
Abstract
Alkaloids are one of the most diverse groups of secondary metabolites found in living organisms. The most economically important alkaloids are the bisindole vinblastine, and vincristine. Unraveling the complexity of the genetic, catalytic and transport processes of monoterpene indole alkaloids biosynthesis is one of the most stimulating intellectual challenges in the plant secondary metabolism field. More than 50 metabolic steps are required to synthesize the most important alkaloids in Catharanthus roseus. Until now about only 20 of the 50 enzymes required for their biosynthesis have been determined and characterized. Hence, there are still a number of important enzymes that need to be characterized, beginning with the isolation and cloning of genes. It is also of fundamental importance to elucidate the regulatory aspects of their biosynthesis, both at the cellular and the molecular level, in order to address the question of their function in the plants that are producing them. In this review, we present an analysis of the state of the art related to the biosynthesis of the monoterpene indole alkaloids.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Alkaloids are one of the most diverse groups of secondary metabolites found in living organisms. While the alkaloids have been traditionally isolated from plants, since around 20% of this kingdom has been found to contain these compounds (De Luca and St Pierre 2000), they have also has been reported in an increasing number of animals, insects, marine invertebrates and microorganisms (Roberts and Wink 1998). A great number of alkaloids have been used in medicine and many of them are prominent components of modern drugs (Schmeller and Wink 1998).
The large family of alkaloid includes around 12,000 characterized products (De Luca and St Pierre 2000) with a large variety of chemical structures that have been classified into four groups according to their biogenetic origin: (1) alkaloids whose carbon skeleton is provided by amino acids such as ornithine/arginine, lysine, histidine, phenylalanine/tyrosine, tryptophan and the acids anthranilic and nicotinic; (2) purinic alkaloids; (3) aminates terpenes and (4) alkaloids poliketides. Among these the tryptophan-derived indol alkaloids that grouped into monoterpene or indol-terpene alkaloids (iridoids) and into numerous sub-groups based on the type of cyclization of the aromatic ring that occurs after removal of a glucose moiety from the 3α(S)-strictosidine. In this case eight sub-groups can be recognized: aspidospermatan, corynanthean, eburnan, ibogan, plumeran, strychnan, vallesiachotaman and vincosan (Van der Heijden et al. 2004).
Terpenoid indole-alkaloids
The terpenoid indole-alkaloids (TIAs) form a family of more than 3,000 members of which only a few have known physiological effects in mammals (Kutchan 1995; Geerlings et al. 2000). This type of alkaloid has been found in several plant families, but mainly in: Apocynaceae, Loganiaceae, Nissaceae and Rubiaceae, which are all of the Gentianales order. Among the better known and studied plants which produce TIAs are Catharanthus roseus, Tabernaemontana divaricata and Rauvolfia serpentina (Waterman 1998; Cordell 1999).
Catharanthus roseus
Catharanthus roseus (L.) G. Don is a perennial plant belonging to the Apocynaceae family, and one of the eight species of the genus Catharanthus (Van der Heijden et al. 2004). The genus comes from Madagascar and India. C. roseus has been cultivated with ornamental aims because it produces flowers of pink or white color for most of the year (Svoboda and Blake 1975). Also it has been used in traditional medicine as a hypoglycemic agent (Singh et al. 2001). The present interest in this plant is due to the fact that is an important source of chemotherapeutic agents with activity against several kinds of cancer (Svoboda and Blake 1975; Schmeller and Wink 1998), and also because it produces a great variety of TIAs (Svoboda and Blake 1975; Van der Heijden et al. 1989, 2004), most of them with pharmacological activity. Among the more important alkaloids produced by C. roseus are those of the bisindole type which include vinblastine, used in the treatment of Hodgkin’s disease (Schmeller and Wink 1998), and vincristine used in the treatment of leukemias (Schmeller and Wink 1998). This plant also produces antihypertensive agents such as ajmalicine and serpentine (Shanks et al. 1998), which are used to combat heart arrhythmias and to improve the blood circulation in the brain (Moreno et al. 1995; Schmeller and Wink 1998). The cost of 1 kg of vinblastine in the market is around a million dollars and the world annual production is near 12 kg. On the other hand, vincristine has reached the price of 3.5 million dollars for 1 kg and its annual production is 1 kg.
The high cost of these alkaloids is due to the fact that they are present in very small amounts in C. roseus leaves (around 0.0005% DW) and their extraction is carried out in the presence of many other compounds with very similar chemical and physical properties (Scott 1979; De Luca and Laflamme 2001).
This problem has created the necessity of finding alternative sources to obtain bisindole alkaloids. At the beginning of the 1980s, it was expected that plant tissue culture could be a useful alternative source of pharmacologically active C. roseus alkaloids but these have only been obtained in very low concentrations even after a substantial amount of research (Van der Heijden et al. 1989; Verpoorte et al. 1993; Moreno et al. 1995; De Luca et al. 1998). This lack of success with tissue cultures has been attributed to the lack of knowledge about the biosynthesis and degradation of these products.
Biosynthesis of TIAs
The biosynthesis of the TIAs in C. roseus is extraordinarily complex and requires the participation of the shikimate and 1-deoxy-d-xylulose-5-phosphate synthase pathways that yield the indole and terpene components required for their elaboration (De Luca 1993; Meijer et al. 1993c).
The enzyme l-amino acid-aromatic carboxy-lyase (tryptamine-forming) (EC 4.1.1.28)Footnote 1 (common name [cn] = tryptophan decarboxylase) catalyzes the conversion of l-tryptophan to l-tryptamine, deriving tryptophan to secondary metabolism. Tryptamine and secologanin, the monoterpene glucoiridoid precursor, are condensed to form strictosidine, the universal glucoalkaloid precursor of all the alkaloids isolated from C. roseus. The enzyme that catalyzes the condensation of tryptamine and secologanin is the 3α(S)-strictosidine tryptamine-liase (EC 4.3.3.2) (cn = strictosidine synthase).
The immediate precursors of TIAs, tryptamine and secologanin, require precursors coming from primary metabolism. In plants, as in animals and microorganisms, the precursors of the aromatic amino acids phenylalanine, tyrosine and tryptophan are derived from the shikimic acid pathway (Berg et al. 1995; Mathews et al. 2000).
Secologanin biosynthesis
The biosynthesis of secologanin involves a much more complex and branching route than the shikimic acid pathway used to supply tryptophan for MIA biosynthesis. Plants have two independent routes of biosynthesis of isopentenyl diphosphate for isoprenoid formation, which work in different cellular compartments such as (1) the classical pathway of acetate/mevalonate for the terpenoid biosynthesis in the cytoplasm, and (2) the route of 1-deoxy-d-xylulose-5-phosphate into the plastids (Fig. 1) (Lange et al. 2000; Lichtenthaler 2001).

Isopentenyl diphosphate biosynthesis pathway (IPP). The numbers inside the circle indicate the enzymes: acetyl-CoA C-acetyltransferase (1); 3-hydroxy-3-methylglutaryl CoA synthase (2); 3-hydroxy-3-methylglutaryl CoA reductase (3); mevalonate kinase (4); 5-phosphomevalonate kinase (5); diphosphomevalonate decarboxylase (6); 1-deoxy-d-xylulose-5-phosphate synthase (7); 1-deoxy-d-xylulose-5-phosphate reductoisomerase (8); 2-C-methyl-d-erytritol 4-phosphate cytidiltransferase (9); 4-(cytidine 5′-diphospho)-2-C-methyl-d-erytritol kinase (10); 2-C-methyl-d-eritritol 2,4-cyclodiphosphate synthase (11); 4-hydroxy-3-methylbut-2-en-1-il diphosphate synthase (12); 4-hydroxy-3-methylbut-2-enyl diphosphate reductase (13); isopentenyl diphosphate isomerase (14) (Rudney 1957; Tchen 1958; Bloch et al. 1959; Durr and Rudney 1960; Stern et al. 1960; Agranoff et al. 1960; Arigoni et al. 1997; McCaskill and Croteau 1998; Adam and Zapp 1998; Lange et al. 2000; Lichtenthaler 2001)
Our understanding of the metabolic route for the biosynthesis of the mevalonate is based on the investigations made on the biosynthesis of sterols, mainly in mammals and yeasts (Gray 1987). In plants these mechanisms are similar; however, the amount of the final products that are synthesized by the vegetables from this molecule is much greater.
The mevalonate pathway initiates when two acetyl CoA molecules are condensed to form acetoacetyl CoA by the acetyl-CoA:acetyl-CoA C-acetyltransferase reaction (EC 2.3.1.9) (cn = acetyl-CoA C-acetyltransferase) (reaction 1, Fig. 1). In the next step acetyl-CoA:acetoacetyl-CoA C-acetyltransferase (EC 2.3.3.10) (cn = hydroxymethylglutaryl-CoA synthase) catalyzes the conversion of acetoacetyl CoA and acetyl-CoA to 3-hydroxy-3-methylglutaryl-CoA (reaction 2, Fig. 1), which is reduced to (R)-mevalonate by the enzyme (R)-mevalonate:NADP+ oxidoreductase (CoA-acylating) (EC 1.1.1.34) (cn = hydroxymethylglutaryl-CoA reductase, HMGR) (reaction 3, Fig. 1). The (R)-mevalonate is phosphorylated two times; first by the ATP:(R)-mevalonate-5-phosphotransferase (EC 2.7.1.36) (cn = mevalonate kinase) (reaction 4, Fig. 1) and then by the ATP:(R)-5-phosphomevalonate phosphotransferase (EC 2.7.4.2) (cn = phosphomevalonate kinase) (reaction 5, Fig. 1) to produce (R)-5-diphosphomevalonate. Finally, the (R)-5-diphosphomevalonate is decarboxylated by the ATP:(R)-5-diphosphomevalonate carboxy-lyase (dehydrating) (EC 4.1.1.33) (cn = diphosphomevalonate decarboxilase) to isopentenyl pyrophosphate (IPP) (reaction 6, Fig. 1).
The IPP is turned into its isomer, dimethylallyl diphosphate (DMAPP) by the isopentenyl-diphosphate Δ3–Δ2 isomerase (EC 5.3.3.2) (cn = isopentenyl-diphosphate Δ-isomerase) (reaction 14, Fig. 1; reaction 1, Fig. 2). The condensation of DMAPP with one molecule of IPP is carried out by the dimethylallyl diphosphate:isopentenyl-diphosphate dimethylallyltranstransferase (EC 2.5.1.1) (cn = dimethylallyltranstransferase) (reaction 2, Fig. 2); this reaction generates the geranyl diphosphate (GPP), initiating the biosynthesis of the monoterpenes. Afterwards, the geranyl-diphosphate:isopentenyl-diphosphate geranyltranstransferase (EC 2.5.1.10) (cn = geranyltranstransferase) catalyzes the bonding of GPP with a molecule of IPP and produces the trans, transfarnesyl diphosphate (reaction 3, Fig. 2) which is channeled to the biosynthesis of sesquiterpenes, triterpenes, diterpenes, etc. (Gershenzon and Croteau 1990) (Fig. 2). Unlike animals, in plants some of the enzymes described for this part of the pathway form multienzymatic complexes. In radish (Raphanus sativus) (Bach et al. 1994) and C. roseus (Van der Heijden et al. 1994) the existence of a membrane associated enzyme that displays the activities of acetoacetyl-CoA C-acetyltransferase and hydroxymethylglutaryl-CoA synthase has been detected.

Until early 90s it was thought that plant isoprenoids were biosynthesized from mevalonate (Bach 1995; Chappell et al. 1995; Chappell 1995a, b), and that the enzyme HMGR could represent a limiting step for the biosynthesis of the vegetal isoprenoids, as happens for the biosynthesis of cholesterol in animals (Goldstein and Brown 1990). Also it was thought that the biosynthesis of IPP was carried out exclusively in the cytoplasm and then transported into the plastids. However, Heintze et al. (1994), observed that isolated plastids from wheat were able to biosynthesize carotenes from [14C]-acetate, which suggests that they possess the complete machinery to synthesize IPP. These experiments led to the discovery of a complete new metabolic pathway for the biosynthesis of the plastidic IPP (Schwender et al. 1996; Lichtenthaler et al. 1997).
The 1-deoxy-d-xylulose-5-phosphate synthase pathway, discovered in Escherichia coli by Rohmer et al. (1993), initiates with the condensation of pyruvate and glyceraldehyde-3-phosphate to produce 1-deoxy-d-xylulose-5-phosphate, in a reaction dependent on thiamine and catalyzed by the 1-deoxy-d-xylulose-5-phosphate synthase (EC 2.2.1.7) (Lichtenthaler 1999) (reaction 7, Fig. 1). This enzyme was isolated for the first time in plants from Mentha x piperita and it has a molecular mass of 71 kDa (Lange et al. 1998). In plants this alternative biosynthetic pathway generates IPP into the plastids for the biosynthesis of isoprenoids (Lange et al. 1998; McCaskill and Croteau 1998; Adam and Zapp 1998; Lichtenthaler 1999).
According to Contin et al. (1998), secologanin proceeds mainly from the IPP produced by the 1-deoxy-d-xylulose-5-phosphate synthase pathway. However, data in the literature suggest that the mevalonate pathway also participates in the biosynthesis of this monoterpene, even if to lesser degree. In our laboratory we determined that C. roseus hairy roots, harboring hamster 3-hydroxy-3-methylglutaryl-CoA reductase cDNA without membrane binding domain were able to modify both alkaloid and sterol biosynthesis (Ayora-Talavera et al. 2002). Clones with the lowest soluble and microsomal HMGR activity produced more ajmalicine and catharanthine than the control but had reduced campesterol concentration. Clones with high soluble HMGR activity produced high concentration of campesterol and five to seven times more serpentine than the control cells but no catharanthine and only low amounts of ajmalicine accumulated (Ayora-Talavera et al. 2002).
The involvement of both pathways has also been proposed in the case of the biosynthesis of other terpenes such as β-carotene, phytol and luteine from C. roseus (Arigoni et al. 1997) and the sesquiterpenes of Matricaria recutita (Adam and Zapp 1998).
In C. roseus only some of the enzymes that are required for the biosynthesis of the secologanin have been determined (Fig. 3); the best known enzymes are: HMGR (reaction 3, Fig. 1), which biosynthesizes mevalonate (Van der Heijden et al. 1989); the geraniol-10-hydroxylase (reaction 3, Fig. 3), which has been determined, isolated and purified from cellular suspensions (Meijer et al. 1993a; Canto-Canché and Loyola-Vargas 2000, 2001); the enzyme NADPH:cytochrome P-450 reductase, a component of geraniol-10-hydroxylase, which was purified to homogeneity by ion exchange chromatography (Madyastha and Coscia 1979a); and a non-specific monoterpene-alcohol oxidoreductase which works in the presence of NAD(P)+ (reaction 4, Fig. 3) and is capable of catalyzing the oxidation of 10-hydroxygeraniol to the correspondent aldehyde (Madyastha and Coscia 1979b).

Biosynthesis of the monoterpene iridoide secologanin. The numbers inside the circle indicate the enzymes: geraniol pyrophosphate synthase (1); phosphatase (2); geraniol-10-hydroxylase (3); NADP+: monoterpene oxidoreductase (4); 10-oxogeranial cyclase (5); 7-deoxyloganine, NADPH:oxygen oxidoreductase (7α-hydrolase) (6 and 8); S-adenosyl-l-methionine:loganate 11-O-methyltransferase (7 and 10); and loganin:oxygen oxidoreductase (9 and 11). The position of a specific phosphatase for the geranyl diphosphate at the beginning of the route proposed by the group of the Dr. R. Verpoorte (Meijer 1993), is indicated. The existence of this enzyme has not been demonstrated yet. The dotted arrows indicate probable or non-characterized steps. Several arrows in the same step indicate the probability of various metabolic steps. (■) Step inhibited by catharanthine. Diagram based of De Luca (1993), Gershenzon and Croteau (1990) and Meijer (1993c)
Recently our group has been able to determine the activity of 10-oxogeranial cyclase from transformed roots of C. roseus, using the true substrate of the enzyme. This enzyme catalyzes the first cyclization in the biosynthetic pathway of secologanin (reaction 5, Fig. 3) (Sánchez-Iturbe et al. 2005).
In addition, the (S)-adenosyl-l-methionine-loganic acid-methyltransferase has been found in seeds of C. roseus; this enzyme catalyzes the formation of loganine from loganic acid or secologanic acid and the (S)-adenosyl-l-methionine (reaction 6, Fig. 3) (Madyastha et al. 1973). Recently, the activity of the secologanin synthase (reaction 7, Fig. 3), a protein of the P450 family, has been characterized (Yamamoto et al. 2000; Irmler et al. 2000). Not all the enzymes have been purified. Geraniol 10-hydroxylase has been found as the only enzyme that is inhibited by an alkaloid, the catharanthine.
