
Abstract
Two centuries after the discovery of the first alkaloids, many enzymes involved in plant alkaloid biosynthesis have been identified. Nevertheless, the biosynthetic pathways for most of the plant alkaloids still remain incompletely characterised and understanding the regulatory mechanisms controlling the onset and flux of alkaloid biosynthesis is virtually inexistent. This information is however crucial to allow modelling of metabolic networks and predictive metabolic engineering. In the postgenomics era, new functional genomics tools, enabling comprehensive investigations of biological systems, are continuously emerging and are now gradually being implemented in the field of plant secondary metabolism as well. Here we discuss the advances these promising new technologies have already brought and may still bring with regard to the dissection of plant alkaloid biosynthesis. Encouraging results were obtained in alkaloid producing species such as Papaver somniferum, Catharanthus roseus and Nicotiana tabacum. Therefore we anticipate that functional genomics and the knowledge it brings along, will eventually allow a better exploitation of the plant biosynthetic machinery.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction: the long road to unwinding alkaloid biosynthesis
About 20% of flowering plant species produce alkaloids and display an enormous structural diversity therein, despite they are practically all derived from a restricted number of universal amino-acid precursors. Since the discovery of morphine, two centuries ago, more than 12,000 alkaloids have been isolated (Facchini 2001). Biosynthesis of morphine but also of other well-known alkaloids such as nicotine or vinblastine has been subjected to extensive research efforts, and consequently many of the enzymes involved in the biosynthesis of these three alkaloids have been identified. Notwithstanding, for each of them the biosynthetic pathway still remains incompletely characterised.
Elucidation of alkaloid biosynthetic pathways has been facing many difficulties, mainly because of the low concentrations of alkaloid biosynthetic enzymes in plant tissues producing them and the limited availability of radiolabelled substrates or cofactors for the development of sensitive enzyme assays. Classically, genes encoding alkaloid biosynthetic enzymes were isolated through knowledge of peptide sequences determined from purified enzymes. Obviously, due to the low concentrations many alkaloid enzymes may be recalcitrant to conventional purification strategies. In an alternative strategy, biochemical purification is avoided and candidate clones are isolated by homology-based screening, often in combination with expression pattern analysis. Thereafter, the isolated genes are expressed in heterologous systems such as bacteria or yeast, and the recombinant proteins tested for the expected enzyme activities. An important breakthrough was achieved by the generation of plant cell suspension cultures with increased rates of alkaloid biosynthesis, which moreover could often receive additional stimuli by the addition of phytohormones or elicitors of biotic or abiotic nature. These research approaches allowed the discovery of tens of new enzymes catalyzing steps in the biosynthesis of diverse alkaloids, including those of the isoquinoline, terpenoid indole, tropane and purine classes (for reviews see Croteau et al. 2000; Facchini 2001; De Luca and Laflamme 2001; Hashimoto and Yamada 2003; Facchini et al. 2004; Facchini and St-Pierre 2005). For instance it led to the full characterization of the berberine biosynthetic pathway, the first alkaloid biosynthetic pathway to be completed. Elucidation of many other alkaloid biosynthetic pathways is still ongoing, with some of them, such as of morphine, approaching completion.
Functional genomics, on the advent of a new breakthrough
For the majority of alkaloids or other secondary metabolites, thorough knowledge of the whole biosynthetic pathway or detailed understanding of the regulatory mechanisms controlling the onset and flux of alkaloid biosynthesis is still lacking. The next important breakthrough in alkaloid biosynthesis research may be prompted by the development of powerful functional genomics tools, allowing comprehensive investigations of biological systems at an accelerated pace (Oksman-Caldentey and Inzé 2004). Indeed, using genomics, we can now identify all genes in a plant; and using transcriptomic and proteomic techniques we can resolve which genes are (in)activated in a particular plant cell or organ under a particular growth or stress condition. Connecting genomics, transcriptomics or proteomics with metabolomics by a systems biology approach may then allow exploring the biochemical machinery of plants, ultimately leading to the discovery of new pathways, the modelling of metabolic networks and predictive metabolic engineering. A crucial prerequisite is of course the availability of gene sequences, either of a complete genome, which is the case for only a few model plant species, or of expressed sequence tag (EST) databases. Given that currently for most medicinal plants extensive sequence resources are not available, only a limited number of such comprehensive studies of complex metabolic systems have been reported so far. This number is however expected to increase rapidly in the coming years.
One of the first and to-date most extensively applied functional genomics approaches to isolate genes implicated in plant secondary metabolism involved random sequencing of cell-specific cDNA libraries. In this approach computational and expression-based analyses are combined, based on the rationale that cDNAs corresponding to genes involved in the biosynthesis of a particular metabolite will be abundant in a cDNA library made from the tissue in which the particular metabolite is produced and thus the ‘biosynthetic’ genes are expressed. One of the first studies reporting the use of this method showed how random sequencing of a peppermint oil gland secretory cell cDNA library could be used to identify the pathway responsible for menthol biosynthesis (Lange et al. 2000). Other successful research exploiting this technological approach, include reports on the isolation of genes involved in taxol biosynthesis in Taxus cuspidata cells (Jennewein et al. 2004) and artemisinin biosynthesis in Artemisia annua trichomes (Teoh et al. 2006).
In general, the same rationale is followed when combining any transcriptome profiling method with metabolic profiling (Yamazaki and Saito 2002; Oksman-Caldentey and Saito 2005; Fridman and Pichersky 2005). For instance, taxol biosynthesis was further dissected using differential display analysis of gene expression in jasmonate-treated Taxus cuspidata cells (Schoendorf et al. 2001). Differential display was also successfully used to isolate anthocyanin biosynthetic genes by comparing red and green forms of Perilla frutescens (Saito et al. 1999; Yamazaki et al. 1999, 2003). Genes involved in the formation of volatile compounds in strawberry (Aharoni et al. 2000) and rose (Guterman et al. 2002) have been identified by using DNA microarray analysis in combination with targeted analysis of volatile metabolites. The same rationale holds as well when proteome analysis is combined with metabolite profiling. Proteomic analysis of different fractions of poppy latex allowed for instance the isolation of two novel poppy alkaloid biosynthetic genes (Ounaroon et al. 2003).
