Ther 293:230–236
Tandon VR, Kapoor B, Bano G et al (2006) P-glycoprotein : pharmacological relevance relevance. Indian J Pharmacol 38:13–24
Tapas AR, Sakarkar DM, Kakde RB (2008) Flavonoids as nutraceuticals : a Review. Trop J Pharm Res 7:1089–1099
Thilakarathna SH, Rupasinghe HPV (2013) Flavonoid bioavailability and attempts for bioavailability enhancement. Nutrients 5:3367–3387
Thomas H, Coley HM (2013) Overcoming multidrug resistance in cancer: an update on the clinical strategy of inhibiting p-glycoprotein. Cancer Control 10:159–165
Toh JY, Tan VMH, Lim PCY et al (2013) Flavonoids from fruit and vegetables: a focus on cardiovascular risk factors. Curr Atheroscler Rep 15:368
Toki K, Saito N, Irie Y et al (2008) 7-O-Methylated anthocyanidin glycosides from Catharanthus roseus. Phytochemistry 69:1215–1219
Tran VH, Marks D, Duke RK et al (2011) Modulation of P-glycoprotein-mediated anticancer drug accumulation, cytotoxicity, and ATPase activity by flavonoid interactions. Nutr Cancer 63:435–443
Trompier D, Baubichon-Cortay H, Chang X-B et al (2003) Multiple flavonoid-binding sites within multidrug resistance protein MRP1. Cell Mol Life Sci 60:2164–2177
Tsuji PA, Stephenson KK, Wade KL et al (2013) Structure-activity analysis of flavonoids: direct and indirect antioxidant, and antiinflammatory potencies and toxicities. Nutr Cancer 65:1014–1025
Udaya Kumar N, Sailendra M, Peddanna K et al (2011) Virtual screening of flavonoids as inhibitory agents of p-glycoprotein. IJABPT 2:130–140
Van der Kolk DM, de Vries EG, van Putten WJ et al (2000) P-glycoprotein and multidrug resistance protein activities in relation to treatment outcome in acute myeloid leukemia. Clin Cancer Res 6:3205–3214
Varma M (2003) P-glycoprotein inhibitors and their screening: a perspective from bioavailability enhancement. Pharmacol Res 48:347–359
Vauzour D, Vafeiadou K, Rodriguez-Mateos A et al (2008) The neuroprotective potential of flavonoids: a multiplicity of effects. Genes Nutr 3:115–126
Volk H, Burkhardt K, Potschka H et al (2004) Neuronal expression of the drug efflux transporter P-glycoprotein in the rat hippocampus after limbic seizures. Neuroscience 123:751–759
Wagner H, Aurnhammer G, Hörhammer L et al (1969) Synthesis of poncirin, an isosakuranetin-7-beta-neohesperidoside from Poncirus trifoliata Raf. Chem Ber 102:785–791
Wang EJ, Casciano CN, Clement RP, Johnson WW (2001) Inhibition of P-glycoprotein transport function by grapefruit juice psoralen. Pharm Res 18:432–438
Wang E, Barecki-Roach M, Johnson WW (2002) Elevation of P-glycoprotein function by a catechin in green tea. Biochem Biophys Res Commun 297:412–418
Wang Y-H, Chao P-DL, Hsiu S-L et al (2004) Lethal quercetin-digoxin interaction in pigs. Life Sci 74:1191–1197
Wang Y-H, Li Y, Yang S-L, Yang L (2005) An in silico approach for screening flavonoids as P-glycoprotein inhibitors based on a Bayesian-regularized neural network. J Comput Aided Mol Des 19:137–147
Wang H, Zhao X, Wang Y, Yin S (2007) Potential toxicities of flavonoids. Wei Sheng Yan Jiu 36:640–642
Wasowski C, Marder M (2012) Flavonoids as GABA A receptor ligands: the whole story? J Exp Pharmacol 4:9–24
Wesołowska O (2011) Interaction of phenothiazines, stilbenes and flavonoids with multidrug resistance-associated transporters, P-glycoprotein and MRP1. Acta Biochim Pol 58:433–448