In general, very little is known about the biosynthesis of secologanin. The pathway has been postulated from experiments carried out using glucose, mevalonato, geraniol or other precursors labeled with 3H or 14C. These experiments have been carried out in suspension cultures of C. roseus, R. serpentina, Gardenia jasminoides, Teucrium marum and Nepetia cataria, as well as in different species of the Lonicera genus (Contin et al. 1998). Geraniol is the first product channeling the carbon skeletons to the biosynthesis of the monoterpenes. It has been proposed that there are at least 11 enzymatic steps between the geraniol and the secologanin as final product. However, only six are known: the geraniol 10-hydroxylase (reaction 3, Fig. 3), the NADP+:monoterpene oxidoreductase (reaction 4, Fig. 3), the oxogeranial (iridodial) cyclase (reaction 5, Fig. 3), the 7-deoxyloganine, NADPH:oxigen oxidoreductase (7α-hydrolase) (EC 1.14.13.74) (reactions 6 and 8, Fig. 3), the (S)-adenosyl-l-methionine:loganate 11-O-methyltransferase (EC 2.1.1.50) (reactions 7 and 10, Fig. 3), and the loganine:oxigen oxidoreductase (EC 1.3.3.9) (reactions 9 and 11, Fig. 3). Most enzymes have been not full characterized.
It has been proposed that the first regulated step in the biosynthesis of loganine is the oxygenation of geraniol. This reaction is catalyzed by the enzyme geraniol 10-hydroxylase (reaction 3, Fig. 3) and it has been suggested that this enzyme compromises the carbon skeletons for the biosynthesis of secologanin. Some of the observations that support the fact that this enzyme represents a regulation point are: its activity is induced under conditions which produce an increase in the alkaloid content, and its activity is inhibited by the addition of phosphates, which causes a decrease in the alkaloid content (Schiel et al. 1987).
Other conditions, which induce an increase or a decrease in the alkaloid content also show a correlation with the activity of geraniol 10-hydroxylase (Meijer et al. 1993a). This enzyme is inhibited by catharanthine (K i = 1 mM) (McFarlane et al. 1975). The physiological significance of this inhibition could be that the accumulation of this particular alkaloid provokes a feedback inhibition (Meijer et al. 1993c).
Geraniol 10-hydroxylase
Geraniol 10-hydroxylase is a monooxygenase of the P450 protein family (Meehan and Coscia 1973) and is localized in the vacuole membrane (Madyastha et al. 1977). This enzyme catalyzes the hydroxylation of geraniol at position 10 and requires O2 and NADPH (reaction 3, Fig. 3) (Meehan and Coscia 1973). The NADPH electrons are transferred to the monooxygenase heme group through the NADPH:cytochrome P450 reductase (CPR, E.C.1.6.2.4) (Donaldson and Luster 1991).
This flavoprotein is absolutely necessary for monooxygenase catalysis and it is formed by two components: cytochrome dependent NADPH dependent P-450 reductase and cytochrome P-450, with its own geraniol 10-hydroxylase activity (Meijer et al. 1993b).
The first component is a membranal flavoprotein that has been purified to homogeneity; its molecular mass is approximately 78 kDa and its cofactors are FMN and FAD (Madyastha et al. 1976, 1977; Madyastha and Coscia 1979a, b). The second component is a protein of molecular mass of 56 kDa that has been purified apparently to homogeneity. It accepts solely geraniol as substrate and not geraniol-P or nerol-P, which could be considered evidence of the existence of a previous dephosphorylation (Meijer et al. 1993a).
This enzyme has as noncompetitive inhibitor catharanthine (K i = 1 μm) that is stimulated by DTT and inhibited by CO, although this inhibition is reverted by light (Meehan and Coscia 1973; Madyastha et al. 1976; Madyastha and Coscia 1979b).
The cDNA of cytochrome P-450 reductase that codifies a peptide of 78.9 kDa has been cloned using a probe of conserved regions between these kind of proteins (Meijer et al. 1993b). Nevertheless, when this cDNA was used to transform Nicotiana tabacum and A. thaliana, the protein was expressed but geraniol 10-hydroxylase activity was not detected. The antibodies generated against the protein that codifies the cDNA crossed with the protein purified previously (Vetter et al. 1992).
We reported the characterization of this polyclonal serum raised against the purified G10H polypeptide. Anti-G10H IgG was able to inhibit the G10H activity and also recognized the G10H polypeptide from C. roseus and other plants producing TIAs (Canto-Canché et al. 2005).
G10H activity has been only detected in cell cultures of species belonging to the Apocynaceae family. The highest amounts of G10H activity (240 pkatal mg protein−1) were found in cell cultures of C. roseus. Further increase of G10H activity (335 pkatal mg protein−1) could be achieved by transferring the C. roseus cell culture to an induction medium known to enhance alkaloid production (Collu et al. 2002). Methyl jasmonate strongly induced G10H gene expression coordinately with other terpenoid indole alkaloid biosynthesis genes in C. roseus cell culture (Collu et al. 2001). Recently it has been found that cytokinin and/or ethylene greatly enhanced the expression of the geraniol-10-hydroxylase gene (Papon et al. 2005).
To obtain basic information about the regulation of the enzyme geraniol 10-hydroxylase, the behavior of this enzyme and its redox partner—the NADPH:Cyt C (P450) reductase—was examined in C. roseus hairy roots subjected to various treatments (abscisic acid, zeatine, P-free medium, transfer to B 5 /2 medium deprived of phosphorous, 0.1% macerozyme, transfer to alkaloid induction medium (B 5 /2 medium plus 8% sucrose) and fungus homogenate addition. For several of these treatments, no concerted responses of either enzyme were observed, but in general, the reductase was more responsive than G10H (Canto-Canché and Loyola-Vargas 2000). CPR activity was enhanced by all treatments, except zeatine, which had no effect (Canto-Canché and Loyola-Vargas 2000). It was shown that G10H activity is correlated with the ability of the cells to accumulate TIAs, although it seemed that increased G10H is not the only requirement for increased alkaloid accumulation (Collu et al. 2002).
NADPH-hemoprotein oxidoreductase
The NADPH-hemoprotein oxidoreductase (EC 1.6.2.4) is the redox partner of classical P450-monooxygenases, which have crucial roles in the metabolism of terpenes, alkaloids, flavonoids, phytoalexins etc. It has become evident that, contrary to the situation in animals and yeast, various CPR isoforms occur in some plants, although their specific physiological functions are largely unknown. C. roseus-CPR has been reported to be encoded by a single gene and early papers concerning the C. roseus-CPR protein also reported a single CPR polypeptide. The observation of diverse CPRs during purification or by immunoblot was attributed to proteolytic degradation. We obtained CPR-immunotype of C. roseus roots using two heterologous antisera directed against the CPR from Sorghum bicolor and Helianthus tuberosus, respectively (Canto-Canché and Loyola-Vargas 2001). Both antisera developed the same immunogenic profile with two cross-reactive polypeptides. The two immunoreactive polypeptides are probably not the result of proteolytic degradation, since increasing protease inhibitor concentration during the extraction and manipulation of the samples did not affect the occurrence of these two CPR forms. Roots from plants growing in the field showed identical CPR-immunotypes with those seen in vitro, indicating that this immunoprofile actually belongs to C. roseus roots. The lectin concanavaline A was able to inhibit the CPR activity from C. roseus hairy roots. Therefore, the immuno-reactive polypeptides probably result from post-translational glycosylation of the original polypeptide. Not only the roots but also the flowers, leaves and the stem showed more than a single CPR form. The different tissues of the plant showed different immuno-reactive bands, which were reproducible even though they came from tissues of plants growing in the field.
Monoterpene alcohol:NADP+ oxidoreductase
It has been established that this enzyme plays an important role in the biosynthesis of the secologanin (De Luca et al. 1998), and catalyzes the reversible oxidation of 10-hydroxygeraniol in the presence of NADP+ to produce 10-oxogeraniol or 10-hydroxygeranial (reaction 4, Fig. 3). Apparently this is an unspecific enzyme in relation to the OH group that is able to oxidize (Uesato et al. 1986). This enzyme has been purified from R. serpentina and its molecular mass is 44 kDa and its pI is 5.4. It is inactivated by iodoacetamide and by N-ethylmaleimide, which suggests that it requires SH groups for its activity (Ikeda et al. 1991). The K m value for the NADP+ and NADPH is 5.5 and 25 μM, respectively, having nerol (oxidation) or neral (reduction) as substrate. This enzyme does not use NAD+ or NADH as cofactors and it can accept primary allyl alcohols with chains greater to six carbons as substrates; nevertheless, it does not accept secondary alcohol, nor ethanol (De Luca et al. 1998).
10-Oxogeranial:iridodial cyclase
The enzyme 10-oxogeranial:iridodial cyclase catalyzes the formation of iridodial starting from 10-oxogeranial and NADPH (reaction 5, Fig. 3). It was isolated, semipurified and characterized from soluble extracts of C. roseus hairy roots (Sánchez-Iturbe et al. 2005) and purified 440 fold from R. serpentina (Uesato et al. 1986; Uesato et al. 1987) and it seems to be a homotetramer with a molecular mass of 118 kDa and an optimal pH 7.0. The C. roseus enzyme has a molecular mass of 66 kDa. The optimum pH for the semipurified enzyme was 7 and its pI was 5.4. The determination of the initial velocity of the 10-oxogeranial:iridodial cyclase in the presence of NADPH as the fixed-variable substrate at different concentrations of 10-oxogeranial follows the typical Michaelis–Menten equation. Calculation of the K m using the double reciprocal method yields K m values for NADPH and 10-oxogeranial of 70 μM and 0.52 mM, respectively (Sánchez-Iturbe et al. 2005).
7-Deoxyloganin, NADPH:oxigen oxidoreductase [7α-hydroxylase] (EC 1.14.13.74)
This enzyme catalyzes the formation of loganin starting from 7-deoxyloganin and NADPH. It can use also 7-deoxyloganate as substrate (reactions 6 and 8, Fig. 3). The reaction is inhibited by carbon monoxide, as well as the diverse inhibitors of the cytochrome P450 proteins, suggesting that this enzyme belongs to the cytochrome P450 family. The K m for 7-deoxyloganin and NADPH is of 170 and 18 μM, respectively (Katano et al. 2001).
Loganin:oxygen oxidoreductase (EC 1.3.3.9) (secologanin synthase)
This enzyme catalyzes the last step of secologanin biosynthesis from loganin, and involves the oxidative rupture of loganin’s methylcyclopentane ring (reaction 11, Fig. 3). The enzyme also recognizes the loganic acid as substrate (reaction 7, Fig. 3). This enzyme is a member of cytochrome P450 family and it is present in the microsomal fraction of cellular suspensions as well as in the leaf epidermis of C. roseus (Irmler et al. 2000). It also has been observed in cellular suspensions of Lonicera japonica (Yamamoto et al. 1999, 2000).
S-adenosyl-l-methionine:loganate 11-O-methyltransferase (EC 2.1.1.50)
This enzyme catalyzes the O-methylation of the carboxyl group of loganic acid. It can also catalyze the transfer of the methyl group from (S)-adenosyl-l-methionine either to loganic acid (reaction 9, Fig. 3) or to secologanic acid (reaction 10, Fig. 3), but it does not accept 7-deoxyloganic acid as a substrate. These data suggest that in secologanin biosynthesis, the hydroxylation precedes the methylation. This enzyme has been purified from C. roseus etiolated seedlings. The K m values for the (S)-adenosyl-l-methionine and loganic acid are 0.06 and 12.5 mM, respectively (Meijer et al. 1993c).
Biosynthesis of the TIAs
Most of the research done to characterize the enzymes of TIA biosynthesis has been carried out by taking into account the enormous complexity of the pathway. Excellent reviews of the subject have been made by De Luca (1993) and Meijer et al. (1993c). The list of the enzymes that have been studied in minor or greater degree include tryptophan decarboxylase, geraniol 10-hydroxylase, NADPH-cytochrome P450 reductase, oxidoreductase NADP+-dependent, 10-oxogeranial:iridodial cyclase, secologanin synthase, SAM-loganate O-methyltransferase, 7-deoxyloganin 7-hydroxylase, strictosidine synthase, strictosidine-β-d-glucosidase, cathenamine reductase, tabersonine 16-hydroxylase, S-adenosyl-l-methionine:16-hydroxytabersonine-16-O-methyltransferase, S-adenosyl-l-methionine:16-metoxy 2,3-dihydro-3-hydroxytabersonine-N-methyltransferase, desacetoxyvindoline-4-hydroxylase, acetylCoA:4-O-desacetylvindoline-4-O-acetyltransferase, tabersonine 17-hydroxylase and minovincinine 19-O-acetyltransferase.
The indol precursor tryptamine is synthesized from tryptophan (reaction 1, Fig. 4) in the reaction catalyzed by the enzyme aromatic-l-amino-acid carboxy-lyase (EC 4.1.1.28) (common name: tryptophan decarboxylase, TDC), which uses pyridoxal phosphate as cofactor. This reaction also can use 5-hydroxytryptophan as a substrate.

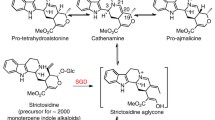
Biosynthesis of strictosidine, the universal precursor of TIAs. The numbers inside the circle indicate the enzymes: tryptophan decarboxylase (1); strictosidine synthase (2); strictosidine glucosidase (3); geissoschizine dehydrogenase (4). The slash arrows show different metabolic steps (De Luca 1993; Meijer et al. 1993c; Verpoorte et al. 1997; Sundberg and Smith 2002)
Both precursors, secologanin and tryptamine, are condensed to produce the strictosidine-O-glucose in the reaction catalyzed by the 3α(S)-strictosidine tryptamine-lyase (EC 4.3.3.2) (common name: strictosidine synthase, STR) (reaction 2, Fig. 4) (Stöckigt and Zenk 1977a). The product of this reaction, the 3α(S) strictosidine, is deglycosilated by the β-d-glucohydrolase (EC 3.2.1.105) (common name: strictosidine β-glucosidase) to yield the strictosidine aglycone (reaction 3, Fig. 4) that is considered the universal precursor of all the indol alkaloids, including vindoline and catharanthine (Stöckigt et al. 1976; Stöckigt and Zenk 1977b). The strictosidine aglycone is converted to 4,21-didehydrogeissoschizine spontaneously, and the last compound can either be transformed to cathenamine, the direct precursor of ajmalicine and serpentine (Stöckigt et al. 1977), or it can be enzymatically reduced to geissoschiizine (reaction 4, Fig. 4).
Since TDC and STR catalyze critical steps in the biosynthesis of TIAs, these enzymes have been amply studied (De Luca and Cutler 1987; Stevens et al. 1993).
TDC has been purified from suspension cultures (Fernandez et al. 1989) and hairy roots of C. roseus (Islas-Flores et al. 1994). The TDC is a cytoplasmic enzyme formed by two identical subunits of 55 kDa, has an approximate molecular mass of 110 kDa, a pI of 5.9, and a pH optimal of 8.5, and is modulated by diverse factors. It has been determined that the activity of the TDC increases in cultures of C. roseus in response to fungal homogenates (Pasquali et al. 1992) or by the addition of hydrolytic enzymes (Moreno-Valenzuela 1999). In the first case, it is known that the increase of its activity is due to the increase in the transcription of the gene and the translation of the messengers. On the other hand, in opposition to the effect of the fungi homogenate, the auxins repress genetic expression of the TDC (Goddijn et al. 1992).
Using a yeast one-hybrid technique three members of the Cys2/His2-type (transcription factor IIIA-type) zinc finger protein family from C. roseus, ZCT1, ZCT2, and ZCT3 were identified after the induction by fungal elicitors such as yeast extract (Pauw et al. 2004). These proteins bind in a sequence-specific manner to the TDC and STR promoters in vitro and repress the activity of these promoters in trans-activation assays. In addition, the ZCT proteins can repress the activating activity of APETALA2/ethylene response factor domain transcription factors, the ORCAs, on the STR promoter. The expression of the ZCT genes is rapidly induced by yeast extract and methyl jasmonate. These results suggest that the ZCT proteins act as repressors in the regulation of elicitor-induced secondary metabolism in C. roseus.
The K m value for tryptophan for this enzyme is 75 and 13 μM for 5-hydroxytryptophan. d-Tryptophan turns out to be a non-competitive inhibitor and tryptamine (the product of the reaction) is a competitive inhibitor (Noé et al. 1984; Islas-Flores et al. 1994).
TDC, besides being regulated by external factors (fungi attacks, etc.), is also subject to tissue specific and developmental regulation as well UV light (Ouwerkerk et al. 1999a, b). In seedlings of C. roseus from seeds germinated in vitro, under dark conditions, the increase in its activity is transitory. It begins to be detected after 3 days, reaches its maximum activity after 5 days, and then it declines (De Luca et al. 1988). In general, in seedlings its activity is greater in the aerial parts (being greater in young leaves than in the mature ones); whereas in the adult plant its activity is greater in the roots (Pasquali et al. 1992).