Pioneering work on a functional genomics approach—in an era where the term ‘functional genomics’ still had to be invented—to study alkaloid pathways was published in 1994 (Hibi et al. 1994). This study described the very first attempt to isolate genes involved in alkaloid—nicotine from common tobacco—biosynthesis by a differential screening approach. Since then integrated functional genomics approaches aimed at elucidation of alkaloid biosynthetic pathways have, to our knowledge, been undertaken for three plant species; the opium poppy (Papaver somniferum), common tobacco (Nicotiana tabacum) and the Madagascar periwinkle (Catharanthus roseus). In this review, we discuss the state-of-the-art of knowledge of alkaloid biogenesis, and in particular the contributions made therein by functional genomics-based research, for these three plant species.
Benzylisoquiniline alkaloids: Papaver somniferum
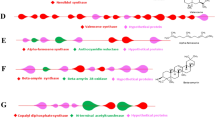
Benzylisoquinoline alkaloids (BIAs) are a large and diverse alkaloid group with over 2500 defined structures (Facchini 2001). BIAs are derived from l-tyrosine, and biosynthesis begins with the condensation of dopamine and 4-hydroxyphenylacetaldehyde by norcoclaurine synthase (Fig. 1). (S)-Norcoclaurine is then, through a number of enzymatic reactions, converted to the central branch-point intermediate (S)-reticuline. At this point, the pathway can branch to the production of morphinans (e.g. thebaine, codeine, morphine) or to the production of sanguinarines (Fig. 1). Papaver somniferum, the opium poppy, synthesises more than 80 alkaloids, and many poppy biosynthetic enzymes, active in multiple cell types, and the corresponding genes have been identified (Facchini 2001; Kutchan 2005).

Structure and biosynthesis of benzylisoquinoline alkaloids in Papaver somniferum. Metabolites are given as full names in lowercase and enzymes as abbreviations in capitals. Full and dashed arrows mark single and multiple conversion steps between intermediates, respectively. Enzymes listed: BBE (berberine bridge enzyme), CFS ((S)-chelanthifoline synthase), CNMT (S-adenosyl-l-methionine:norcoclaurine 6-O-methyltransferase), COR (codeinone reductase), CYP719A1 ((S)-canadine synthase), CYP80B1 ((S)-N-methylcoclaurine 3′-hydroxylase), DBOX (dihydrobenzophenanthridine oxidase), DRR (1,2-dehydroreticuline reductase), DRS (dehydroreticuline synthase), MSH ((S)-cis-N-methylstylopine 14-hydroxylase), NCS (norcoclaurine synthase), 4′OMT ((R,S)-3′-hydroxy-N-methylcoclaurine 4′-O-methyltransferase), 6OMT (norcoclaurine 6-O-methyltransferase), 7OMT ((R,S)-reticuline 7-O-methyltransferase), P6H (protopine-6-hydroxylase), SAT (salutaridinol 7-O-acetyltransferase), SOMT (scoulerine N-methyltransferase), SOR (salutaridine:NADPH 7-oxidoreductase), SPS ((S)-stylopine synthase), STOX ((S)-tetrahydroprotoberberine oxidase), STS (salutaridine synthase), TNMT (tetrahydroprotoberberine-cis-N-methyltransferase), TYDC (tyrosine/dopa decarboxylase)
The first report on a functional genomics approach to study poppy alkaloid pathways described a two-dimensional SDS-PAGE based proteomics approach (Decker et al. 2000). In this study, poppy latex was separated into different fractions: the cytosolic serum and the vesicle sediment, the latter further subsectioned by density centrifugation. For each fraction the soluble proteins were separated by two-dimensional SDS-PAGE and their sequences analysed by internal peptide microsequencing. Codeinone reductase, a representative of the morphinan-specific branch (Fig. 1), could be detected within the cytosolic serum fraction. Furthermore, 75 serum-specific protein spots were analysed and for 69 of them, internal peptide microsequencing, followed by homology-based database searching, allowed assigning a putative function. Similarly, for 23 vesicle-specific protein spots a putative function could be assigned. In an extension of this work, proteomic analysis of the latex allowed isolating two novel alkaloid biosynthetic genes: (R,S)-reticuline 7-O-methyltransferases and norcoclaurine 6-O-methyltransferase (Ounaroon et al. 2003; see also Fig. 1). By microsequencing of protein spots in the size range expected for plant methyltransferase monomers, the authors were capable of isolating gel spots whose partial amino acid sequences were homologous to those of plant O-methyltransferases. Cloning of the corresponding genes and functional analysis of the recombinant proteins in a heterologous system established that the isolated genes indeed encoded O-methyltransferases and were involved in poppy alkaloid biosynthesis.
Very recently, two sequencing projects from P. somniferum seedlings were initiated (Ziegler et al. 2005; Millgate et al. 2004). In the first, an AFLP-like sequencing approach was employed to obtain 849 ‘UniGenes’ that were printed on macroarrays (Ziegler et al. 2005). This gene resource was then employed in a comparative macroarray-based expression analysis between morphine-containing P. somniferum plants and eight other, morphine-free, Papaver species and led to the identification of a novel O-methyltransferase involved in benzylisoquinoline biosynthesis, i.e. S-adenosyl-l-methionine:(R,S)-3′-hydroxy-N-methylcoclaurine-4′-O-methyltransferase (see Fig. 1). The second P. somniferum gene resource consists of a 17,000-gene poppy microarray, printed with ESTs derived by random sequencing from a P. somniferum shoot cDNA library (Millgate et al. 2004). This microarray was used for global expression analysis of the poppy top1 mutant, which does not complete morphinan biosynthesis into morphine and codeine but instead accumulates the precursors thebaine and oripavine (Millgate et al. 2004). This expression analysis allowed the identification of 10 genes, significantly differentially underexpressed in top1, seven of which were highly homologous to known genes of other species. None of these genes has however been proved to be responsible for the top1 phenotype and their potential role in the morphine biosynthesis pathway is not yet understood. Nevertheless, these kinds of resources clearly open new avenues for elucidating the poppy alkaloid metabolic network and, eventually, modifying it.