Wesołowska O, Hendrich AB, Łaniapietrzak B et al (2009) Perturbation of the lipid phase of a membrane is not involved in the modulation of MRP1 transport activity by flavonoids. Cell Mol Biol Lett 14:199–221
Yang K, Wu J, Li X (2008) Recent advances in the research of P-glycoprotein inhibitors. Biosci Trends 2:137–146
Yang SH, Lee JH, Lee DY et al (2011) Effects of morin on the pharmacokinetics of docetaxel in rats with 7,12-dimethylbenz[a]anthracene (DMBA)-induced mammary tumors. Arch Pharm Res 34:1729–1734
Yang J, Qian D, Guo J et al (2013) Identification of the major metabolites of hyperoside produced by the human intestinal bacteria using ultra performance liquid chromatography/quadrupole-time-of-flight mass spectrometry. J Ethnopharmacol 147:174–179
Yeum C-H, Choi J-S (2006) Effect of naringin pretreatment on bioavailability of verapamil in rabbits. Arch Pharm Res 29:102–107
Yoo HH, Lee M, Chung HJ et al (2007) Effects of diosmin, a flavonoid glycoside in citrus fruits, on P-glycoprotein-mediated drug efflux in human intestinal Caco-2 cells. J Agric Food Chem 55:7620–7625
Zhang S, Morris ME (2003a) Effects of the flavonoids biochanin A, morin, phloretin, and silymarin on P-Glycoprotein-mediated transport. J Pharmacol Exp Ther 304:1258–1267
Zhang S, Morris ME (2003b) Effect of the flavonoids biochanin A and silymarin on the P-glycoprotein-mediated transport of digoxin and vinblastine in human intestinal Caco-2 cells. Pharm Res 20:1184–1191
Zhang L, Strong JM, Qiu W et al (2006) Scientific perspectives on drug transporters and their role in drug interactions. Mol Pharm 3:62–69
Zhang J, Yang G, Lin R, Hu Z (2011) Determination of paeoniflorin, calycosin-7-O-β-D-glucoside, ononin, calycosin and formononetin in rat plasma after oral administration of Buyang Huanwu decoction for their pharmacokinetic study by liquid chromatography-mass spectrometry. Biomed Chromatogr 25:450–457
Zhang C, Kwan P, Zuo Z, Baum L (2012) The transport of antiepileptic drugs by P-glycoprotein. Adv Drug Deliv Rev 64:930–942
Zhou G-X, Lu C-L, Wang H-S, Yao X-S (2009) An acetyl flavonol from Nervilia fordii (Hance) Schltr. J Asian Nat Prod Res 11:498–502
Zhu L, Zhao L, Wang H et al (2013) Oroxylin A reverses P-glycoprotein-mediated multidrug resistance of MCF7/ADR cells by G2/M arrest. Toxicol Lett 219:107–115
Zou P, Xing L, Tang Q et al (2012) Comparative evaluation of the teratogenicity of genistein and genistin using rat whole embryo culture and limbud micromass culture methods. Food Chem Toxicol 50:2831–2836
Acknowledgments
The authors thank the support of Fundação para a Ciência e a Tecnologia (FCT, Portugal) through the fellowship SFHR/BD/84936/2012, involving the POPH—QREN which is co-funded by FSE and MEC. The authors also thank the funding through the Strategic Project PEst-C/SAU/UI0709/2011.
Conflict of interest
The authors have declared no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Ferreira, A., Pousinho, S., Fortuna, A. et al. Flavonoid compounds as reversal agents of the P-glycoprotein-mediated multidrug resistance: biology, chemistry and pharmacology. Phytochem Rev 14, 233–272 (2015). https://doi.org/10.1007/s11101-014-9358-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11101-014-9358-0