Although the TDC derives tryptophan from the processes of secondary metabolism, the overexpression of the TDC gene in C. roseus does not result in a greater production of TIAs. Despite this fact an increase in the amount of the protein with TDC activity and in the production of tryptamine takes place (Moreno et al. 1995). The oversynthesis of tryptamine is not reflected in a greater biosynthesis of alkaloids (Morris et al. 1985; Van der Heijden et al. 1989; Meijer et al. 1993c). This result suggests that TDC is not the only regulating step in the biosynthesis of these metabolites, confirming the previous results obtained with suspension cultures (Knobloch and Berlin 1983; Eilert et al. 1987a; Mérillon et al. 1989). On the other hand, as would be expected, the activity of TDC can diminish without affecting the total content of TIAs. Moreno-Valenzuela et al. (1998), dedifferentiated a hairy root culture into a suspension culture, and later they redifferentiated the cells to roots. During this process the TDC activity was dramatically altered. In the passage from roots to cells, the activity of the TDC diminished an order of magnitude or more and the TIAs diminished from 10 to 2 mg × g−1 of dry weight during this process. In the redifferentiated roots, the amount of TDC activity was similar to that detected in suspension cultures, whereas the production of alkaloids recovered to 90% with respect to the initial production. These data suggest that the cells do not require elevated amounts of TDC activity to produce the TIAs. This result recently has been verified using transgenic cultures overexpressing the TDC activity (Whitmer et al. 2002b).
A genomic clone of TDC from the C. roseus genome has been isolated and characterized. The gene for this enzyme is present in a single copy (Goddijn et al. 1993). The quantification of the amounts of TDC in seedlings and cellular suspensions shows that the expression of the gene as much as the enzymatic activity is induced in transient form, suggesting that the activity of the TDC is regulated at the transcriptional, translational and post-translational levels (Knobloch et al. 1981; De Luca et al. 1986; Eilert et al. 1987a; Pasquali et al. 1992).
Another enzyme, STR, catalyzes the stereospecific condensation of the secologanin with tryptamine to produce the 3α(S)-strictosidine. The enzyme is highly specific for its two substrates, i. e. it does not accept secologanic acid or tryptophan as substrates, but it shows a slight activity with analogs substituted in positions 5, 6 or 7 of the tryptamine. Until recently there were no indications that this enzyme is inhibited by alkaloids, the end products of this metabolic route (Mizukami et al. 1979; Treimer and Zenk 1979a; Treimer and Zenk 1979b).
STR was purified and characterized several years ago. It is a soluble enzyme of molecular mass between 35 and 38 kDa (Mizukami et al. 1979). Ten years later, the identification of four different forms of STR in suspension cells and leaves of C. roseus were reported (Pfitzner and Zenk 1989). De Waal et al. (1995) determined that the number of STRs is higher. They purified six different STRs, distinguishable by electrophoresis using gels of high concentration (acrylamide 20%).
In C. roseus seven STR isoenzymes have been detected, four of which can be separated by their catalytic activities (different value of K m) and by their pI (between 4.3 and 4.8). Pfizer and Zenk (1989) reported that since STR is a glycosylate enzyme, the difference among the isoenzymes is the length of the hydrocarbon chain. On the other hand, there is evidence that shows that the occurrence of isoforms is not related to the developmental stage nor to some tissue-specific regulation since it has been found that isoforms express as much in suspension cultures as in leaves, stems and roots (De Waal et al. 1995). These data suggest that the different isoforms could be the result of different patterns of glucosylation of the original protein, although the types of carbohydrates present have not yet been characterized (De Waal et al. 1995). The functions of the different isoforms of STR are not known.
Unlike TDC, which shows tissue specific regulation, STR presents similar amounts of activity in the different organs of the plant (De Luca et al. 1988), and its activity is not altered when the in vitro cultures of C. roseus are transferred into a production medium of alkaloids (8% sucrose) (Knobloch et al. 1981). However, a cDNA encoding bHLH transcription factor was isolated by the yeast one-hybrid system from a C. roseus cDNA library using the G-box element of the STR gene promoter as bait. The corresponding protein (named CrMYC1) was shown to bind specifically to the G-box in yeast. In C. roseus suspension cells CrMYC1 mRNA concentrations are induced by fungal elicitors and jasmonate suggesting that CrMYC1 may be involved in the regulation of gene expression in response to these signals (Chatel et al. 2003).
In C. roseus the Str gene was shown to be regulated by a wide variety of signals including auxin, methyl jasmonate and fungal elicitors in cell suspension cultures and by tissue-specific control in plant organs (Menke et al. 1999; Van der Fits et al. 2000; Sibéril et al. 2001). The Str promoter contains a functional G-box (CACGTG) cis-regulatory sequence. CrGBF1 had a high binding specificity for class I G-boxes including the Str G-box. CrGBF1 showed a lower affinity for class II G-boxes and for the G-box-like element (AACGTG) found in the TDC gene which encodes another enzyme involved in TIA biosynthesis. CrGBF2 showed a high affinity for all types of G-boxes tested and to a lesser extent for the Tdc G-box-like element (Sibéril et al. 2001).
This enzyme does not seem to be regulated by the development process (De Luca et al. 1988). Nevertheless, during the process described by Moreno-Valenzuela et al. (1998), dedifferentiation-rededifferentiation of C. roseus hairy roots, the activity of STR diminished by 60% when the roots were dedifferentiated to cells in suspension. The activity of the STR only recovered to half, whereas the pattern of isoforms changed when the cells were rededifferentiated to roots. Thus it is not yet clear if this enzyme is regulated by the development process (Moreno-Valenzuela et al. 1998).
Evidence exists that suggests that STR does not constitute a limiting step in the indole alkaloids biosynthesis in seedlings of C. roseus. It has been found that when suspension cells of C. roseus were transferred from a maintenance medium into an induction medium in order to produce alkaloids, the increase in the accumulation of alkaloids does not correlate with an increase in the activity of STR. Nevertheless, in callus of C. roseus transformed with the STR gene, a direct correlation was observed between the increase of the enzymatic activity and the accumulation of alkaloids (Kutchan 1993; Canel et al. 1998). Similar results have been obtained in Cinchona ledgeriana (Aerts et al. 1990, 1994) and seedlings of C. roseus (Aerts et al. 1994). The previous results suggest the possibility that the STR is a rate-limiting step of this pathway. Nevertheless, the information is imprecise and sometimes contradictory, which emphasizes the necessity to expand the investigations that allow us to establish their real role in the TIAs biosynthesis.
The STR is encoded by a single gene, suggesting that isoforms are the result of post-translational modifications of a single precursor (McKnight et al. 1990; Pasquali et al. 1999). This enzyme was the first of the TIAs’ metabolic enzymes to be cloned, first from R. serpentina (Kutchan et al. 1988) and then from C. roseus (McKnight et al. 1990). The messenger RNA for STR presents a region that encodes for a signal peptide of 31 amino acids that directs it to the vacuole (Pasquali et al. 1992), which supports diverse tests that suggesting that STR is a vacuolar enzyme (McKnight et al. 1991; Stevens et al. 1993).
Doireau et al. (1987), and Stevens et al. (1992) have measured the activity of TDC and STR, as well as the concentrations of tryptamine in suspension cultures of C. roseus and other species. These authors observed a tryptamine accumulation in spite of having a high activity of STR. These results support the hypothesis that the rate-limiting step for the biosynthesis of strictosidine and their derivatives is the presence of secologanin. The results of our group (Moreno-Valenzuela et al. 1998) also suggest that the main regulating points for the strictosidine production are located in some of the enzymes that participate in secologanin biosynthesis.
Biosynthesis of the first TIAs
After synthesis of strictosidine, the following step in the biosynthesis of the TIAs is the deglucosilation of strictosidine by strictosidine ß-d-glucohydrolase (EC 3.2.1.105) to produce strictosidine-aglycone (reaction 3, Fig. 4). This is an unstable product which, after several rearrangements which are not known accurately, is transformed into 4,21-dehydrogeissoschizine, which is reduced to geissoschizine by geissoschizine:NADP 4,21 oxidoreductase (EC 1.3.1.36) (reaction 4, Fig. 4) (Pfitzner and Stöckigt 1982).
Strictosidine ß-d-glucohydrolase is a glucoprotein localized in the endoplasmic reticulum (Geerlings et al. 2000). It is highly specific for strictosidine or its 10-methoxyderivative and in its native form seems to be aggregated, in which each 63 kDa subunit is united to the others by hydrophobic interactions (Luijendijk et al. 1998). This enzyme has been recently functionally expressed in Escherichia coli, purified to homogeneity and crystallized (Barleben et al. 2005).
It has been demonstrated that strictosidine ß-d-glucohydrolase is codified by a single gene that has been cloned recently and it is expressed at different levels in flowers, stems, leaves and roots, which suggests tissue-specific regulation. In addition, the quantified enzymatic activity in each organ is proportional to the mRNA amount detected (Geerlings et al. 2000). Since the level of expression of strictosidine ß-d-glucohydrolase is induced by methyl jasmonate (MeJa), and its activity is elevated in cultures that accumulate TIAs (Stevens et al. 1992) is considered a key step in the biosynthesis of these compounds.
On the other hand, the information on the geissoschizine:NADP 4.21 oxidoreductase is poor, with only one report of a partial purification from cellular suspensions of C. roseus. In comparison with other enzymes of this pathway its specific activity is very low (Pfitzner and Stöckigt 1982).
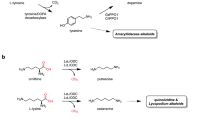
Likewise, 4,21-dehydrogeissoschizine, the biosynthetic pathway that leads to the biosynthesis of ajmalicine, catharanthine and tabersonine, is little documented. The ajmalicine biosynthetic pathway begins when the 4,21-dehydrogeissoschizine is converted to cathenamine (reaction 1, Fig. 5) by cathenamine synthase (Meijer et al. 1993c). The cathenamine is reduced to ajmalicine by cathenamine reductase (reaction 2, Fig. 5). The enzymes involved in this part of the pathway have been studied very little and it is known only that cathenamine reductase is NADPH dependent (Felix et al. 1981). Until now two isoenzymes have been separated, with one of them taking part in ajmalicine biosynthesis and the other one converting the iminium form of cathenamine to tetrahydroalstonine (Meijer et al. 1993c). It is not known how these two enzymes are regulated.

Ajmalicine and serpentine biosynthesis. The numbers inside the circle indicate the enzymes: cathenamine synthase (1); catenamina reductase (2); unspecific peroxidase (3); and tetrahydroalstonine synthase (4). The slash arrows show different metabolic steps. There is the possibility that the pathway does not start with 4,21-dehydrogeissoschizine, but with the 20,21-aldehyde of dehydrocorinanteine (Verpoorte et al. 1997; Roberts 1998; Sundberg and Smith 2002)
Cathenamine reductase (reaction 2, Fig. 5), which catalyzes cathenamine reduction to ajmalicine, has an optimum pH 6.6 and a molecular mass of 81 kDa. The characterization of the enzyme tetrahydroalstonine synthase (reaction 4, Fig. 5), which catalyzes the formation of tetrahydroalstonine from the iminium form of cathenamine, shows that both enzymes have the same optimum pH and the same molecular mass. The partially purified tetrahydroalstonine synthase has K m of 62 μM for cathenamine and utilizes NADPH but not NADH as cofactor (Hemscheidt and Zenk 1985).
Ajmalicine is oxidized to serpentine by peroxidases which are inside the vacuole (Blom et al. 1991) and it is known that its production is strongly influenced by light (Loyola-Vargas et al. 1992). Until now these peroxidases have not been purified and there is no data on their specificity, since ajmalicine is oxidized to serpentine in the presence of peroxidases from radish. Apparently this conversion allows the plant cells to maintain ajmalicine stored inside the vacuole. In cellular suspensions of C. roseus the oxidation of ajmalicine seems to be regulated by light because in its presence the cultures produce a great amount of serpentine, but when cultivated in the dark they only accumulate ajmalicine (Blom et al. 1991; Loyola-Vargas et al. 1992).
De Luca (1993) has suggested that geissoschizine reduction by geissoschizine NADP+ 4,21 oxidoreductase is reversible (reaction 4, Fig. 4) and that the ramification of the pathway can occur from a different modification than cathenamine cyclization. This last product is reduced to form ajmalicine, whereas the iminium form also is reduced to form tetrahydroalstonine (reaction 4, Fig. 5) (Hemscheidt and Zenk 1985).
Catharanthine has been detected in most of the in vitro cultures (Drapeau et al. 1987; Hong et al. 1997; Lee and Shuler 2000; Zhao et al. 2001c), but vindoline until recently had been found only in trace concentration (Misawa and Goodbody 1996; O’Keefe et al. 1997). The selection of the line of calli of C. roseus able to accumulate up to 0.45 mg−1 g DW of vindoline when incubated in the light has recently been reported (Zhao et al. 2001b).
The biosynthesis and accumulation of vindoline is associated with the presence of light and the developmental stage of the plant, and the expression of enzymes for its biosynthesis is organ-specific (Fahn et al. 1985a, b; De Luca et al. 1986; De Luca and Cutler 1987; De Luca et al. 1988, 1989; De Luca 1993; Vázquez-Flota et al. 1997; Vázquez-Flota and De Luca 1998a; De Luca and St Pierre 2000; Vázquez-Flota et al. 2002).
The 4,21-didehydrogeissoschizine is also the precursor of ajmaline biosynthesis. It is a highly complex molecule that contains nine chiral carbon atoms (Dogru et al. 2000). The biosynthesis of ajmaline involves nearly ten enzymatic steps (Fig. 6). The first metabolic step that derives 4,21-didehydrogeissoschizine to ajmaline biosynthesis is the oxidation of this compound to geissoschizine, in the reaction catalyzed by the geissoschizine dehydrogenase (EC 1.3.1.36) (reaction 1, Fig. 6), which adds stereospecifically hydrogen atom at carbon 21 in position α. The enzyme has been purified 35 times from cellular suspensions of C. roseus. The enzyme’s optimal pH 7.6 and its K m values for geissoschizine and for NADP+ are 83 μM and 77 nM, respectively (Pfitzner and Stöckigt 1982).

Biosynthesis of ajmaline. The numbers inside the circle indicate the enzymes: geissoschizine dehydrogenase (1); vomilenine reductase (2); polyneuridine aldehyde esterase (3); vinorine synthase (4); vinorine hydroxylase (5); 1,2-dihydrovomilenine:NADP+ oxidoreductase (6). The dotted arrows indicate unknown enzymes. Several arrows in the same step indicate the probability of various metabolic steps (Dogru et al. 2000; von Schumann et al. 2002)
The polyneuridine-aldehyde esterase (EC 3.1.1.78) catalyzes the conversion of aldehyde polyneuridine to 16-epi-vellosimine (reaction 3, Fig. 6) (Dogru et al. 2000). It produces an immediate precursor to the biosynthesis of the ajmalane skeleton that leads to ajmaline biosynthesis. This enzyme has been recently purified 423 times to homogeneity from cellular suspensions of R. serpentine. It has a molecular mass of 30 kDa and pI of 5.9 and it has been cloned through a cDNA library. The pure enzyme has a specific activity of 160.8 nkatal mg protein−1 and is highly specific for polyneuridine aldehyde as substrate for which it has a K m of 0.83 mM and belongs to the family of α/β-hydrolases (Dogru et al. 2000).
The following step in the ajmaline biosynthetic pathway, is vinorine biosynthesis and it is catalyzed by acyl CoA:16-epiveilosimine O-acetyltransferase (cyclizing) (cn: vinorine synthase) (EC 2.3.1.160) (reaction 4, Fig. 6). This enzyme has been purified 160 times from cellular suspensions of R. serpentine. Among its more interesting properties is its high optimum pH (8.5) and optimal temperature (35°C), a pI of 4.4 and a molecular mass of 31 kDa. The K ms for its substrates: the 16-epi-veilosimine and acetyl-CoA are of 19.4 and 64 μM, respectively, and like other enzymes in the ajmaline biosynthetic pathway it is highly specific for its substrates (Pfitzner et al. 1986). Based on partial peptide sequences a cDNA clone from R. serpentina was isolated and functionally expressed in Escherichia coli. The amino acid sequence of vinorine synthase has highest level of identity (28–31%) to that of Papaver salutaridinol acetyltransferase, Fragaria alcohol acyltransferase, and Catharanthus deacetylvindoline acetyltransferase involved in morphine, flavor, and vindoline biosynthesis, respectively (Bayer et al. 2004).