Interestingly, a virus-induced gene silencing (VIGS) approach for P. somniferum has been developed recently (Hileman et al. 2005). VIGS was originally established as a Solanaceae-specific approach for determining the biological function of gene products and takes advantage of the fact that plants induce homology-dependent defence mechanisms in response to virus attacks. VIGS results in the degradation of endogenous RNA with extensive sequence complementarity to for instance exogenous transgenes introduced into virus-based vectors (Ruiz et al. 1998). The extension of the VIGS method to a non-Solanaceae species, although still in an early developmental phase, may be a promising tool to perform large-scale functional screenings of gene tags from medicinal plants, for instance of the ESTs generated in the P. somniferum sequencing projects mentioned above.
A recent study (Allen et al. 2004) confronted us with our current limited understanding of (poppy) alkaloid biosynthesis. Allen and coworkers used RNA interference to successfully block codeinone reductase expression (see Fig. 1), and a substantial reduction of morphine content was indeed observed in transgenic poppies. Surprisingly however, instead of an increased accumulation of immediate morphine-type precursors normally present in the latex, the transgenic poppy latex accumulated rare alkaloids, including the upstream precursor reticuline, located seven enzymatic steps before the codeinone reductase-mediated reaction in the biosynthesis pathway. The reasons for these unexpected results and the drastic switch in the alkaloid pattern were not clear. These findings clearly underscored the argument that knowledge of gene sequences and biosynthetic enzymes alone is not sufficient to design a rational approach towards alkaloid pathway modification and that in this regard understanding of the entire metabolic and cellular system is required.
Systems biology integrates gene expression sensu latu and therefore goes beyond the transcriptome and proteome. For instance, when investigating metabolic networks, systems-level data on metabolite accumulation (metabolomics), metabolite transport and metabolic flux should be included. But systems biology also may incorporate data from e.g. the promoterome, the genome-wide study of promoter activities or the localisome, the genome-wide analysis of protein localisation in intracellular structures or different organs or tissues. Obviously, due to the present lack of extensive full-length gene collections and efficient plant transformation protocols, for alkaloid producing medicinal plants this cannot yet be performed on a genome-wide scale. Notwithstanding, in the case of morphine biosynthesis in the opium poppy, the road is being paved to achieve this tantalising goal. In situ hybridisation of known alkaloid transcripts in poppy reflects the promoterome and immunocytochemical localisation of several known alkaloid biosynthetic enzymes confirmed the existence of an extensive compartmentalisation of morphinan biosynthesis (Bird et al. 2003; Weid et al. 2004). It awaits exploitation of high-throughput poppy transformation protocols to extend this kind of research to a genome-wide scale, by which tagged genes can be employed to verify localisation (e.g. by means of GFP tagging) or protein–protein interactions (e.g. by yeast two-hybrid or tap tag technology). Correspondingly, successful protocols for Agrobacterium tumefaciens-mediated transformation of opium poppy have been developed (Park and Facchini 2000; Chitty et al. 2003).
Nicotine: Nicotiana tabacum
Secondary metabolism in tobacco is quite well studied and an excellent review was published by Nugroho and Verpoorte (2002). More than 2500 compounds have been identified in tobacco, with the most prominent group formed by the alkaloids, including nicotine but also other compounds such as nornicotine, anabasine, anatabine and anatalline. The biosynthetic origin of nicotine begins with the plant polyamine putrescine, which can be formed directly by decarboxylation of ornithine or indirectly from arginine in a reaction initiated also by a decarboxylase (Fig. 2). Putrescine in tobacco can serve as a precursor in the biosynthesis of higher polyamines or can be converted to N-methylputrescine by the action of putrescine N-methyltransferase (PMT), the first committed step in nicotine biosynthesis (Hashimoto and Yamada 1994). In other Solanaceae species, but not tobacco, N-methylputrescine can also be further converted to tropane alkaloids, such as hyoscyamine and scopolamine, or calystegins.

Structure and biosynthesis of pyridine alkaloids in Nicotiana tabacum. Metabolites are given as full names in lowercase and enzymes as abbreviations in capitals. Enzymes listed: ADC (arginine decarboxylase), AIH (agmatine deiminase), AP (aspartate oxidase), AS (arginase), CYP82E4 (nicotine N-demethylase), LDC (lysine decarboxylase), MPO (methylputrescine oxidase), NCPAH (N-carbamoylputrescine amidohydrolase), ODC (ornithine decarboxylase), PMT (putrescine N-methyltransferase), QPRTase (quinolinate phosphoribosyltransferase)
As mentioned above, the very first report on a ‘functional genomics’ approach to study alkaloid biogenesis in tobacco was published in 1994 by Hibi and coworkers. They exploited the availability of N. tabacum cultivars with low nicotine contents, the Burley nic1 and nic2 mutants (Legg and Collins 1971). The Nic1 and Nic2 loci (also referred to as the A and B loci) are thought to be regulatory genes because the mutant alleles decrease nicotine accumulation and nicotine biosynthesis enzyme levels. Differential screening by subtraction hybridisation of cDNA libraries of wild-type and mutant cultured roots allowed isolation of two genes, A411 and A622, encoding PMT and a protein with homology to isoflavone reductase, respectively (Hibi et al. 1994). It has been speculated that A622 may encode the enzyme catalysing the condensation between the 1-methyl-Δ1-pyrrolinium cation and nicotinic acid to nicotine (see Fig. 1) but conclusive evidence to sustain this hypothesis has not been obtained yet. Similarly, the exact identity of the regulatory Nic1 and Nic2 genes still remains elusive, despite the attention they received.
Following this pioneering report, further genome-wide expression analysis related with tobacco alkaloid biosynthesis had to await the true breakthrough of functional genomics. The first follow-up to be reported was a characterisation of cDNAs differentially expressed in tobacco roots during the early stages of alkaloid biosynthesis by subtractive hybridisation screening (Wang et al. 2000). Sixty differentially expressed genes were isolated, amongst which many encoding known nicotine biosynthetic enzymes, but no novel gene products (directly) involved in alkaloid biosynthesis were identified within this set. In contrast, we launched a successful gene discovery program based on an experimental approach, in which alkaloid targeted metabolite profiling was combined with cDNA-amplified fragment length polymorphism-based transcript profiling of tobacco BY-2 cells treated with methyl jasmonate, an efficient elicitor of nicotine biosynthesis in tobacco (Goossens et al. 2003; De Sutter et al. 2005). The profiling data suggested an extensive jasmonate-mediated genetic reprogramming of metabolism; out of 20,000 visualized transcript tags a transcriptome of 459 jasmonate-modulated ESTs could be composed that was compared with the observed jasmonate-induced shifts in biosynthesis of tobacco metabolites (Goossens et al. 2003). The generated gene inventory revealed the presence of all, except one, of the genes known to be involved in the biosynthesis of nicotine alkaloids in Nicotiana species. Most of these were co-induced in time by methyl jasmonate in the elicited tobacco BY-2 cells and clustered together with novel genes or genes encoding proteins with still unknown functions, which were postulated to be probable candidates to encode missing links in nicotine biosynthesis, either at the structural or regulatory level. Indeed, in a rationally designed screen to discover potential regulators of genes coding for key enzymes in nicotine biosynthesis, we found two novel tobacco AP2-domain transcription factors, NtORC1 and NtJAP1, that positively regulate the PMT promoter (De Sutter et al. 2005). Simultaneously, by further exploration of the profiling data and gene inventory we could forward novel gene functions, not only with regard to jasmonate-mediated activation of alkaloid biosynthesis (De Sutter et al. 2005) but also with regard to the links between jasmonate and cell cycle-regulated cytokinin metabolism (Kwade et al. 2005) or the de novo biosynthesis of vitamin C (Wolucka et al. 2005). Another successful study employed a microarray-based strategy to identify genes that are differentially regulated between closely related tobacco lines that accumulate either nicotine or nornicotine as the predominant leaf alkaloid (Siminszky et al. 2005). Transcript profiling was achieved in three phases. Firstly, EST databases, each representing a compilation of more than > 11,000 sequencing runs of randomly selected clones, were generated by using cDNA libraries from senescing leaves of two isogenic tobacco lines, accumulating either nicotine or nornicotine as the predominant leaf alkaloid. Secondly, two types of DNA chips were synthesized, one representing the nornicotine line (4,992 cDNAs), and another representing the complete non-redundant unigene set of the combined EST databases (6,963 cDNAs). Finally, hybridizations were conducted with RNA isolated from different tobacco genotypes that had been subjected to different treatments known to enhance the metabolic conversion of nicotine to nornicotine. These experiments led to the identification of a closely related family of tobacco CYP450 genes, of which at least one member, CYP82E4 encodes an enzyme with nicotine demethylase activity (Siminszky et al. 2005; see also Fig. 2).
An alternative functional genomics approach to investigate tobacco alkaloid biosynthesis was described recently (Rogers et al. 2003). In a way to access the genomic potential of a plant, the creation of a ‘functional genomics library’ was pursued by generating large populations of tobacco cell cultures, in which each clone represents a ‘gain of function’ mutation, in this case by means of T-DNA activation tagging. T-DNA activation tagging is a method used to generate dominant mutations in plant cells by the insertion of a T-DNA that carries constitutive enhancer elements that can cause transcriptional activation of flanking plant genes. Using this approach, one clonal culture (out of 10,000 screened) was obtained that overproduced nicotine. Intact plants regenerated from this clone continued to overproduce nicotine at fivefold higher levels than wild-type N. tabacum (Littleton et al. 2005). However, despite T-DNA tagging may allow rapid gene rescue, the exact nature of the gene responsible for the observed increase in nicotine biosynthesis was not revealed. Accordingly, subsequent cloning of the gene potentially responsible for this phenotype and reintroduction of the gene into wild-type plants is needed to confirm the phenotype. Still, this T-DNA activation tagging approach represents an attractive alternative and has for instance also been successfully applied to identify transcription factors regulating terpenoid indole alkaloid biosynthesis in Catharanthus roseus (van der Fits and Memelink 2000; van der Fits et al. 2001).
Lately, many more ‘omics’ tools and resources are being developed specifically for tobacco. We list here a number of them. First, a resource that in the near future will become available due to the efforts of the Tobacco Genome Initiative is the full N. tabacum genome sequence (http://www.tobaccogenome.org). This initiative is coordinated at the Plant Pathology Department of the North Carolina State University, is supported by Philip Morris USA, Inc., and aims to sequence and annotate more than 90% of the open reading frames in the genome of N. tabacum. Other genetic resources that are already publicly available include for instance large EST collections of N. sylvestris roots and leaves (representing more than 3,500 genes; Katoh et al. 2003) and N. tabacum BY-2 cells (representing about 7,000 genes; Matsuoka et al. 2004). In total, more than 450,000 ESTs are currently available for Nicotiana and other species of the Solanaceae family, allowing comparative transcriptome analysis across species (Rensink et al. 2005). Construction of N. tabacum protein databases has also been initiated, for instance of leaf trichomes and BY-2 cells (Amme et al 2005; Laukens et al. 2004). As mentioned in the beginning of this section, a catalogue of more than 2,500 tobacco metabolites is available (Nugroho and Verpoorte 2002). For metabolome analysis however, the intrinsic structural variety imposes most challenges at the analytical level, both for profiling multiple metabolites in parallel as for the quantitative analysis of selected metabolites (Oksman-Caldentey and Saito 2005). Performing non-targeted metabolome analysis methods for tobacco have been developed recently, either based on Nuclear Magnetic Resonance (Choi et al. 2004) or Fourier Transform Ion Cyclotron Mass Spectrometry (Aharoni et al. 2002; Mungur et al. 2005). These methods are suitable for metabolomic fingerprinting or rapid screening of differential tobacco samples and will therefore represent invaluable tools to study the tobacco metabolic network and dissect it at the molecular level.