Vinorine is hydroxylated to vomilenine by vinorine, NADPH:oxygen oxidoreductase (21α-hydrolylating) (EC 1.14.13.75) (reaction 5, Fig. 6). The enzyme is completely NADPH and oxygen dependent, it is located in the microsomal fraction and is inhibited by typical inhibitors of cytochrome P-450 proteins, which is the reason why this monooxygenase is surely a cytochrome P-450 protein. The enzyme has an optimum pH 8.3 and its K m for NADPH and vinorine is 3 and 26 μM, respectively (Falkenhagen and Stöckigt 1995).
The last known step in ajmaline biosynthesis is the saturation of the indolmine double bond of vomilenine in stereospecific form to produce 2β(R)-1,2-dihydrovomilenine by 1,2-dihydrovomilenine:NADP+ oxidoreductase (EC 1.5.1.32) (reaction 6, Fig. 6). The enzyme catalyzes the reduction reaction is NADPH dependent and has been isolated recently from cellular suspensions of R. serpentine. It has a molecular mass of 43 kDa (von Schumann et al. 2002).
Biosynthesis of catharanthine and tabersonine
The biosynthesis of catharanthine as well as of tabersonine is initiated from the 20,21-aldehyde of dehydrocorinanteine and possibly proceeds through estemmadenine (El-Sayed et al. 2004) and dehydrosecodine cathenamine pathway, the precursor of the alkaloids with skeletons iboga or Aspidosperma (Fig. 7) (Sundberg and Smith 2002). Nothing is known about the nature of the enzymes involved in the conversion of cathenamine to catharanthine and tabersonine.

Catharanthine and tabersonine biosynthesis. The dotted arrows indicate unknown enzymes and metabolic steps. Several arrows in the same step indicate the probability of various metabolic steps (Sundberg and Smith 2002)
Vázquez-Flota et al. (1994) have determined that cultures of transformed roots challenged with MeJa increase the production of catharanthine. These results suggest that some of the enzymes participating in the biosynthesis of this alkaloid are subject to regulation and can be used for the study of the route of catharanthine biosynthesis (Fig. 7).
Biosynthesis of vindoline
Cellular suspensions of C. roseus have the capacity to synthesize catharanthine and tabersonine (Verpoorte et al. 1993; Moreno et al. 1995) and transform tabersonine in 16-metoxytabersonine (St-Pierre and De Luca 1995), but they do not have the enzymatic activity necessary for the vindoline biosynthesis (De Luca and Cutler 1987; De Carolis et al. 1990; Vázquez-Flota and De Luca 1998a). Therefore, a great amount of attention has been devoted to the study of the enzymes and regulatory mechanisms involved in vindoline biosynthesis.
The biosynthesis of vindoline from tabersonine is composed of six enzymatic steps (Fig. 8) catalyzed by the tabersonine NADPH:oxygen oxidoreductase (16-hydroxylating) (EC 1.14.13.73) (common name: tabersonine 16-hydroxylase), S-adenosyl-l-methionine:11-O-demethyl-17-O-deacetylvindoline 11-O-methyltransferase (EC 2.1.1.94) (common name: 11-O-demethyl-17-O-deacetylvindoline O-methyltransferase), a hydroxylase not characterized, S-adenosyl-L-methionine:16-metoxy-2,3-dihydro-3-hydroxy-tabersonine N-methyltransferase (EC 2.1.1.99) (common name: 16-metoxy-2,3-dihydro-3-hydroxy-tabersonine N-methyltransferase), desacetoxyvindoline,2-oxoglutarato:oxygen oxidoreductase (4β-hydroxylating (EC 1.14.11.20) (common name: desacetoxyvindoline-4-hydroxylase), and acetylCoA:17-O-deacetylvindoline 17-O-acetyltransferase (EC 2.3.1.107) (common name: 17-O-deacetylvindoline O-acetyltransferase) (Fahn et al. 1985a, b; De Luca et al. 1986; De Luca and Cutler 1987; De Carolis et al. 1990; De Luca et al. 1992; De Carolis and De Luca 1993; St-Pierre and De Luca 1995; Vázquez-Flota and De Luca 1998a).

Vindoline biosynthesis. The numbers inside the circle indicate the enzymes: tabersonine 16-hydroxylase (1); 11-O-demethyl-17-O-deacetylvindoline O-methyltransferase (2); uncharacterized hydroxylase (3); 16-metoxy-2,3-dihydro-3-hydroxy-tabersonine N-methyltransferase (4); deacetoxylvindoline 4-hydroxylase (5); 17-O-deacetylvindoline O-acetyltransferase (6) (Fahn et al. 1985a, b; De Luca et al. 1986; De Luca and Cutler 1987; De Carolis et al. 1990; De Luca et al. 1992; De Carolis and De Luca 1993; St-Pierre and De Luca 1995; De Luca et al. 1998; Roberts 1998; Vázquez-Flota and De Luca 1998a; Schröder et al. 1999; Vázquez-Flota et al. 2000b; Laflamme et al. 2001)
The results reported suggest that the route of vindoline biosynthesis is regulated by the processes of development and is restricted to the aerial parts of the adult plant, mainly in young leaves and cotyledons. In these tissues high amount of mRNA have been detected, as well as activity of the above enzymes (Fig. 11) (De Luca et al. 1985; St-Pierre and De Luca 1995; Vázquez-Flota and De Luca 1998a). This activity diminishes with age and organ type, and is already low in adult leaves, floral stems, roots and tips. Tabersonine 16-hydroxylase and 11-O-demethyl-17-O-deacetylvindoline O-methyltransferase, but not 16-metoxy-2,3-dihydro-3-hydroxy-tabersonine N-methyltransferase, desacetoxyvindoline-4-hydroxylase and 17-O-deacetylvindoline O-acetyltransferase have been detected in cell cultures of C. roseus (De Luca and Cutler 1987; St-Pierre and De Luca 1995; Vázquez-Flota et al. 2002).
Regulation of this route takes place by means of complex mechanisms at different levels. This regulation includes factors that affect the presence of these enzymes in transcriptional, post-transcriptional and post-translational levels to its location within the cell, the type of cell and, as was mentioned previously, the age of the tissue.
The enzymes tabersonine 16-hydroxylase (16-metoxy-2, 3-dihydro-3-hydroxytabersonine) N-methyltransferase, desacetoxyvindoline-4-hydroxylase and 17-O-desacetylvindoline O-acetyltransferase have been reported from plantlets and reach their maximum activity 6 days after the germination was initiated (De Luca et al. 1986). When these plantlets are placed in the light, the enzymes tabersonine 6-hydroxylase and desacetoxyvindoline-4-hydroxylase increase their activity six-fold, whereas 17-O-desacetylvindoline-17-O-acetyltransferase increase 10-fold (De Luca et al. 1988; De Carolis et al. 1990; St-Pierre and De Luca 1995). The effect of light on the desacetoxyvindoline-4-hydroxylase and 17-O-desacetylvindoline-17-O-acetyltransferase activity is mediated by phytochrome, since they are activated by a pulse of red light (660 nm) and this activation is reverted by a pulse of far red light (710 nm) (Aerts and De Luca 1992; Vázquez-Flota and De Luca 1998b). The 16-methoxy-2,3-dihydro-3-hydroxy-tabersonine-N-methyltransferase is the only enzyme of this pathway whose activity is not affected by the light (De Luca et al. 1988). Of all enzymes involved in vindoline biosynthesis, the desacetoxivindoline-4-hydroxylase is the most interesting. In plantlets of C. roseus, the gene d4h transcripts and the protein itself already are present in the dark. After exposing plantlets to light, the enzymatic amount and activity of desacetoxyvindoline-4-hydroxylase and concentration of gene d4h transcripts increase significantly. The reported results suggest that this increase in the activity is due to one isoenzyme of pI 4.7. The enzyme is inactive in the dark, which in the presence of the light, undergoes post-translational modifications that make it activate (Vázquez-Flota et al. 1997; Vázquez-Flota and De Luca 1998b). In a coordinated way, the light also induces dat gene trascription and its product, 17-O-desacetylvindoline-17-O-acetyltransferase increases its enzymatic activity (St-Pierre et al. 1998).
TDC and STR in these plantlets exhibit a peak of activity 5 days after imbibition in all tissues and neither of the two enzymes are stimulated by light (De Luca et al. 1986, 1988). MeJa induces the vindoline accumulation and increases the enzymatic activities of TDC, STR, desacetoxyvindoline-4-hydroxylase and 17-O-desacetoxyvindoline-17-O-acetyltransferase (Aerts et al. 1994).
Tabersonine NADPH:oxygen oxidoreductase (16-hydroxylating) (EC 1.14.13.73)
This enzyme is a P-450 monoxygenase located in the endoplasmic reticulum that requires NADPH, O2 and tabersonine to carry out its function (reaction 1, Fig. 8). This enzyme is inhibited by CO, clotrimazol, miconazol and cytochrome C (St-Pierre and De Luca 1995). This enzyme is detected preferentially in epidermis cells (Murata and De Luca 2005).
S-adenosyl- l -methionine:11-O-demethyl-17-O-deacetylvindoline 11-O-methyltransferase (EC 2.1.1.94)
This methyltransferase has been found both in cellular suspensions of C. roseus (Fahn et al. 1985b) and in plants of the same species (De Luca et al. 1986). The enzyme requires (S)-adenosyl-l-methionine as donor of methyl groups and is highly specific for 16-hydroxytabersonine (reaction 2, Fig. 8).
S-adenosyl- l -methionine:16-metoxy-2,3-dihydro-3-hydroxy-tabersonine N-methyltransferase (EC 2.1.1.99)
This enzyme is located in the chloroplast thylakoids (De Luca and Cutler 1987). It requires (S)-adenosyl-l-methionine as the donor of methyl groups (reaction 4, Fig. 8) and is highly specific for its substrate, the 16-methoxy-2,3-dihydro-3-hydroxy-tabersonine (De Luca et al. 1987). The enzyme has been partially purified, and has an apparent molecular mass of 60 kDa and is absolutely dependent on the presence of a double bond in position 2 (Dethier and De Luca 1993).
Desacetoxyvindoline, 2-oxoglutarato:oxygen oxidoreductase (4β-hydroxylating) (EC 1.14.11.20)
This enzyme is a dioxygenase α-ketoglutarate dependent, and catalyzes, in the cytoplasm, the addition of O2 to desacetoxyvindoline and α-ketoglutarate producing deacetylvindoline, succinate and CO2. This is the next to last reaction of the biosynthesis of vindoline (reaction 5, Fig. 8) (De Carolis et al. 1990; De Carolis and De Luca 1993; Vázquez-Flota and De Luca 1998a). This enzyme is highly specific, only hydrolize the C-4 of the N(1)-methyl-desacetoxyvindoline to form N(1)-methyl desacetylvindoline. The addition of ascorbic acid and ferrous ions increase its activity. The enzyme, purified to apparent homogeneity from young leaves of C. roseus, is a monomer with a molecular mass of approximately 45 kDa and it has three isoforms with pI of 4.6, 4.7 and 4.8.
The interaction kinetics of the substrate with the enzyme and studies of inhibition by the product suggest that the reaction is carried out by means of an ordered Ter–Ter mechanism. In this mechanism the first substrate is 2-oxoglutarate, followed by O2, and finally desacetoxyvindoline. The K m values for the 2-oxoglutarate, the O2 and desacetoxyvindoline are 45, 45 and 0.03 μM, respectively (De Carolis et al. 1990; De Carolis and De Luca 1993, 1994a, b).
Although this enzyme is found at trace amounts in etiolated plantlets of C. roseus, its activity increases substantially in the presence of light, simultaneously with the biosynthesis of vindoline (Vázquez-Flota and De Luca 1998a; Vázquez-Flota et al. 2000b). Etiolated plantlets begin to accumulate significant amounts of transcripts and protein of the hydroxylase, but there is practically no change in activity during the different stages of development. On the contrary, treatment with light produces an important increase in enzyme activity, but the increase in the transcript concentrations and protein is very small. These data suggest that the point at which light regulates the hydroxylase process is at the post-traslational level (Vázquez-Flota and De Luca 1998a; Vázquez-Flota et al. 2000b).
Using degenerate oligonucleotides for to scrutinize a cDNA library from C. roseus plantlets, three clones were obtained, of which two represent dimorphic alleles whose sequences differ solely in the untranslated 3′ end (Vázquez-Flota et al. 1997).
Acetyl-CoA:17-O-deacetylvindoline 17-O-acetyltransferase (EC 2.3.1.107)
The last step in the vindoline biosynthesis is catalyzed by 17-deacetylvindoline-O-acetyltransferase, dependent on acetyl-CoA (reaction 6, Fig. 8) (De Luca et al. 1985; Fahn et al. 1985a). The appearance of the enzyme is regulated by development in C. roseus plantlets and the enzymatic activity appears only after a treatment with light of the etiolated plantlets (De Luca et al. 1985). This acetyltransferase is a dimeric enzyme that has been purified to homogeneity and has a molecular mass of 32/21 kDa (Power et al. 1990) or 26/20 kDa (Fahn and Stöckigt 1990). The pure enzyme is formed by five isoforms with pI in the range of 4.3–5.4 (Fahn and Stöckigt 1990; Power et al. 1990). The K m values for acetyl-CoA and the deacetylvindoline are 5.4 and 6.5 μM, and 0.7 and 1.3 μM, for the enzymes partially purified (De Luca et al. 1985) and purified to homogeneity, respectively (Fahn and Stöckigt 1990; Power et al. 1990). Inhibition studies show that CoA is a powerful competitive inhibitor of deacetylvindoline (K i = 8 μM) (De Luca et al. 1985), similar to tabersonine (50% of inhibition at 45 μM) (Fahn and Stöckigt 1990), whereas the enzyme is not inhibited significantly by vindoline until concentrations of 2 mM (De Luca et al. 1985; Fahn and Stöckigt 1990).
The use of degenerate oligonucleotides for the amino and carboxyl terminal ends, and the later amplification of its product by PCR, allowed us to obtain a probe. This probe was used to carry out the scrutiny of a genomic DNA library of C. roseus. A clone was obtained that code for a protein of 439 amino acids with the predicted molecular mass of 49,890 Da. The clone, containing an extension of six histidines in the amino terminal, was expressed in E. coli and showed elevated activity. The activity of the recombinant enzyme is completely dependent on the presence of deacetylvindoline or acetyl-CoA. The analysis by Western blot of the recombinant protein and leaves of C. roseus yields a single band with a molecular mass of 50 kDa, which suggests that the isolated heterodimer during the purification is probably an artifact of the technique used (De Luca et al. 1998).
Although the determination of enzyme kinetics of vindoline biosynthesis has not yet been carried out, it is possible to say that the regulatory steps of this pathway are the two hydroxylation reactions (reactions 1 and 5, Fig. 8) (De Luca et al. 1998).
Bisindole alkaloid biosynthesis
The bisindole alkaloid biosynthesis begins with the catharanthine and vindoline condensation to form 3′,4′-anhydrovinblastine, the precursor of vinblastine, vincristine, leurosine and catharine (Fig. 9). This reaction, like the following oxidation, seems to be carried out by vacuolar cationic peroxidases, which suggest that the limiting step of the biosynthesis of this type of alkaloid is the availability of monomeric precursors (Kutney et al. 1981, 1988; Endo et al. 1988; Misawa et al. 1988; Sottomayor et al. 1996, 1997, 1998; Sottomayor and Barceló 2003; Smith et al. 2003; Sottomayor et al. 2004).

Biosynthesis of bisindole alkaloids vincristine and vinblastine. The numbers inside the circle indicate the enzymes: α-3′,4′-anhydrovinblastine synthase (1) (Kutney et al. 1981, 1988; Endo et al. 1988; Misawa et al. 1988; Verpoorte et al. 1997; Sottomayor et al. 1998; Smith et al. 2003). The dotted arrows indicate unknown enzymes and metabolic steps
Recently, an enzyme has been purified which catalyzes this step and has been termed α-3′,4′-anhydrovinblastine synthase (AVLBS) (reaction 1, Fig. 9). This enzyme is dependent of H2O2, and has been purified to apparent homogeneity (192×) from C. roseus leaves. It has a molecular mass of 45.4 kDa and an optimal pH 6.5 although also it presents substantial activity at a pH 4–5, which is the pH commonly found in the vacuoles. In addition, the pure enzyme of AVLBS has peroxidase activity. Its visible spectrum of absorbance shows maximums of absorption at 404, 501 and 633 nm suggesting that it is a protein with iron in hemo group and that belongs to the peroxidase family (Sottomayor et al. 1998).
El-Sayed and Verpoorte (2005) showed that treatment with MeJa can induce formation of bisindole alkaloids as the result of catabolism of the monomeric alkaloids catharanthine and vindoline. Serpentine also increased in the same period under the treatment as the catabolic product of ajmalicine. Basic peroxidases that are responsible for the formation of anhydrovinblastine and serpentine showed high activity at days 8 and 21 in treated leaves, causing the increase in anhydrovinblastine and serpentine.