Terpenoid indole alkaloids: Catharanthus roseus
The biosynthesis of terpenoid indole alkaloids (TIAs) proceeds via the central precursor strictosidine, which constitutes a fusion product of the shikimate pathway-derived tryptamine moiety and of the plastidic non-mevalonate pathway-derived secologanin moiety (Fig. 3). Altogether more than 120 TIAs have been isolated from Catharanthus during the past decades (van der Heijden et al. 2004). Besides the monomeric TIAs produced in several branches of the pathway, the pharmaceutically more interesting bisindole alkaloids are formed from the condensation of tabersonine-derived vindoline and catharanthine. Starting from the amino acid tryptophan and the monoterpenoid geraniol, the biosynthesis of bisindole alkaloids in C. roseus involves at least 35 intermediates and 30 enzymes (van der Heijden et al. 2004). Extensive intra- and intercellular translocation is involved in TIA biosynthesis (St-Pierre et al. 1999; Irmler et al. 2000; Burlat et al. 2004) and several transcription factors, in particular members of the plant-specific AP2-domain family, have been shown to coordinate the expression of TIA biosynthetic genes in response to external and internal signals (Memelink et al. 2001).

Structure and biosynthesis of terpenoid indole alkaloids in Catharanthus roseus. Metabolites are given as full names in lowercase and enzymes as abbreviations in capitals. Full and dashed arrows mark single and multiple conversion steps between intermediates, respectively. Enzymes listed: AS (anthranilate synthase), CMK (4-diphosphocytidyl-2C-methyl-d-erythrol kinase), CMS (4-diphosphocytidyl-2C-methyl-d-erythrol-4-phosphate synthase), CPR (cytochrome P450 reductase), DAT (deacetylvindoline 4-O-acetyltransferase), D4H (desacetoxyvindoline 4-hydroxylase), DXR (1-deoxy-d-xylulose-5-phosphate reductoisomerase), DXS (1-deoxy-d-xylulose-5-phosphate synthase), G10H (geraniol 10-hydroxylase), GPPS (geranyl diphosphate synthase), HDR (1-hydroxy-2-methyl-2-(E)-butenyl-4-diphosphate reductase), HDS (1-hydroxy-2-methyl-2-(E)-butenyl-4-diphosphate synthase), 10HGO (10-hydroxygeraniol oxidoreductase), IPPI (isopentenylpyrophosphate isomerase), MAT (acetyl-CoA:minovincine-O-acetyltransferase), MECS (2C-methyl-d-erythrol-2,4-cyclodiphosphate synthase), NMT (N-methyltransferase), OMT (O-methyltransferase), SGD (strictosidine β-d-glucosidase), SLS (secologanin synthase), STR (strictosidine synthase), TDC (tryptophan decarboxylase), T16H (tabersonine 16-hydroxylase)
Whereas the genes encoding TIA biosynthetic enzymes usually have been identified one-by-one by the classical approaches, the transcription factors that regulate TIA biosynthesis were isolated through T-DNA activation tagging and yeast one-hybrid (Y1H) screenings (Memelink et al. 2001). In an attempt to isolate master regulators of TIA biosynthetic genes the T-DNA tagging approach was applied to C. roseus suspension cells (van der Fits and Memelink, 2000; van der Fits et al. 2001). T-DNA tagged cells were screened for resistance to a toxic substrate of one of the TIA biosynthetic enzymes: tryptophan decarboxylase (see Fig. 3). This screening yielded several interesting leads, one of them ORCA3, an AP2-domain transcription factor and so far the most potent transcriptional regulator of TIA metabolism (van der Fits and Memelink 2000). In another screening for TIA regulatory genes, Memelink and coworkers employed the Y1H technique, a method to isolate novel genes encoding proteins that bind to a target cis-acting regulatory DNA element. In this case promoter elements of another TIA gene, strictosidine synthase (Str, see Fig. 3), were used as bait DNA sequences. This method proved extremely successful and moreover suggested a considerable degree of complexity in the control of TIA biosynthesis. In fact, multiple transcription factors of different classes, including AP2, MYB, bHLH and zinc finger-type proteins, turned out to show binding specificity to Str promoter elements (Menke et al. 1999; van der Fits et al. 2000; Chatel et al. 2003; Pauw et al. 2005).
The first comprehensive functional genomics profiling approach to cover TIA biosynthesis in C. roseus employed systematic proteome analysis of a cell suspension culture accumulating strictosidine, ajmalicine and vindolinine (Jacobs et al. 2005). Two-dimensional SDS-PAGE was used to select 88 protein spots with a differential accumulation pattern, corresponding to the accumulation pattern of the alkaloids in the investigated culture. These protein spots were subsequently further characterised by mass spectrometry and for 58 of them a functional annotation based on sequence homology searching could be provided. Only two spots within this set, representing STR isoforms, were known to be involved in TIA biosynthesis. Other unique sequences were found, which may relate to unidentified biosynthetic proteins. The main limitations that were observed with this approach in Catharanthus, as well as within the similar poppy proteomic study (Decker et al. 2000), were (i) the generally low abundance of secondary metabolism proteins, preventing their isolation, and (ii) the lack of sequence data of both genes and proteins from Catharanthus, hampering identification of protein spots.
One method that may either circumvent or address these limitations of proteomic studies of non-model medicinal plant systems such as C. roseus, is cDNA-AFLP-based transcript profiling (Breyne et al. 2003). Large-scale gene discovery programs in non-model plants are hampered enormously by the fact that standard transcript profiling methods, such as serial analysis of gene expression or microarray analysis, are not applicable because of the lack of large sequence repertoires. In contrast, the cDNA-AFLP technology can be used to identify genes in such plants and acquire quantitative expression profiles at the same time. By applying this technique on jasmonate-elicited TIA producing C. roseus cells grown under diverse auxin regimes, we determined the quantitative temporal accumulation patterns of 10,790 transcript tags (Rischer et al. 2006). In total, good-quality sequences for 417 differentially expressed transcript tags were obtained. When blasting these 417 tag sequences with the 236 C. roseus EMBL entries publicly available prior to this study, less than 10% gave a perfect match. Thus, the vast majority of the tags identified represented novel C. roseus sequence information. Here, we further compared this transcriptome sequence data set with the sequences of the C. roseus proteome data set (Jacobs et al. 2005). For nine of the protein spots a potential corresponding transcript tag could be identified (Table 1), including the two STR isoforms and the so-called PS protein, suggested to be associated with TIA biosynthesis (Leménager et al. 2005). Importantly, and in contrast to the proteomics approach, we could identify most of the so far known structural and regulatory genes involved in TIA biosynthesis and successfully monitor them within a single cDNA-AFLP experiment (Rischer et al. 2006). In parallel we performed non-targeted metabolic profiling of the same samples, leading to a final set of 178 known and novel metabolites, including nine known TIA molecules (Rischer et al. 2006). Integration and correlation analysis of the expression profiles of the 417 gene tags and the accumulation profiles of the 178 metabolite peaks allowed depicting novel gene-to-gene or gene-to-metabolite networks for TIA biosynthesis in C. roseus cells (Rischer et al. 2006). These networks revealed for instance that the different branches of TIA biosynthesis were subject to differing hormonal regulation. On the basis of these networks, a select number of novel genes and metabolites likely to be involved in TIA biosynthesis could be forwarded; candidates that may deserve priority in diverse types of future functional screenings such as the one successfully employed for the gene inventory obtained by cDNA-AFLP analysis of elicited tobacco cells (De Sutter et al. 2005). Further extension of the metabolite profiling data with quantification of metabolic fluxes in an organisational approach (Morgan and Shanks 2002) could help elucidate the function, pathway structures and pathway fluxes for novel and known TIA compounds.
Conclusions and perspectives
Recently, several biochemical pathway knowledge databases for Arabidopsis thaliana have been developed, for instance AraCyc, MAPMAN and BioPathAt (Mueller et al. 2003; Thimm et al. 2004; Lange and Ghassemian 2005). These databases are powerful tools for integrative analysis of biochemical pathways and facilitate enormously analysis of ‘omics’-scale experiments. Although we are still somewhat remote from establishing similar biochemical pathway knowledge databases for alkaloid-producing medicinal plants, the rapid pace at which new and ever more potent functional genomics technologies are being developed, will offer unprecedented opportunities to map biosynthesis of alkaloids in non-model medicinal plants. Indeed, encouraging results have been obtained already for a number of alkaloid-producing plant species, in particular by integrated transcriptome–metabolome profiling approaches. Therefore we anticipate that functional genomics, and the knowledge it brings along, will allow a better exploitation of plant biochemical capacities, leading to increased production of interesting alkaloids or the design of novel alkaloids with novel or superior biological activities.
Abbreviations
- BIA:
-
benzylisoquinoline alkaloid
- AFLP:
-
amplified fragment length polymorphism
- EST:
-
expressed sequence tag
- PMT:
-
putrescine N-methyltransferase
- STR:
-
strictosidine synthase
- TIA:
-
terpenoid indole alkaloid
- VIGS:
-
virus-induced gene silencing
- Y1H:
-
yeast one-hybrid
References
Aharoni A, Keizer LCP, Bouwmeester HJ, Sun Z, Alvarez-Huerta M, Verhoeven HA, Blaas J, van Houwelingen AMML, De Vos RCH, van der Voet H, Jansen RC, Guis M, Mol J, Davis RW, Schena M, van Tunen AJ, O’Connell AP (2000) Identification of the SAAT gene involved in strawberry flavor biogenesis by use of DNA microarrays. Plant Cell 12:647–662
Aharoni A, De Vos CHR, Verhoeven HA, Maliepaard CA, Kruppa G, Bino R, Goodenowe DB (2002) Nontargeted metabolome analysis by use of Fourier Transform Ion Cyclotron Mass Spectrometry. OMICS 6:217–234
Allen RS, Millgate AG, Chitty JA, Thisleton J, Miller JA, Fist AJ, Gerlach WL, Larkin PJ (2004) RNAi-mediated replacement of morphine with the nonnarcotic alkaloid reticuline in opium poppy. Nat Biotechnol 22:1559–1566
Amme S, Rutten T, Melzer M, Sonsmann G, Vissers JP, Schlesier B, Mock HP (2005) A proteome approach defines protective functions of tobacco leaf trichomes. Proteomics 5:2508–2518
Bird DA, Franceschi VR, Facchini PJ (2003) A tale of three cell types: alkaloid biosynthesis is localized to sieve elements in opium poppy. Plant Cell 15:2626–2635
Breyne P, Dreesen R, Cannoot B, Rombaut D, Vandepoele K, Rombauts S, Vanderhaeghen R, Inzé D, Zabeau M (2003) Quantitative cDNA-AFLP analysis for genome-wide expression studies. Mol Genet Genomics 269:173–179
Burlat V, Oudin A, Courtois M, Rideau M, St-Pierre B (2004) Co-expression of three MEP pathway genes and geraniol 10-hydroxylase in internal phloem parenchyma of Catharanthus roseus implicates multicellular translocation of intermediates during the biosynthesis of monoterpene indole alkaloids and isoprenoid-derived primary metabolites. Plant J 38:131–141
Chatel G, Montiel G, Pré M, Memelink J, Thiersault M, St-Pierre B, Doireau P, Gantet P (2003) CrMYC1, a Catharanthus roseus elicitor- and jasmonate-responsive bHLH transcription factor that binds the G-box element of the strictosidine synthase gene promoter. J Exp Bot 54:2587–2588
Chitty JA, Allen RS, Fist AJ, Larkin PJ (2003) Genetic transformation in commercial Tasmanian cultivars of opium poppy, Papaver somniferum, and movement of transgenic pollen in the field. Funct Plant Biol 30:1045–1058
Choi HK, Choi YH, Verberne M, Lefeber AW, Erkelens C, Verpoorte R (2004) Metabolic fingerprinting of wild-type and transgenic tobacco plants by 1H NMR and multivariate analysis technique. Phytochemistry 65:857–864
Croteau R, Kutchan TM, Lewis NG (2000) Natural products (Secondary metabolites). In: Buchanan B, Gruissem W, Jones R (eds) Biochemistry and molecular biology of plants. American Society of Plant Physiologists, Rockville pp 1250–1318
Decker G, Wanner G, Zenk MH, Lottspeich F (2000) Characterization of proteins in latex of the opium poppy (Papaver somniferum) using two-dimensional gel electrophoresis and microsequencing. Electrophoresis 21:3500–3516
De Luca V, Laflamme P (2001) The expanding universe of alkaloid biosynthesis. Curr Opin Plant Biol 4:225–233
De Sutter V, Vanderhaeghen R, Tilleman S, Lammertyn F, Vanhoutte I, Karimi M, Inzé D, Goossens A, Hilson P (2005) Exploration of jasmonate signalling via automated and standardized transient expression assays in tobacco cells. Plant J 44:1065–1076
Facchini PJ (2001) Alkaloid biosynthesis in plants: biochemistry, cell biology, molecular regulation, and metabolic engineering applications. Annu Rev Plant Physiol Plant Mol Biol 52:29–66
Facchini PJ, Bird DA, St-Pierre B (2004) Can Arabidopsis make complex alkaloids? Trends Plant Sci 9:116–122