Compartmentalization
The uses of in situ RNA hybridization and immunolocalization have revealed that TIA biosynthesis is a highly dynamic, complex and compartmentalized process in C. roseus (Kutchan 2005). The enzymes are localized in different cell types in the leaves and in root tips, as well as in different cellular compartments into the cells (Murata and De Luca 2005). The compartmentalization and localization involved in this metabolic pathway can be considered another regulatory mechanism, since this location requires the transport of different metabolites from one point to another for its transformation. Recently it has been suggested that different types of pumps are involved in the trafficking of natural products biosynthetic intermediates (Yazaki 2005, 2006).
Using Northern blot and in situ hybridization, Burlat et al. (2004) showed that three MEP pathway genes: 1-deoxy-d-xylulose-5-phosphate synthase, 1-deoxy-d-xylulose-5-phosphate reductoisomerase and 2-C-methyl-d-erytritol 2,4-cyclodiphosphate synthase as well as the geraniol 10-hydroxylase gene display identical cell-specific expression patterns. The authors predict the translocation of pathway intermediates from the internal phloem parenchyma to the epidermis and, ultimately, to laticifers and idioblasts during TIAs biosynthesis. Similarly, the translocation of intermediates from the phloem parenchyma is probably also required during the biosynthesis of hormones and photosynthetic primary metabolites derived from the MEP pathway.
In C. roseus dedifferentiated cell culture, CaaX-prenyltransferases (CaaX-PTases), including protein farnesyltransferase (PFT) and type I protein geranylgeranyltransferase (PGGT-I), catalyze a post-translational prenylation of proteins. Both CaaX-PTase activities are required for the expression of genes involved in monoterpenoid biosynthesis, including the first two genes of the 2-C-methylerythritol 4-phosphate (MEP) pathway. Using prenyltransferase inhibitor Courdavault (2005) showed that protein prenylation is also essential for the proper expression of MEP pathway genes in C. roseus hairy roots.
Almost all the cell compartments and different cellular types take part in the process; e.g., the messenger RNA for STR displays a region that codifies for a signal peptide of 31 amino acids that directs it to the vacuole (McKnight et al. 1991; Pasquali et al. 1992). This fact is supported by different assays that show that this enzyme is inside of the vacuole (McKnight et al. 1991; Stevens et al. 1993). On the other hand, strictosidine glucosidase and tabersonine 16-hydroxylase are located in the endoplasmic reticulum (St-Pierre and De Luca 1995; Geerlings et al. 2000).
The mRNAs for TDC and STR are located in the epidermal cells of stems, leaves and floral buds (St-Pierre et al. 1999). In the roots, the enzymatic activity of TDC is located in the cytosol and in the apoplastic region of the roots’ meristematic cells with a slight enrichment in the epidermal cells of the root cap and in the meristematic region. In the enlargement zone, TDC was localized only in the first three layers of the cortex. In the maturation zone, the enzyme was not present (St-Pierre et al. 1999; Moreno-Valenzuela et al. 2003).
The reactions catalyzed by the enzymes TDC, 11-O-demethyl-17-O-deacetylvindoline O-methyltransferase, desacetoxyvindoline-4-hydroxylase and 17-O-desacetylvindoline O-acetyltransferase are carried out in the cytoplasm (De Luca and Cutler 1987; St-Pierre and De Luca 1995; Vázquez-Flota et al. 1997). The reaction catalyzed by the enzyme AVLBS is carried out in the vacuole (Sottomayor et al. 1996). The enzyme 16-metoxy-2,3-dihydro-3-hydroxy-tabersonine N-methyltransferase is in the chloroplast thylakoids (Fig. 10) (De Luca and Cutler 1987).

Compartmentalization of the TIA biosynthesis in C. roseus. Redrawn from Meijer et al. (1993c) and Verpoorte et al. (2000). For ease of reading the figure presents all the steps in the same cell. However, the reader must take into account that different cell types and organs are participating in the biosynthesis process. Ray arrows indicate enzymes regulated by light. The dotted arrows indicate reactions not well characterized. Trp = tryptophan; G = geraniol; MV = mevalonate; DAV = dacetylvindoline; MOH = 16-metoxy-2,3-dihydro-3-hydroxytabersonine; 16 MT = 16-metoxytabersonine; 16-OHT = 16-hydroxytabersonine; TAB = tabersonine; EG = strictosidine glycone; DG = strictosidine aglycone; 10-OHG = 10-hydroxygeraniol; VLB = vinblastine; VCR = vincristine; G10OH = geraniol 10-hydroxylase; MIC = 10-oxogeranial cyclase; LMT = (S)-adenosyl-l-methionine:acid loganic methyl transferase; ES = strictosidine synthase; PDX = basic peroxidase; SG = strictosidine glucosidase; CR = cathenamine reductase; THA = tetrahydroalstonine synthase; GDH = geizssoschizine deshydrogenase; T16OH = tabersonine 16-hydroxylase; OMT = 11-O-demethyl-17-O-deacetylvindoline O-methyltransferase; NMT = 16-metoxy-2,3-dihydro-3-hydroxy-tabersonine N-methyltransferase; D4H = deacetoxylvindoline-4-hydroxylase; DAT = 17-O-deacetylvindoline O-acetyltransferase. The question marks denote enzymes that are still not known or for which contradictory reports exist at the moment
The mRNA for desacetoxyvindoline-4-hydroxylase and 17-O-desacetylvindoline O-acetyltransferase is located only in the idioblasts and laticifers cells of leaves, stems and floral buds (St-Pierre et al. 1999). This suggests that must be translocation mechanisms of the intermediaries from one cell to another, in order that the biosynthesis of these metabolites can be carried out (St-Pierre et al. 1999). More important still is the fact that the first steps in the vindoline biosynthesis are carried out in different cellular compartments from those of the last steps (Vázquez-Flota et al. 2000b).
Tabersonine gives rise to end products that are in different tissues in the plant (Fig. 11) (Laflamme et al. 2001; Rodriguez et al. 2003). In this case, the mRNA for the minovincinine 19-O-acetyltransferase (reaction 8, Fig. 11) is only expressed in the cortical cells of the radical meristems. Tabersonine 6,7-epoxidase activity (reaction 9, Fig. 11) converts tabersonine to lochnericine by selective epoxidation at positions 6 and 7 via a reaction dependent of NADPH and molecular oxygen. This activity was found in microsomes of transformed hairy root cultures of C. roseus (Rodriguez et al. 2003).

Compartmentalization of the vindoline biosynthesis and other TIAs. The numbers inside the circle indicate the enzymes: tabersonine 16-hydroxylase (1); S-adenosyl-l-methionine-16-hydroxytabersonine-O-methyltransferase (2); noncharacterized hydroxylase (3); S-adenosyl-l-methionine: 2,3-dihydro-3-hydroxytabersonine-N-methyltransferase (4); deacetoxylvindoline 4-hydroxylase (5); acetyl CoA: desacetylvindoline 4-O-acetyltransferase (6); tabersonine 19-hydroxylase (7) and (10); minovincinine 19-hydroxy-O-acetyltransferase (8); tabersonine 6,7-epoxidase (9), (11) and (12); acetyl transferase (13) (Kutney et al. 1980; Shanks et al. 1998; De Luca et al. 1998; Schröder et al. 1999; Vázquez-Flota et al. 2000b; Laflamme et al. 2001; Rodriguez et al. 2003)
In summary, the biosynthesis and storage of these complex molecules require a combination of phloem, parenchyma, laticifers and epidermal cells (Kutchan 2005). Tryptamine and secologanine precursors produce 16-methoxytabersonine in the leaf epidermal cells (Murata and De Luca 2005; Mahroug et al. 2006). This compound or its 2,3-dihydro-derivative is then transported to different cells (mesophyll/idioblast/laticifer) within the leaves for the final steps of the making of vindoline (Murata and De Luca 2005).
Regulation
Studies made with the indolic fraction, using cellular suspension of C. roseus, suggest that a direct relation does not exist between the values of maximum activity of TDC and the alkaloid production (Merillon et al. 1986; Moreno-Valenzuela et al. 1998; Whitmer et al. 2002a). Data of Eilert et al. (1987b), using fungal elicitors, also support this conclusion. Nevertheless it has also been reported that by varying the concentration of nutrients, a positive correlation between the activity of TDC and the alkaloid accumulation is observed (Knobloch et al. 1981). It is possible that the discrepancy between these results is due to the cellular line under study (Toivonen 1992).
The addition of geraniol, 10-hydroxygeraniol and secologanine, precursors of the terpenic pathway, increases the alkaloid content, specially tabersonine, in cellular suspensions, which seems to suggest that the production of alkaloids can be limited by one or more steps of the terpenic pathway (Merillon et al. 1986; Schiel et al. 1987; Doireau et al. 1987; Facchini and DiCosmo 1991; Shanks et al. 1998; Morgan and Shanks 2000). This conclusion cannot be generalized since contradictory evidence exists, such as the increase in the alkaloid production by means of the tryptamine addition to the culture medium (Zenk and Deus 1982). On the other hand, the addition of mevalonate does not have any effect in TIA biosynthesis (Shanks et al. 1998). Nevertheless, it is important to take into account a recent demonstration that the results depend fundamentally on two factors: the concentration of products added and the stage of the receiving culture. The addition of tryptophan in low concentrations in the early stage of the stationary phase produces an increase in the alkaloid biosynthesis but the addition of diverse precursors in a late stage of the culture cycle does not produce any effect (Morgan and Shanks 2000).
Different transgenic lines of C. roseus, which overexpress TDC and STR, have been used to carry out experiments of precursor feeding (Whitmer et al. 1998; Whitmer et al. 2002a, b). In general, the addition of secologanine at the time of the inoculation produces an important increase in the amount of TIAs, but the addition of tryptophan or tryptamine together with loganin produces a higher increase in the TIA biosynthesis in both transgenic lines.
The addition of external effectors like fungal homogenates, changes in pH of culture medium, etc., also modifies, in some cases substantially, the TIA biosynthesis (Godoy-Hernández and Loyola-Vargas 1991; Sáenz-Carbonell et al. 1993; Vázquez-Flota et al. 1994; Vázquez-Flota and Loyola-Vargas 1994; Godoy-Hernández and Loyola-Vargas 1997; Moreno-Valenzuela et al. 1999; Godoy-Hernández et al. 2000; Vázquez-Flota et al. 2000a; Zhao et al. 2001d).
The addition of pectinase increases the tabersonine amount 2.5 times whereas the addition of chitin elevates the ajmalicine concentration in 50% and the addition of jasmonic acid increases the lochnericine amounts and horhammericine, but not those of tabersonine (Shanks et al. 1998). The addition of fungal homogenates from mycelia fungi, as well as filtrates from fungi cultures, increases TIA accumulation (Godoy-Hernández and Loyola-Vargas 1991; Vázquez-Flota et al. 1994; Zhao et al. 2001d).
Other environmental factors also modify the TIA biosynthesis, among the most important of which are the osmotic and salt stresses (Godoy-Hernández and Loyola-Vargas 1991; Vázquez-Flota and Loyola-Vargas 1994; Zhao et al. 2001a). The addition of KCl, NaCl, manitol and sorbitol increases the TIA accumulation. When some of these effectors are used jointly, and the precursors of the TIAs biosynthesis like tryptamine and tryptophan are added, the TIA production improves substantially (Zhao et al. 2001a).
The biosynthesis of catharanthine and vindoline is regulated differentially. The vindoline biosynthesis is regulated at four different levels: tissue, cellular, by development and by the environmental conditions, whereas the catharanthine is not.
The tissue-specific location of the enzymes for the TIAs biosynthesis, the ontogenic regulation of the vindoline biosynthesis as a result of the coordinated expression of enzymes involved in the process, and the presence of specific alkaloids in the roots and the aerial part of the plants confirm that the biosynthetic routes are expressed in a tissue-specific form.
During the development of the plantlets, the activities of TDC and STR are expressed 36–48 h before those of 17-O-desacetylvindoline O-acetyltransferase (De Luca et al. 1988). The activation of these two latter enzymes concurs with the quantitative transformation of tabersonine in vindoline and it requires the participation of light (Balsevich et al. 1986; De Luca et al. 1988; Aerts and De Luca 1992; Vázquez-Flota and De Luca 1998a; Vázquez-Flota et al. 2000b). It has been also determined that a basipetal gradient of expression of TDC, STR, desacetoxyvindoline-4-hydroxylase and 17-O-desacetylvindoline O-acetyltransferase exists, which suggest that the expression of vindoline biosynthesis occurs transiently during the early development of leaves, stems and roots (St-Pierre et al. 1999; De Luca and St Pierre 2000).
The light plays a central role in the biosynthetic regulation of some alkaloids. Particularly, the light activates some of the last steps of the vindoline biosynthesis. The light does not participate in the formation of idioblasts nor of laticifer cells, so the activation by the light of the enzymes that carry out the induction of the TIA biosynthesis is still under study (Vázquez-Flota et al. 2000b).
In summary, catharanthine is biosynthesized throughout all plant tissues whereas vindoline is biosynthesized only in the aerial parts. The reactions catalyzed by TDC and STR are located in the upper and lower epidermis of the leaves, in the protoderma of the root and in the cortical cells around the apical tip. Desacetoxyvindoline-4-hydroxylase and 17-O-desacetylvindoline O-acetyltransferase are localized in the idioblasts and laticifer cells. It is possible that the root biosynthesizes catharanthine and tabersonine and that these compounds are transported via xylem to laticifer cells associated to traqueid elements. These cells can transform tabersonine in vindoline and this compound with catharanthine forms the bisindole alkaloids.
Cellular differentiation
The development process produces, in addition to morphogenesis, the biochemical specialization of cells, resulting in the biosynthesis and/or accumulation of secondary metabolites such as alkaloids of different biosynthetic origin (Robinson 1974; Robinson 1981; Flores and Filner 1985; Hashimoto and Yamada 1994; Facchini and De Luca 1995; Moreno-Valenzuela et al. 1998; Facchini 2001).
It has been shown that the hyoscyamine content, alkaloid derived from tropane, in transformed roots of Hyosciamus muticus is similar to that found in plant roots. Nevertheless, when the roots are dedifferentiated to callus, the amount of alkaloids is diminished at trace levels, but when the calluses are redifferentiated to roots, the content of alkaloids returns to the original concentrations. This protocol can be repeated many times with the same result (Flores and Filner 1985).
In other processes it has also been determined that a direct correlation between the cellular differentiation and the biosynthesis of secondary metabolites exists; e.g., the monoterpenes biosynthesis in the epidermic glands of Mentha piperita (Dörnenburg and Knorr 1995). In C. roseus it has been determined that the laticifer cells’ presence is essential for the production of monoterpene-indole alkaloids (De Luca et al. 1986). Thus, vindoline, one of the two monomeric units of the bisindol alkaloids, is synthesized in shoots but not in the calli (Constabel et al. 1982). Similar results have been obtained for vinblastine, a product accumulated in shoots regenerated from calli of C. roseus but not in calli or cellular suspensions (Endo et al. 1987; Datta and Srivastava 1997). On the other hand, the catharanthine biosynthesis practically takes place in the entire plant (Westekemper et al. 1980; Deus-Neumann et al. 1987; Balsevich and Bishop 1989).
Final remarks
The complexity of genetic and catalytic processes and those of transport of the TIA biosynthesis is one of the more stimulating intellectual challenges in the area of the secondary metabolites. The present knowledge of the TIA biosynthesis is still fragmented, whereas the biosynthetic route from tabersonine to vindoline is reasonable well known, including the cloning of the genes that codify for the required enzymes (De Luca et al. 1998). We are still far from understanding the biosynthesis of secologanine, ajmalicine, tabersonine, catharanthine and several of the intermediaries of the TIA pathway. However, today with new approaches (Magnotta et al. 2006) and the use of new techniques such as gene-to-metabolite networks (Rischer et al. 2006), metabolomics analysis (Kim et al. 2006; Oresic et al. 2006), expressed sequence tags (Murata et al. 2006), and transcriptome (Shukla et al. 2006) and proteome analysis (Jacobs et al. 2005) this is changing. During the coming years these new techniques will help to elucidate the biosynthetic pathway of the major intermediates in the TIA biosynthesis.
Another aspect of great importance that must be resolved is the transport of the molecules involved in the TIA biosynthesis. Since this biosynthesis involves different organelles and tissues within the plant, the mobilization of the components of the metabolic pathway must involve a complex transport network. This will become an active research area in the near future. It is also necessary to elucidate the diverse aspects of TIA biosynthesis regulation at the cellular as well as at the molecular level, in order to be in a position to determine the TIA function in plants.
Finally, an important issue that must be solved in the near future is how the TIA are degraded and, the most important question, what is the role of these compounds in the biology of the plant?
Notes
The first time that an enzyme in named, the systematic name and the Enzyme Commission Number (EC) assigned by IUBMB (www.chem.qmul.ac.uk/iubmb/enzyme/) is given. The common name is given in parenthesis; after the first time, and in all figure legends, the common name is used.
Abbreviations
- AVLBS:
-
α-3′, 4′-Anhydrovinblastine synthase
- CaaX-PTases:
-
CaaX-prenyltransferases
- CPR:
-
Cytochrome P450 reductase
- DMAPP:
-
Dimethylallyl diphosphate
- GPP:
-
Geranyl diphosphate
- IPP:
-
Isopentenyl pyrophosphate
- MeJa:
-
Methyl jasmonate
- MEP:
-
2-C-methylerythritol 4-phosphate
- PFT:
-
Protein farnesyltransferas
- PGGT-I:
-
Type I protein geranylgeranyltransferase
- STR:
-
Strictosidine synthase
- TDC:
-
Tryptophan decarboxylase
- TIAs:
-
Terpenoid indole-alkaloids
References
Adam KP, Zapp J (1998) Biosynthesis of the isoprene units of chamomile sesquiterpenes. Phytochemistry 48:953–959
Aerts RJ, De Luca V (1992) Phytochrome is involved in the light-regulation of vindoline biosynthesis in Catharanthus. Plant Physiol 100:1029–1032
Aerts RJ, Gisi D, De Carolis E et al (1994) Methyl jasmonate vapor increases the developmentally controlled synthesis of alkaloid in Catharanthus and Cinchona seedling. Plant J 5:635–643
Aerts RJ, Van der Leer T, Van der Heijden R et al (1990) Developmental regulation of alkaloid production in Cinchona seedlings. J Plant Physiol 136:86–91
Agranoff BW, Eggerer H, Henning U et al (1960) Biosynthesis of terpenes. VII. Isopentenyl pyrophosphate isomerase. J Biol Chem 235:326–332
Arigoni D, Sagner S, Latzel C et al (1997) Terpenoid biosynthesis from 1-deoxy-d-xylulose in higher plants by intramolecular skeletal rearrangement. Proc Natl Acad Sci (USA) 94:10600–10605
Ayora-Talavera T, Chappell J, Lozoya-Gloria E et al (2002) Overexpression in Catharanthus roseus hairy roots of a trucated hamster 3-hydroxy-3-methylglutaryl-CoA reductase gene. Appl Biochem Biotechnol 97:135–145
Bach TJ (1995) Some new aspects of isoprenoid biosynthesis in plants. A review. Lipids 30:191–202
Bach TJ, Raudot V, Vollack K-U et al (1994) Further studies on the enzymatic conversion of acetyl-coenzyme a into 3-hydroxy-3-methylglutaryl-coenzyme A in radish. Plant Physiol Biochem 32: 775–783
Balsevich J, Bishop G (1989) Distribution of catharanthine, vindoline, 3′-4′-anhydrovinblastine in the aerial parts of some Catharanthus roseus plants and the significance thereof in relation to alkaloid production in cultured cells. In: Kurz WGW (ed) Primary and secondary metabolism of plant cell cultures II. Springer-Verlag, Berlin, pp 149–153
Balsevich J, De Luca V, Kurz WGW (1986) Altered alkaloid pattern in dark grown seedlings of Catharanthus roseus. The isolation and characterization of 4-desacetoxyvindoline+: a novel indole alkaloid and proposed precursor of vindoline. Heterocycles 24:2415–2421
Banthorpe DV, Bucknall GA, Doonan HJ et al (1976) Biosynthesis of geraniol and nerol in cell-free extracts of Tanacetum vulgare. Phytochemistry 15:91–100
Barleben L, Ma X, Koepke J et al (2005) Expression, purification, crystallization and preliminary X-ray analysis of strictosidine glucosidase, an enzyme initiating biosynthetic pathways to a unique diversity of indole alkaloid skeletons. Biochim Biophys Acta Prot Prot 1747:89–92
Bayer A, Ma X, Stockigt J (2004) Acetyltransfer in natural product biosynthesis—functional cloning and molecular analysis of vinorine synthase. Bioorg Med Chem 12:2787–2795
Berg JM, Tymoczko JL, Stryer L (1995) Biochemistry. W.H. Freeman & Company, New York
Bloch K, Chaykin S, Phillips AH et al (1959) Mevalonic acid pyrophosphate and isopentenylpyrophosphate. J Biol Chem 234:2595–2604
Blom TJM, Sierra M, Van Vliet TB et al (1991) Uptake and accumulation of ajmalicine into isolated vacuoles of cultured cells of Catharanthus roseus (L.) G. Don. and its conversion into serpentine. Planta 183:170–177
Burlat V, Oudin A, Courtois M et al (2004) Co-expression of three MEP pathway genes and geraniol 10-hydroxylase internal phloem parenchyma of Catharanthus roseus implicates multicellular translocation of intermediates during the biosynthesis of monoterpene indole alkaloids and isoprene-derived primary metabolites. Plant J 38:131–141
Canel C, Lopes-Cardoso MI, Whitmer S et al (1998) Effects of over-expression of strictosidine synthase and tryptophan decarboxylase on alkaloid production by cell cultures of Catharanthus roseus. Planta 205:414–419
Canto-Canché B, Loyola-Vargas VM (2000) Non-coordinated response of cytochrome P450-dependent geraniol 10-hydroxylase and NADPH: Cyt C (P-450) reductase in Catharanthus roseus hairy roots under different conditions. Phyton 66:183–190
Canto-Canché B, Loyola-Vargas VM (2001) Multiple forms of NADPH-cytochrome P450 oxidoreductase in the Madagascar periwinkle Catharanthus roseus. In Vitro Cell Dev Biol Plant 37:622–628
Canto-Canché B, Meijer AH, Collu G et al (2005) Characterization of a polyclonal antiserum against the monoterpene monooxygenase, geraniol 10-hydroxylase from Catharanthus roseus. J Plant Physiol 162:393–402
Chappell J (1995a) Biochemistry and molecular biology of the isoprenoid biosynthetic pathway in plants. Annu Rev Plant Physiol Plant Mol Biol 46:521–547
Chappell J (1995b) The biochemistry and molecular biology of isoprenoid metabolism. Plant Physiol 107:1–6
Chappell J, Wolf F, Proulx J et al (1995) Is the reaction catalyzed by 3-hydroxy-3-methylglutaryl coenzyme A reductase a rate-limiting step for isoprenoid biosynthesis in plants? Plant Physiol 109:1337–1343
Chatel G, Montiel G, Pré M et al (2003) CrMYC1, a Catharanthus roseus elicitor- and jasmonate-responsive bHLH transcription factor that binds the G-box element of the strictosidine synthase gene promoter. J Exp Bot 54:2587–2588
Collu G, Alonso-García A, Van der Heijden R et al (2002) Activity of the cytochrome P450 enzyme geraniol 10-hydroxylase and alkaloid production in plant cell cultures. Plant Sci 162:165–172
Collu G, Unver N, Peltenburg-Looman A et al (2001) Geraniol 10-hydroxylase, a cytochrome P450 enzyme involved in terpenoid indole alkaloid biosynthesis. FEBS Lett 508:215–220
Constabel F, Gaudet-LaPrairie P, Kurz WGW et al (1982) Alkaloid production in Catharanthus roseus cell cultures. XII. Biosynthetic capacity of callus from original explants and regenerated shoots. Plant Cell Rep 1:139–142
Contin A, Van der Heijden R, Lefeber AW et al (1998) The iridoid glucoside secologanin is derived from the novel triose phosphate/pyruvate pathway in a Catharanthus roseus cell culture. FEBS Lett 434:413–416
Cordell GA (1999) The monoterpene alkaloids. In: Cordell GA (ed) The alkaloids. Academic Press, San Diego, pp 261–376
Courdavault V, Burlat V, St-Pierre B et al (2005) Characterisation of CaaX-prenyltransferases in Catharanthus roseus: relationships with the expression of genes involved in the early stages of monoterpenoid biosynthetic pathway. Plant Sci 168:1097–1107
Datta A, Srivastava PS (1997) Variation in vinblastine production by Catharanthus roseus during in vivo and in vitro differentiation. Phytochemistry 46:135–137
De Carolis E, Chan F, Balsevich J et al (1990) Isolation and characterization of a 2-oxoglutarate dependent dioxygenase involved in the second-to-last step in vindoline biosynthesis. Plant Physiol 94:1323–1329
De Carolis E, De Luca V (1993) Purification, characterization, and kinetic analysis of a 2-oxoglutarate-dependent dioxygenase involved in vindoline biosynthesis from Catharanthus roseus. J Biol Chem 268:5504–5511
De Carolis E, De Luca V (1994a) 2-oxoglutarate-dependent dioxygenase and related enzymes: biochemical characterization. Phytochemistry 36:1093–1107
De Carolis E, De Luca V (1994b) A novel 2-oxoglutarate-dependent dioxygenase involved in vindoline biosynthesis: characterization, purification and kinetic properties. Plant Cell Tiss Org Cult 38:281–287
De Luca V (1993) Enzymology of indole alkaloid biosynthesis. In: Lea PJ (ed) Methods in plant biochemistry, vol 9. Academic Press Limited, London, pp 345–368
De Luca V, Aerts RJ, Chavadej S et al (1992) The biosynthesis of monoterpenoid indole alkaloids in Catharanthus roseus. In: Singh BK, Flores HE, Shannon JC (eds) Biosynthesis and molecular regulation of amino acids in plants. American Society of Plant Physiology, Rockville, pp 275–284
De Luca V, Balsevich J, Kurz WGW (1985) Acetyl coenzyme A: deacetylvindoline O-acetyltransferase, a novel enzyme from Catharanthus. J Plant Physiol 121:417–428
De Luca V, Balsevich J, Tyler RT et al (1986) Biosynthesis of indole alkaloids: developmental regulation of the biosynthetic pathway from tabersonine to vindoline in Catharanthus roseus. J Plant Physiol 125:147–156
De Luca V, Balsevich J, Tyler RT et al (1987) Characterization of a novel N-methyltransferase (NMT) from Catharanthus roseus plants. Plant Cell Rep 6:458–461
De Luca V, Brisson N, Kurz WGW (1989) Regulation of vindoline biosynthesis in Catharanthus roseus: molecular cloning of the first and the last steps in biosynthesis. In: Kurz WGW (ed) Primary and secondary metabolism of plants cell cultures II. Springer Verlag, Berlin, pp 154–161
De Luca V, Cutler AJ (1987) Subcellular localization of enzymes involved in indole alkaloid biosynthesis in Catharanthus roseus. Plant Physiol 85:1099–1102
De Luca V, Fernández AJ, Campbell D et al (1988) Developmental regulation of enzymes of indole alkaloid biosynthesis in Catharanthus roseus. Plant Physiol 86:447–450
De Luca V, Laflamme P (2001) The expanding universe of alkaloid biosynthesis. Curr Opi Plant Biol 4:225–233
De Luca V, St Pierre B (2000) The cell and developmental biology of alkaloid biosynthesis. Trends Plant Sci 5:168–173
De Luca V, St-Pierre B, Vázquez-Flota F et al (1998) Indole alkaloid biosynthesis in Catharanthus roseus: the establishment of a model system. In: Lo Chiavo F, Last RL, Morelli G et al (eds) Cellular integration of signalling pathways in plant development. Springer-Verlag, Berlin, pp 171–187
De Waal A, Meijer AH, Verpoorte R (1995) Strictosidine synthase from Catharanthus roseus: purification and characterization of multiple forms. Biochem J 306:571–580
Dethier M, De Luca V (1993) Partial purification of an N-methyltransferase involved in vindoline biosynthesis in Catharanthus roseus. Phytochemistry 32:673–678
Deus-Neumann B, Stöckigt J, Zenk MH (1987) Radioimmunoassay for the quantitative determination of catharanthine. Planta Med 53:184–188
Dogru E, Warzecha H, Seibel F et al (2000) The gene encoding polyneuride aldehyde esterase of monoterpenoid indole alkaloid biosynthesis in plants is an ortholog of the α/b hydroxylase super family. Eur J Biochem 267:1397–1406
Doireau P, Mérillon JM, Guillot A et al (1987) Time-course studies on indole alkaloid accumulation and changes in tryptophan decarboxylase and strictosidine synthase activities: a comparison in three strains of Catharanthus roseus cells. Planta Med 53:364–367
Donaldson RP, Luster DG (1991) Multiple forms of plant cytochromes P-450. Plant Physiol 96:669–674
Dörnenburg H, Knorr D (1995) Strategies for the improvement of secondary metabolite production in plant cell cultures. Enzyme Microb Technol 17:674–684
Drapeau D, Blanch HW, Wilke CR (1987) Ajmalicine, serpentine, and catharanthine accumulation in Catharanthus roseus bioreactor cultures. Planta Med 53:373–376
Durr IF, Rudney H (1960) The reduction of β-hydroxy-β-methylglutaryl coenzyme A to mevalonic acid. J Biol Chem 235:2572–2578
Eilert U, De Luca V, Constabel F et al (1987a) Elicitor-mediated induction of tryptophan decarboxylase and strictosidine synthase activities in cell suspension cultures of Catharanthus roseus. Arch Biochem Biophys 254:491–497
Eilert U, Kurz WGW, Constabel F (1987b) Alkaloid accumulation in plant cell cultures upon treatment with elicitors. In: Green CE, Somers DA, Hackett WP et al (eds) Plant biology, vol 3. Plant tissue and cell culture. Alan R. Liss, Co., New York, pp 213–219
El-Sayed M, Choi YH, Frédérich M et al (2004) Alkaloid accumulation in Catharanthus roseus cell suspension cultures fed with stemmadenine. Biotechnol Lett 26:793–798
El-Sayed M, Verpoorte R (2005) Methyljasmonate accelerates catabolism of monoterpenoid indole alkaloids in Catharanthus roseus during leaf processing. Fitoterapia 76:83–90
Endo T, Goodbody AE, Misawa M (1987) Alkaloid production in root and shoot cultures of Catharanthus roseus. Planta Med 53:479–482
Endo T, Goodbody AE, Vukovic J et al (1988) Enzymes from Catharanthus roseus cell suspension cultures that couple vindoline and catharanthine to form 3′,4′-anhydrovinblastine. Phytochemistry 27:2147–2149
Facchini PJ (2001) Alkaloid biosynthesis in plants: Biochemistry, cell biology, molecular regulation, and metabolic engineering applications. Annu Rev Plant Physiol Plant Mol Biol 52:29–66
Facchini PJ, De Luca V (1995) Phloem-specific expression of tyrosine/dopa decarboxylase genes and the biosynthesis of isoquinoline alkaloids in opium poppy. Plant Cell 7:1811–1821
Facchini PJ, DiCosmo F (1991) Secondary metabolite biosynthesis in cultured cells of Catharanthus roseus (L.) G. Don immobilized by adhesion to glass fibres. Appl Microbiol Biotechnol 35:382–392
Fahn W, Gundlach H, Deus-Neumann B et al (1985a) Late enzymes of vindoline biosynthesis. acetyl-CoA:17-O-acetyl-transferase. Plant Cell Rep 4:333–336
Fahn W, LauBermair E, Deus-Neumann B et al (1985b) Late enzymes of vindoline biosynthesis. S-adenosyl-l-methionine:11-O-demethyl-17-O-deacetylvindoline 11-O-methyltransferase and unspecific acetylesterase. Plant Cell Rep 4:337–340
Fahn W, Stöckigt J (1990) Purification of acetyl-CoA: 17-O-deacetylvindoline 17-O-acetyltransferase from Catharanthus roseus leaves. Plant Cell Rep 8:613–616
Falkenhagen H, Stöckigt J (1995) Enzymatic biosynthesis of vomilenine, a key intermediate of the ajmaline pathway, catalyzed by a novel cytochrome P450-dependent enzyme from plant cell cultures of Rauwolfia serpentina. Z Naturforsch [C] 50:45–53
Felix H, Brodelius P, Mosbach K (1981) Enzyme activities of the primary and secondary metabolism of simultaneously permeabilized and immobilized plant cells. Anal Biochem 116:462–470
Fernandez JA, Kurz WGW, De Luca V (1989) Conformation-dependent inactivation of tryptophan decarboxylase from Catharanthus roseus. Biochem Cell Biol 67:730–734
Flores HE, Filner P (1985) Metabolic relationships of putrescine, GABA and alkaloids in cell and root cultures of Solanaceae. In: Neumann KH, Barz W, Reinhard E (eds) Primary and secondary metabolism in plant cell cultures. Springer-Verlag, Heidelberg, pp 174–186
Geerlings A, Ibañez MML, Memelink J et al (2000) Molecular cloning and analysis of strictosidine β-d-glucosidase, an enzyme in terpenoid indole alkaloid biosynthesis in Catharanthus roseus. J Biol Chem 275:3051–3056
Gershenzon J, Croteau R (1990) Regulation of monoterpene biosynthesis in higher plants. Rec Advan Phytochem 24:99–160