Facchini PJ, St-Pierre B (2005) Synthesis and trafficking of alkaloid biosynthetic enzymes. Curr Opin Plant Biol 8:657–666
Fridman E, Pichersky E (2005) Metabolomics, genomics, proteomics, and the identification of enzymes and their substrates and products. Curr Opin Plant Biol 8:242–248
Goossens A, Häkkinen ST, Laakso I, Seppänen-Laakso T, Biondi S, De Sutter V, Lammertyn F, Nuutila AM, Söderlund H, Zabeau M, Inzé D, Oksman-Caldentey KM (2003) A functional genomics approach toward the understanding of secondary metabolism in plant cells. Proc Natl Acad Sci USA 100:8595–8600
Guterman I, Shalit M, Menda N, Piestun D, Dafny-Yelin M, Shalev G, Bar E, Davydov O, Ovadis M, Emanuel M, Wang J, Adam Z, Pichersky E, Lewinsohn E, Zamir D, Vainstein A, Weiss D (2002) Rose scent: genomics approach to discovering novel floral fragrance-related genes. Plant Cell 14:2325–2338
Hashimoto T, Yamada Y (1994) Alkaloid biogenesis—molecular aspects. Annu Rev Plant Physiol Plant Mol Biol 45:257–285
Hashimoto T, Yamada Y (2003) New genes in alkaloid metabolism and transport. Curr Opin Biotechnol 14:163–168
Hibi N, Higashiguchi S, Hashimoto T, Yamada Y (1994) Gene expression in tobacco low-nicotine mutants. Plant Cell 6:723–735
Hileman LC, Drea S, Martino G, Litt A, Irish VF (2005) Virus-induced gene silencing is an effective tool for assaying gene function in the basal eudicot species Papaver somniferum (opium poppy). Plant J 44:334–341
Irmler S, Schröder G, St-Pierre B, Crouch NP, Hotze M, Schmidt J, Strack D, Matern U, Schröder J (2000) Indole alkaloid biosynthesis in Catharanthus roseus: new enzyme activities and identification of cytochrome P450 CYP72A1 as secologanin synthase. Plant J 24:797–804
Jacobs DI, Gaspari M, van der Greef J, van der Heijden R, Verpoorte R (2005) Proteome analysis of the medicinal plant Catharanthus roseus. Planta 221:690–704
Jennewein S, Wildung MR, Chau M, Walker K, Croteau R (2004) Random sequencing of an induced Taxus cell cDNA library for identification of clones involved in Taxol biosynthesis. Proc Natl Acad Sci USA 101:9149–9154
Katoh A, Yamaguchi Y, Sano H, Hashimoto T (2003) Analysis of expression sequence tags from Nicotiana sylvestris. Proc Jpn Acad B-Phys 79:151–154
Kutchan TM (2005) A role for intra- and intercellular translocation in natural product biosynthesis. Curr Opin Plant Biol 8:292–300
Kwade Z, Świ
 tek A, Azmi A, Goossens A, Inzé D, Van Onckelen H, Roef L (2005) Identification of four adenosine kinase isoforms in tobacco BY-2 cells and their putative role in the cell cycle-regulated cytokinin metabolism. J Biol Chem 280:17512–17519
tek A, Azmi A, Goossens A, Inzé D, Van Onckelen H, Roef L (2005) Identification of four adenosine kinase isoforms in tobacco BY-2 cells and their putative role in the cell cycle-regulated cytokinin metabolism. J Biol Chem 280:17512–17519Lange BM, Wildung MR, Stauber EJ, Sanchez C, Pouchnik D, Croteau R (2000) Probing essential oil biosynthesis and secretion by functional evaluation of expressed sequence tags from mint glandular trichomes. Proc Natl Acad Sci USA 97:2934–2939
Lange BM, Ghassemian M (2005) Comprehensive post-genomic data analysis approaches integrating biochemical pathway maps. Phytochemistry 66:413–451
Laukens K, Deckers P, Esmans E, Van Onckelen H, Witters E (2004) Construction of a two-dimensional gel electrophoresis protein database for the Nicotiana tabacum cv. Bright Yellow-2 cell suspension culture. Proteomics 4:720–727
Legg PD, Collins GB (1971) Inheritance of percent total alkaloids in Nicotiana tabacum L. II. Genetic effect of two loci in Burley 21 × LA Burley 21 populations. Can J Genet Cytol 13:287–291
Leménager D, Ouelhazi L, Mahroug S, Veau B, St-Pierre B, Rideau M, Aguirreolea J, Burlat V, Clastre M (2005) Purification, molecular cloning, and cell specific gene expression of the alkaloid-accumulation associated protein CrPS in Catharanthus roseus. J Exp Bot 56:1221–1228
Littleton J, Rogers T, Falcone D (2005) Novel approaches to plant drug discovery based on high throughput pharmacological screening and genetic manipulation. Life Sci 78(5):467–475
Matsuoka K, Demura T, Gális I, Horiguchi T, Sasaki M, Tashiro G, Fukuda H (2004) A comprehensive gene expression analysis toward the understanding of growth and differentiation of tobacco BY-2 cells. Plant Cell Physiol 45:1280–1289
Memelink J, Verpoorte R, Kijne JW (2001) ORCAnization of jasmonate-responsive gene expression in alkaloid metabolism. Trends Plant Sci 6:212–219
Menke FLH, Champion A, Kijne JW, Memelink J (1999) A novel jasmonate- and elicitor-responsive element in the periwinkle secondary metabolite biosynthetic gene Str interacts with a jasmonate- and elicitor-inducible AP2-domain transcription factor, ORCA2. EMBO J 18:4455–4463
Millgate AG, Pogson BJ, Wilson IW, Kutchan TM, Zenk MH, Gerlach WL, Fist AJ, Larkin PJ (2004) Analgesia: morphine-pathway block in top1 poppies. Nature 431:413–414