Goddijn OJM, De Kam RJ, Zanetti A et al (1992) Auxin rapidly down-regulates transcription of the tryptophan decarboxylase gene from Catharanthus roseus. Plant Mol Biol 18:1113–1120
Goddijn OJM, Van der Duyn Schouten PM, Schilperoort RA et al (1993) A chimaeric tryptophan decarboxylase gene as a novel selectable marker in plant cells. Plant Mol Biol 22:907–912
Godoy-Hernández G, Loyola-Vargas VM (1991) Effect of fungal homogenate, enzyme inhibitors and osmotic stress on alkaloid content of Catharanthus roseus cell suspension cultures. Plant Cell Rep 10:537–540
Godoy-Hernández G, Loyola-Vargas VM (1997) Effect of acetylsalicylic acid on secondary metabolism of Catharanthus roseus tumor suspension culture. Plant Cell Rep 16:287–290
Godoy-Hernández GC, Vázquez-Flota FA, Loyola-Vargas VM (2000) The exposure to trans-cinnamic acid of osmotically stressed Catharanthus roseus cells cultured in a 14-L bioreactor increases alkaloid accumulation. Biotechnol Lett 22:921–925
Goldstein JL, Brown MS (1990) Regulation of the mevalonate pathway. Nature 343:425–430
Gray JC (1987) Control of isoprenoid biosynthesis in higher plants. In: Callow JA (ed) Advances in botanical research, vol 14. Academic Press, New York pp 25–91
Hashimoto T, Yamada Y (1994) Alkaloid biogenesis: molecular aspects. Annu Rev Plant Physiol Plant Mol Biol 45:257–285
Heintze A, Riedel A, Aydogdu S et al (1994) Formation of chloroplast isoprenoids from pyruvate and acetate by chloroplasts from young spinach plants. Evidence for a mevalonate pathway in immature chloroplasts. Plant Physiol Biochem 32:791–797
Hemscheidt T, Zenk MH (1985) Partial purification and characterization of a NADPH dependent tetrahydroalstonine synthase from Catharanthus roseus cell suspension cultures. Plant Cell Rep 4:216–219
Hong J, Lee J, Lee H et al (1997) Enhancement of catharanthine production by the addition of paper pulp waste liquors to Catharanthus roseus in chemostat cultivation. Biotechnol Lett 19:967–969
Ikeda H, Esaki N, Nakai S et al (1991) Acyclic monoterpene primary alcohol: NADP super(+)oxidoreductase of Rauwolfia serpentina cells: the key enzyme in biosynthesis of monoterpene alcohols. J Biochem (Tokyo) 109:341–347
Irmler S, Schröder G, St-Pierre B et al (2000) Indole alkaloid biosynthesis in Catharanthus roseus: new enzyme activities and identification of cytochrome P450 CYP72A1 as secologanin synthase. Plant J 24:797–804
Islas-Flores IR, Loyola-Vargas VM, Miranda-Ham ML (1994) Tryptophan decarboxylase activity in transformed roots from Catharanthus roseus and its relationship to tryptamine, ajmalicine, and catharanthine accumulation during the culture cycle. In Vitro Cell Dev Biol Plant 30:81–83
Jacobs DI, Gaspari M, van der Greef J et al (2005) Proteome analysis of the medicinal plant Catharanthus roseus. Planta 221:690–704
Katano N, Yamamoto H, Iio R et al (2001) 7-Deoxyloganin 7-hydroxylase in Lonicera japonica cell cultures. Phytochemistry 58:53–58
Kim H, Choi Y, Verpoorte R (2006) Metabolomic analysis of Catharanthus roseus using NMR and principal component analysis. In: Saito K, Dixon R, Willmitzer L (eds) Plant metabolomics. Springer, Berlin, pp 261–276
Knobloch K-H, Berlin J (1983) Influence of phosphate on the formation of the indole alkaloids and phenolic compounds in cell suspension cultures of Catharanthus roseus. I. Comparison of enzyme activities and product accumulation. Plant Cell Tiss Org Cult 2:333–340
Knobloch K-H, Hansen B, Berlin J (1981) Medium-induced formation of indole alkaloids and concomitant changes of interrelated enzyme activities in cell suspension cultures of Catharanthus roseus. Z Naturforsch [C] 36c:40–43
Kutchan TM (1993) Strictosidine: from alkaloid to enzyme to gene. Phytochemistry 32:493–506
Kutchan TM (1995) Alkaloid biosynthesis—the basis for metabolic engineering of medicinal plants. Plant Cell 7:1059–1070
Kutchan TM (2005) A role for intra- and intercellular translocation in natural product biosynthesis. Curr Opi Plant Biol 8:292–300
Kutchan TM, Hampp N, Lottspeich F et al (1988) The cDNA clone for strictosidine synthase from Rauvolfia serpentina. FEBS Lett 237:40–44
Kutney JP, Aweryn B, Choi LSL et al (1981) Alkaloid production in Catharanthus roseus cell cultures. IX. Biotransformation studies with 3′,4′-dehydrovinblastine. Heterocycles 16:1169–1171
Kutney JP, Boulet CA, Choi LSL (1988) Alkaloid production in Catharanthus roseus (L.) G. Don cell cultures. XV. Synthesis of bisindole alkaloids by use of immobilized enzyme systems. Heterocycles 27:621–628
Kutney JP, Choi LSL, Kolodziejczyk P et al (1980) Alkaloid production in Catharanthus roseus cell cultures: Isolation and characterization of alkaloids from one cell line. Phytochemistry 19:2589–2595
Laflamme P, St Pierre B, De Luca V (2001) Molecular and biochemical analysis of a Madagascar periwinkle root-specific minovincinine-19-hydroxy-O-acetyltransferase. Plant Physiol 125:189–198
Lange BM, Rujan T, Martin W et al (2000) Isoprenoid biosynthesis: the evolution of two ancient and distinct pathways across genomes. Proc Natl Acad Sci (USA) 97:13172–13177
Lange BM, Wildung MR, McCaskill D et al (1998) A family of transketolases that directs isoprenoid biosynthesis via a mevalonate-independent pathway. Proc Natl Acad Sci (USA) 95:2100–2104
Lee CWT, Shuler ML (2000) The effect of inoculum density and conditioned medium on the production of ajmalicine and catharanthine from immobilized Catharanthus roseus cells. Biotechnol Bioeng 67:61–71
Lichtenthaler HK (1999) The 1-deoxy-d-xylulose-5-phosphate pathway of isoprenoid biosynthesis in plants. Annu Rev Plant Physiol Plant Mol Biol 50:47–65
Lichtenthaler HK (2001) Discovery of the two parallel pathways for isoprenoid biosynthesis in plants. In: Kung S-D, Yang SF (eds) Discoveries in Plant Biology, London, pp 141–161
Lichtenthaler HK, Rohmer M, Schwender J et al (1997) A novel mevalonate-independent pathway for the biosynthesis of carotenoids, phytol and prenyl chain of plastoquinone-9 in green algae and higher plants. In: Williams JP, Khan MU, Lem NW (eds) Physiology, biochemistry and molecular biology of plant lipids. Kluwer Academy Publishers, Dordrecht, pp 177–179
Loyola-Vargas VM, Méndez-Zeel M, Monforte-González M et al (1992) Serpentine accumulation during greening in normal and tumor tissues of Catharanthus roseus. J Plant Physiol 140:213–217
Luijendijk TJC, Stevens LH, Verpoorte R (1998) Purification and characterisation of strictosidine β-D-glucosidase from Catharanthus roseus cell suspension cultures. Plant Physiol Biochem 36:419–425
Madyastha KM, Coscia CJ (1979a) Detergent-solubilized NADPH-cytochrome C (P-450) reductase from the higher plant, Catharanthus roseus. J Biol Chem 254:2419–2427
Madyastha KM, Coscia CJ (1979b) Enzymology of indole alkaloid biosynthesis. Rec Advan Phytochem 13:85–129
Madyastha KM, Guarnaccia R, Baxter C et al (1973) S-Adenosyl-l-methionine: loganic acid methyltransferase. A carboxyl alkylating enzyme from Vinca rosea. J Biol Chem 248:2497–2501
Madyastha KM, Meehan TD, Coscia CJ (1976) Characterization of a cytochrome P-450 dependent monoterpene hydroxylase from the higher plant Vinca rosea. Biochemistry 15:1097–1102
Madyastha KM, Ridgway JE, Dwyer JG et al (1977) Subcellular localization of a cytochrome P-450-dependent monooxygenase in vesicles of the higher plant Catharanthus roseus. J Cell Biol 72:302–313
Magnotta M, Murata J, Chen J et al (2006) Identification of a low vindoline accumulating cultivar of Catharanthus roseus (L.) G. Don by alkaloid and enzymatic profiling. Phytochemistry 67:1758–1764
Mahroug S, Courdavault V, Thiersault M et al (2006) Epidermis is a pivotal site of at least four secondary metabolic pathways in Catharanthus roseus aerial organs. Planta 223:1191–1200
Mathews CK, Van Holde KE, Ahern KG (2000) Biochemistry. Addison Wesley Longman, San Francisco
McCaskill D, Croteau R (1998) Some caveats for bioengineering terpenoid metabolism in plants. Trends Biotechnol 16:349–355
McFarlane J, Madyastha KM, Coscia CJ (1975) Regulation of secondary metabolism in higher plants. Effect of alkaloids on cytochrome P-450 dependent monooxygenase. Biochem Biophys Res Commun 66:1363–1369
McKnight TD, Bergey DR, Burnett RJ et al (1991) Expression of enzymatically active and correctly targeted strictosidine synthase in transgenic tobacco plants. Planta 185:148–152
McKnight TD, Roessner CA, Devagupta R et al (1990) Nucleotide sequence of a cDNA encoding the vacuolar protein strictosidine synthase from Catharanthus roseus. Nucleic Acids Res 18:4939
Meehan TD, Coscia CJ (1973) Hydroxylation of geraniol and nerol by a monooxygenase from Vinca rosea. Biochem Biophys Res Commun 53:1043–1048
Meijer AH (1993) Cytochrome P-450 and secondary metabolism in Catharanthus roseus. Ph. D. Thesis: Faculteit der Godgeleerdheid, Gravenhage, pp 1–151
Meijer AH, De Waal A, Verpoorte R (1993a) Purification of the cytochrome P-450 enzyme geraniol 10-hydroxylase from cell cultures of Catharanthus roseus. J Chromatogr 635:237–249
Meijer AH, Lopes-Cardoso MI, Verpoorte R et al (1993b) Isolation and characterization of a cDNA clone from Catharanthus roseus encoding NADPH: cytochrome P-450 reductase, an enzymes essential for reactions catalysed by cytochrome P-450 mono-oxygenases in plants. Plant J 4:47–60
Meijer AH, Verpoorte R, Hoge JHC (1993c) Regulation of enzymes and genes involved in terpenoid indole alkaloid biosynthesis in Catharanthus roseus. J Plant Res 3:145–164
Menke FL, Parchmann S, Mueller MJ et al (1999) Involvement of the octadecanoid pathway and protein phosphorylation in fungal elicitor-induced expression of terpenoid indole alkaloid biosynthetic genes in Catharanthus roseus. Plant Physiol 119:1289–1296
Mérillon JM, Ouelhazi L, Doireau P et al (1989) Metabolic changes and alkaloid production in habituated and non-habituated cells of Catharanthus roseus grown in hormone-free medium. Comparing hormone-deprived non-habituated cells with habituated cells. J Plant Physiol 134:54–60
Merillon JM, Ramawat KG, Chenieux J-C et al (1986) Hormonal autotrophy and production of indole alkaloids in Catharanthus roseus cell cultures. In: Somers DA (ed) VI International congress plant tissue and cell culture. International Association for Plant Tissue Culture, Minneapolis, p 370
Misawa M, Endo T, Goodbody AE et al (1988) Synthesis of dimeric indole alkaloids by cell free extracts from cell suspension cultures of Catharanthus roseus. Phytochemistry 27:1355–1359
Misawa M, Goodbody AE (1996) Production of antitumor compounds by plant cell cultures. In: DiCosmo F, Misawa M (eds) Plant cell culture secondary metabolism toward industrial application. CRC Press, Boca Raton, FL, pp 123–138
Mizukami H, Nordlov H, Lee S-L et al (1979) Purification and properties of strictosidine synthetase (an enzyme condensing tryptamine and secologanin) from Catharanthus roseus cultured cells. Biochemistry 18:3760–3763
Moreno PRH, Van der Heijden R, Verpoorte R (1995) Cell and tissue cultures of Catharanthus roseus: a literature survey. 2. Updating from 1988 to 1993. Plant Cell Tiss Org Cult 42:1–25
Moreno-Valenzuela OA (1999) Regulación de la vía de síntesis de los alcaloides indólicos en raíces transformadas de Catharanthus roseus. Centro de Investigación Científica de Yucatán, Mérida, Yucatán
Moreno-Valenzuela OA, Galaz-Avalos RM, Minero-García Y et al (1998) Effect of differentiation on the regulation of indole alkaloid production in Catharanthus roseus hairy root. Plant Cell Rep 18:99–104
Moreno-Valenzuela OA, Minero-García Y, Brito-Argáez L et al (2003) Immunocytolocalization of tryptophan decarboxylase in Catharanthus roseus hairy roots. Mol Biotechnol 23:11–18
Moreno-Valenzuela OA, Monforte-González M, Muñoz-Sánchez JA et al (1999) Effect of macerozyme on secondary metabolism plant product production and phospholipase C activity in Catharanthus roseus hairy roots. J Plant Physiol 155:447–452
Morgan JA, Shanks JV (2000) Determination of metabolic rate-limitations by precursor feeding in Catharanthus roseus hairy root cultures. J Biotechnol 79:137–145
Morris P, Scragg AH, Smart NJ et al (1985) Secondary product formation by cell suspension cultures. In: Dixon RA (ed) Plant cell culture. A Practical Approach. IRL Press, Oxford, pp 127–167
Murata J, Bienzle D, Brandle JE et al (2006) Expressed sequence tags from Madagascar periwinkle (Catharanthus roseus). FEBS Lett 580:4501–4507
Murata J, De Luca V (2005) Localization of tabersonine 16-hydroxylase and 16-OH tabersonine-16-O-methyltransferase to leaf epidermal cells defines them as a major site of precursor biosynthesis in the vindoline pathway in Catharanthus roseus. Plant J 44:581–594
Noé W, Mollenschott C, Berlin J (1984) Tryptophan decarboxylase from Catharanthus roseus cell suspension cultures: purification, molecular and kinetic data of the homogenous protein. Plant Mol Biol 3:281–288
O’Keefe BR, Mahady GB, Gills JJ et al (1997) Stable vindoline production in transformed cell cultures of Catharanthus roseus. J Nat Prod 60:261–264
Oresic M, Rischer H, Oksman-Caldentey K (2006) Metabolomics of plant secondary compounds: profiling of Catharanthus cell cultures. In: Saito K, Dixon R, Willmitzer L (eds) Plant metabolomics. Springer, Berlin, pp 277–289
Ouwerkerk PBF, Hallard D, Verpoorte R et al (1999a) Identification of UV-B light-responsive regions in the promoter of the tryptophan decarboxylase gene from Catharanthus roseus. Plant Mol Biol 41:491–503
Ouwerkerk PBF, Trimborn TO, Hilliou F et al (1999b) Nuclear factors GT-1 and 3AF1 interact with multiple sequences within the promoter of the Tdc gene from Madagascar periwinkle: GT-1 is involved in UV light-induced expression. Mol Gen Genet 261:610–622
Papon N, Bremer J, Vansiri A et al (2005) Cytokinin and ethylene control indole alkaloid production at the level of the MEP/terpenoid pathway in Catharanthus roseus suspension cells. Planta Med 71:572–574
Pasquali G, Erven AS, Ouwerkerk PB et al (1999) The promoter of the strictosidine synthase gene from periwinkle confers elicitor-inducible expression in transgenic tobacco and binds nuclear factors GT-1 and GBF. Plant Mol Biol 39:1299–1310
Pasquali G, Goddijn OJM, De Waal A et al (1992) Coordinated regulation of two indole alkaloid biosynthetic genes from Catharanthus roseus by auxin and elicitors. Plant Mol Biol 18:1121–1131
Pauw B, Hilliout FAO, Martin VS et al (2004) Zinc finger proteins act as transcriptional repressors of alkaloid biosynthesis genes in Catharanthus roseus. J Biol Chem 279:52940–52948
Pfitzner A, Polz L, Stöckigt J (1986) Properties of vinorine synthase—the Rauwolfia enzyme involved in the formation of the ajmaline skeleton. Z Naturforsch [C] 41:103–114
Pfitzner A, Stöckigt J (1982) Partial purification and characterization of geissoschizine dehydrogenase from suspension cultures of Catharanthus roseus. Phytochemistry 21:1585–1588
Pfitzner U, Zenk MH (1989) Homogeneous strictosidine synthase isoenzymes from cell suspension cultures of Catharanthus roseus. Planta Med 55:525–530
Power R, Kurz WGW, De Luca V (1990) Purification and characterization of acetylcoenzyme A: deacetylvindoline 4-O-acetyltransferase from Catharanthus roseus. Arch Biochem Biophys 279:370–376
Reed BC, Rilling HC (1975) Crystallization and partial characterization of prenyltransferase from avian liver. Biochemistry 14:50–54
Rischer H, Oresic M, Seppanen-Laakso T et al (2006) Gene-to-metabolite networks for terpenoid indole alkaloid biosynthesis in Catharanthus roseus cells. Proc Natl Acad Sci (USA) 103:5614–5619
Roberts MF (1998) Enzymology of alkaloids biosynthesis. In: Roberts MF, Wink M (eds) Alkaloids. Biochemistry, ecology, and medicinal applications. Plenum Press, New York, pp 109–146
Roberts MF, Wink M (1998) Introduction. In: Roberts MF, Wink M (eds) Alkaloids. Biochemistry, ecology, and medicinal applications. Plenum Press, New York, pp 1–7
Robinson T (1974) Metabolism and function of alkaloids in plants. Science 184:430–435
Robinson T (1981) The biochemistry of alkaloids. Springer-Verlag
Rodriguez S, Compagnon V, Crouch NP et al (2003) Jasmonate-induced epoxidation of tabersonine by cytochrome P-450 in hairy root cultures of Catharanthus roseus. Phytochemistry 64:401–409
Rohmer M, Knani M, Simonin P et al (1993) Isoprenoid biosynthesis in bacteria: A novel pathway for the early steps leading to isopentenyl diphosphate. Biochem J 295:517–524
Rudney H (1957) The biosynthesis of β-hydroxy-β-methylglutaric acid. J Biol Chem 227:363–377
Sáenz-Carbonell L, Santamaría JM, Villanueva MA et al (1993) Changes in the alkaloid content of plants of Catharanthus roseus L. (Don) as a result of water stress and treatment with abscisic acid. J Plant Physiol 142:244–247
Sánchez-Iturbe P, Galaz-Avalos RM, Loyola-Vargas VM (2005) Determination and partial purification of a monoterpene cyclase from Catharanthus roseus hairy roots. Phyton 55–69
Schiel O, Witte L, Berlin J (1987) Geraniol-10-hydroxylase activity and its relation to monoterpene indole alkaloid accumulation in cell suspension cultures of Catharanthus roseus. Z Naturforsch [C] 42c:1075–1081
Schmeller T, Wink M (1998) Utilization of alkaloids in modern medicine. In: Roberts MF, Wink M (eds) Alkaloids. Biochemistry, ecology, and medicinal applications. Plenum Press, New York, pp 435–459
Schröder G, Unterbusch E, Kaltenbach M et al (1999) Light-induced cytochrome P450-dependent enzyme in indole alkaloid biosynthesis: tabersonine 16-hydroxylase. FEBS Lett 458:97–102
Schwender J, Seemann M, Lichtenthaler HK et al (1996) Biosynthesis of isoprenoids (carotenoids, sterols, prenyl side-chains of chlorophylls and plastoquinone) via a novel pyruvate/glyceraldehyde 3-phosphate non-mevalonate pathway in the green alga Scenedesmus obliquus. Biochem J 316:73–80
Scott AI (1979) Biosynthesis of indole alkaloids. Acc Chem Res 3:151
Shanks JV, Bhadra R, Morgan J et al (1998) Quantification of metabolites in the indole alkaloid pathways of Catharanthus roseus: implications for metabolic engineering. Biotechnol Bioeng 58:333–338
Shukla AK, Shasany AK, Gupta MM et al (2006) Transcriptome analysis in Catharanthus roseus leaves and roots for comparative terpenoid indole alkaloid profiles. J Exp Bot 57:3921–3932
Sibéril Y, Benhamron S, Memelink J et al (2001) Catharanthus roseus G-box binding factors 1 and 2 act as repressors of strictosidine synthase gene expression in cell cultures. Plant Mol Biol 45:477–488
Singh SN, Vats P, Suri S et al (2001) Effect of an antidiabetic extract of Catharanthus roseus on enzymatic activities in streptozotocin induced diabetic rats. J Ethnopharmacol 76:269–277
Smith JI, Amouzou E, Yamaguchi A et al (2003) Peroxidase from bioreactor cultivated Catharanthus roseus cell cultures mediates biosynthesis of α-3′,4′-anhydrovinblastine. Biotechnol Appl Biochem 10:568–575
Sottomayor M, Barceló AR (2003) Peroxidase from Catharanthus roseus (L.) G. Don and the biosynthesis of α-3′,4′-anhidrovinblastine: a specific role for multifunctional enzyme. Protoplasma 222:97–105
Sottomayor M, De Pinto MC, Salema R et al (1996) The vacuolar localization of a basic peroxidase isoenzyme responsible for the synthesis of α-3′,4′-anhydrovinblastine in Catharanthus roseus (L) G. Don leaves. Plant Cell Environ 19:761–767
Sottomayor M, DiCosmo F, Barceló AR (1997) On the fate of catharanthine and vindoline during the peroxidase-mediated enzymatic synthesis of α-3′,4′-anhydrovinblastine. Enzyme Microb Technol 21:543–549
Sottomayor M, Lopes-Cardoso I, Pereira LG et al (2004) Peroxidase and the biosynthesis of terpenoid indole alkaloids in the medicinal plant Catharanthus roseus (L.) G. Don. Phytochem Rev 3:159–171
Sottomayor M, López-Serrano M, DiCosmo F et al (1998) Purification and characterization of α-3′,4′-anhydrovinblastine synthase (peroxidase-like) from Catharanthus roseus (L) G. Don. FEBS Lett 428:299–303
St-Pierre B, De Luca V (1995) A cytochrome P-450 monooxygenase catalyzes the first step in the conversion of tabersonine to vindoline in Catharanthus roseus. Plant Physiol 109:131–139
St-Pierre B, Laflamme P, Alarco AM et al (1998) The terminal O-acetyltransferase involved in vindoline biosynthesis defines a new class of proteins responsible for coenzyme A-dependent acyl transfer. Plant J 14:703–713
St-Pierre B, Vázquez-Flota FA, De Luca V (1999) Multicellular compartmentation of Catharanthus roseus alkaloid biosynthesis predicts intercellular translocation of a pathway intermediate. Plant Cell 11:887–900
Stern JR, Drummond GI, Coon MJ et al (1960) Enzymes of ketone body metabolism. I. Purification of an acetoacetate-synthesizing enzyme from ox liver. J Biol Chem 235:313–317
Stevens LH, Blom TJM, Verpoorte R (1993) Subcellular localization of tryptophan decarboxylase, strictosidine synthase and strictosidine glucosidase in suspension cultured cells of Catharanthus roseus and Tabernaemontana divaricata. Plant Cell Rep 12:573–576
Stevens LH, Schripsema J, Pennings EJM et al (1992) Activities of enzymes involved in indole alkaloid biosynthesis in suspension cultures of Catharanthus, Cinchona and Tabernaemontana species. Plant Physiol Biochem 30:675–681
Stöckigt J, Husson H-P, Kan-fan Ch et al (1977) Cathenamine, a central intermediate in the cell free biosynthesis of ajmalicine and related indole alkaloids. J Chem Soc Chem Commun 164–166
Stöckigt J, Treimer JF, Zenk MH (1976) Synthesis of ajmalicine and related indole alkaloids by cell free extracts of Catharanthus roseus cell suspension cultures. FEBS Lett 70:267–270
Stöckigt J, Zenk MH (1977a) Isovincoside (strictosidine), the key intermediate in the enzymatic formation of indole alkaloids. FEBS Lett 79:233–237
Stöckigt J, Zenk MH (1977b) Strictosidine (isovincoside): the key intermediate in the biosynthesis of monoterpenoid indole alkaloids. J Chem Soc Chem Commun 646–648
Sundberg RJ, Smith SQ (2002) The iboga alkaloids and their role as precursors of anti-neoplastic bisindole Catharanthus alkaloids. In: Cordell GA (ed) The alkaloids. Academic Press, San Diego, pp 281–376
Svoboda GH, Blake DA (1975) The phytochemistry and pharmacology of Catharanthus roseus (L.) G. Don. In: Taylor WI, Farnsworth NR (eds) The Catharanthus alkaloids. Marcel Dekker, Inc., New York, pp 45–83
Tchen TT (1958) Mevalonic kinase: purification and properties. J Biol Chem 233:1100–1103
Toivonen L (1992) Utilization of Catharanthus roseus hairy root and cell suspension cultures in plant biotechnology. Ph.D. Thesis, Helsinki University of Technology, Helsinki, pp 1–93
Treimer JF, Zenk MH (1979a) Purification and properties of strictosidine synthase, the key enzyme in indole alkaloid formation. Eur J Biochem 101:225–233
Treimer JF, Zenk MH (1979b) Strictosidine synthase from cell cultures of Apocynaceae plants. FEBS Lett 97:159–162
Uesato S, Ikeda H, Fujita T et al (1987) Elucidation of iridodial formation mechanism partial purification and characterization of the novel monoterpene cyclase from Rauwolfia serpentina cell suspension cultures. Tetrahedron Lett 28:4431–4434
Uesato S, Ogawa Y, Inouya H et al (1986) Synthesis of iridodial by cell free extracts from Rauwolfia serpentina cell suspension cultures. Tetrahedron Lett 13:2893–2896
Van der Fits L, Zhang H, Menke FLH et al (2000) A Catharanthus roseus BPF-1 homologue interacts with an elicitor-responsive region of the secondary metabolite biosynthetic gene Str and is induced by elicitor via a JA-independent signal transduction pathway. Plant Mol Biol 44:675–685
Van der Heijden R, De Boer-Hlupá V, Verpoorte R et al (1994) Enzymes involved in the metabolism of 3-hydroxy-3-methylglutaryl-coenzyme A in Catharanthus roseus. Plant Cell Tiss Org Cult 38:345–349
Van der Heijden R, Jacobs DI, Snoeijer W et al (2004) The Catharanthus alkaloids: Pharmacognosy and biotechnology. Curr Med Chem 11:1241–1253
Van der Heijden R, Verpoorte R, Ten Hoopen HJG (1989) Cell and tissue cultures of Catharanthus roseus (L.) G. Don: a literature survey. Plant Cell Tiss Org Cult 18:231–280
Vázquez-Flota F, De Carolis E, Alarco AM et al (1997) Molecular cloning and characterization of desacetoxyvindoline-4-hydroxylase, a 2-oxoglutarate dependent dioxygenase involved in the biosynthesis of vindoline in Catharanthus roseus (L) G. Don. Plant Mol Biol 34:935–948
Vázquez-Flota F, De Luca V (1998a) Developmental and light regulation of desacetoxyvindoline 4-hydroxylase in Catharanthus roseus (L.) G. Don. Evidence of a multilevel regulatory mechanism. Plant Physiol 117:1351–1361
Vázquez-Flota F, De Luca V, Carrillo-Pech MR et al (2002) Vindoline biosynthesis is transcriptionally blocked in Catharanthus roseus cell suspension cultures. Mol Biotechnol 22:1–8
Vázquez-Flota F, Loyola-Vargas VM (1994) A Catharanthus roseus salt tolerant line. II. Alkaloid production. J Plant Physiol 144:613–616
Vázquez-Flota F, Monforte-González M, Méndez-Zeel M et al (2000a) Effects of nitrogen source on alkaloid metabolism in callus cultures of Catharanthus roseus (L.) G Don. Phyton 66:155–164
Vázquez-Flota F, Moreno-Valenzuela OA, Miranda-Ham ML et al (1994) Catharanthine and ajmalicine synthesis in Catharanthus roseus hairy root cultures. Medium optimization and elicitation. Plant Cell Tiss Org Cult 38:273–279
Vázquez-Flota FA, De Luca V (1998b) Jasmonate modulates development- and light-regulated alkaloid biosynthesis in Catharanthus roseus. Phytochemistry 49:395–402
Vázquez-Flota FA, St Pierre B, De Luca V (2000b) Light activation of vindoline biosynthesis does not require cytomorphogenesis in Catharanthus roseus seedlings. Phytochemistry 55:531–536
Verpoorte R, Van der Heijden R, Memelink J (2000) Engineering the plant cell factory for secondary metabolite production. Transg Res 9:323–343
Verpoorte R, Van der Heijden R, Moreno PRH (1997) Biosynthesis of terpenoid indole alkaloids in Catharanthus roseus cells. In: Cordell GA (ed) The alkaliods. Academic Press, San Diego, pp 221–299
Verpoorte R, Van der Heijden R, Schripsema J et al (1993) Plant cell biotechnology for the production of alkaloids: present status and prospects. J Nat Prod 56:186–207
Vetter H-P, Mangold U, Schröder G et al (1992) Molecular analysis and heterologous expression of an inducible cytochrome P-450 protein from periwinkle (Catharanthus roseus L.). Plant Physiol 100:998–1007
von Schumann G, Gao S, Stöckigt J (2002) Vomilenina reductasa a novel enzyme catalyzing a crucial step in the biosynthesis of the therapeutically applied antiarrhythmic alkaloid ajmaline. Bioorg Med Chem 10:1913–1918
Waterman PG (1998) Chemical taxonomy of alkaloids. In: Roberts MF, Wink M (eds) Alkaloids. Biochemistry, ecology, and medicinal applications. Plenum Press, New York, pp 87–107
Westekemper P, Wieczorek U, Gueritte F et al (1980) Radioimmunoassay for the determination of the indole alkaloid vindoline in Catharanthus. Planta Med 39:24–37
Whitmer S, Canel C, Hallard D et al (1998) Influence of precursor availability on alkaloid accumulation by transgenic cell line of Catharanthus roseus. Plant Physiol 116:853–857
Whitmer S, Van der Heijden R, Verpoorte R (2002a) Effect of precursor feeding on alkaloid accumulation by a strictosidine synthase over-expressing transgenic cell line S1 of Catharanthus roseus. Plant Cell Tiss Org Cult 69:85–93
Whitmer S, Van der Heijden R, Verpoorte R (2002b) Effect of precursor feeding on alkaloid accumulation by a tryptophan decarboxylase over-expressing transgenic cell line T22 of Catharanthus roseus. J Biotechnol 96:193–203
Yamamoto H, Katano N, Ooi A et al (2000) Secologanin synthase which catalyzes the oxidative cleavage of loganin into secologanin is a cytochrome P450. Phytochemistry 53:7–12
Yamamoto H, Katano N, Ooi Y et al (1999) Transformation of loganin and 7-deoxyloganin into secologanin by Lonicera japonica cell suspension cultures. Phytochemistry 50:417–422
Yazaki K (2005) Transporters of secondary metabolites. Curr Opi Plant Biol 8:301–307
Yazaki K (2006) ABC transporters involved in the transport of plant secondary metabolites. FEBS Lett 580:1183–1191
Zenk MH, Deus B (1982) Natural product synthesis by plant cell cultures. In: Fujiwara A (ed) Proceedings 5th international congress of plant tissue and cell culture. The Japanese Association for Plant Tissue Culture, Japan, pp 391–394
Zhao J, Hu Q, Guo YQ et al (2001a) Effects of stress factors, bioregulators, and synthetic precursors on indole alkaloid production in compact callus clusters cultures of Catharanthus roseus. Appl Microbiol Biotechnol 55:693–698
Zhao J, Zhu WH, Hu Q (2001b) Effects of light and plant growth regulators on the biosynthesis of vindoline and other indole alkaloids in Catharanthus roseus callus cultures. Plant Growth Regul 33:43–49
Zhao J, Zhu WH, Hu Q (2001c) Enhanced catharanthine production in Catharanthus roseus cell cultures by combined elicitor treatment in shake flasks and bioreactors. Enzyme Microb Technol 28:673–681
Zhao J, Zhu WH, Hu Q (2001d) Selection of fungal elicitors to increase indole alkaloid accumulation in Catharanthus roseus suspension cell culture. Enzyme Microb Technol 28:666–672
Acknowledgements
The authors thank Emily Wortman-Wunder, Clelia De la Peña and Elan Alford for English correction of the manuscript as well as to the anonymous reviewers. For the nomenclature of the enzymes, the rules of the International Union of Biochemistry and Molecular Biology (http://www.chem.qmw.ac.uk/iubmb/enzyme/) were used.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Loyola-Vargas, V.M., Galaz-Ávalos, R.M. & Kú-Cauich, R. Catharanthus biosynthetic enzymes: the road ahead. Phytochem Rev 6, 307–339 (2007). https://doi.org/10.1007/s11101-007-9064-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11101-007-9064-2