Morgan JA, Shanks JV (2002) Quantification of metabolic flux in plant secondary metabolism by a biogenetic organizational approach. Metab Eng 4:257–262
Mueller LA, Zhang P, Rhee SY (2003) AraCyc: a biochemical pathway database for Arabidopsis. Plant Physiol 132:453–460
Mungur R, Glass AD, Goodenow DB, Lightfoot DA (2005) Metabolite fingerprinting in transgenic Nicotiana tabacum altered by the Escherichia coli glutamate dehydrogenase gene. J Biomed Biotechnol 2005:198–214
Nugroho LH, Verpoorte R (2002) Secondary metabolism in tobacco. Plant Cell Tissue Org 68:105–125
Oksman-Caldentey K-M, Inzé D (2004) Plant cell factories in the post-genomic era: new ways to produce designer secondary metabolites. Trends Plant Sci 9:433–440
Oksman-Caldentey K-M, Saito K (2005) Integrating genomics and metabolomics for engineering plant metabolic pathways. Curr Opin Biotechnol 16:174–179
Ounaroon A, Decker G, Schmidt J, Lottspeich F, Kutchan TM (2003) (R,S)-Reticuline 7-O-methyltransferase and (R,S)-norcoclaurine 6-O-methyltransferase of Papaver somniferum—cDNA cloning and characterization of methyl transfer enzymes of alkaloid biosynthesis in opium poppy. Plant J 36:808–819
Park SU, Facchini PJ (2000) Agrobacterium-mediated transformation of opium poppy. Papaver somniferum, via shoot organogenesis. J Plant Physiol 157:207–214
Pauw B, Hilliou FAO, Martin VS, Chatel G, de Wolf CJF, Champion A, Pré M, van Duijn B, Kijne JW, van der Fits L, Memelink J (2005) Zinc finger proteins act as transcriptional repressors of alkaloid biosynthesis genes in Catharanthus roseus. J Biol Chem 279:52940–52948
Rensink WA, Lee Y, Liu J, Iobst S, Ouyang S, Buell CR (2005) Comparative analyses of six solanaceous transcriptomes reveal a high degree of sequence conservation and species-specific transcripts. BMC Genomics 6:124
Rischer H, Orešič M, Seppänen-Laakso T, Katajamaa M, Lammertyn F, Ardiles-Diaz W, Van Montagu MCE, Inzé D, Oksman-Caldentey K-M, Goossens A (2006) Gene-to-metabolite networks for terpenoid indole alkaloid biosynthesis in Catharanthus roseus cells. Proc Natl Acad Sci USA 103:5614–5619
Rogers DT, Falcone DF, Littleton JM (2003) A functional genomics strategy for plant drug discovery. Pharmagenomics 14:26–34
Ruiz MT, Voinnet O, Baulcombe DC (1998) Initiation and maintenance of virus-induced gene silencing. Plant Cell 10:937–946
Saito K, Kobayashi M, Gong Z, Tanaka Y, Yamazaki M (1999) Direct evidence for anthocyanidin synthase as a 2-oxoglutarate-dependent oxygenase: molecular cloning and functional expression of cDNA from a red forma of Perilla frutescens. Plant J 17:181–189
Schoendorf A, Rithner CD, Williams RM, Croteau RB (2001) Molecular cloning of a cytochrome P450 taxane 10 β-hydroxylase cDNA from Taxus and functional expression in yeast. Proc Natl Acad Sci USA 98:1501–1506
Siminszky B, Gavilano L, Bowen SW, Dewey RE (2005) Conversion of nicotine to nornicotine in Nicotiana tabacum is mediated by CYP82E4, a cytochrome P450 monooxygenase. Proc Natl Acad Sci USA 102:14919–14924
St-Pierre B, Vazquez-Flota FA, De Luca V (1999) Multicellular compartmentation of Catharanthus roseus alkaloid biosynthesis predicts intercellular translocation of a pathway intermediate. Plant Cell 11:887–900
Teoh KH, Polichuk DR, Reed DW, Nowak G, Covello PS (2006) Artemisia annua L. (Asteraceae) trichome-specific cDNAs reveal CYP71AV1, a cytochrome P450 with a key role in the biosynthesis of the antimalarial sesquiterpene lactone artemisinin. FEBS Lett 580:1411–1416
Thimm O, Bläsing O, Gibon Y, Nagel A, Meyer S, Krüger P, Selbig J, Müller LA, Rhee SY, Stitt M (2004) MAPMAN: a user-driven tool to display genomics data sets onto diagrams of metabolic pathways and other biological processes. Plant J 37:914–939
van der Fits L, Memelink J (2000) ORCA3, a jasmonate-responsive transcriptional regulator of plant primary and secondary metabolism. Science 289:295–297
van der Fits L, Zhang H, Menke FLH, Deneka M, Memelink J (2000) A Catharanthus roseus BPF-1 homologue interacts with an elicitor-responsive region of the secondary metabolite biosynthetic gene Str and is induced by elicitor via a JA-independent signal transduction pathway. Plant Mol Biol 44:675–685
van der Fits L, Hilliou F, Memelink J (2001) T-DNA activation tagging as a tool to isolate regulators of a metabolic pathway from a genetically non-tractable plant species. Transgenic Res 10:513–521
van der Heijden R, Jacobs DI, Snoeijer W, Hallard D, Verpoorte R (2004) The Catharanthus alkaloids: pharmacognosy and biotechnology. Curr Med Chem 11:607–628
Wang J, Sheehan M, Brookman H, Timko MP (2000) Characterization of cDNAs differentially expressed in roots of tobacco (Nicotiana tabacum cv Burley 21) during the early stages of alkaloid biosynthesis. Plant Sci 158:19–32
Weid M, Ziegler J, Kutchan TM (2004) The roles of latex and the vascular bundle in morphine biosynthesis in the opium poppy, Papaver somniferum. Proc Natl Acad Sci USA 101:13957–13962
Wolucka BA, Goossens A, Inzé D (2005) Methyl jasmonate stimulates the de novo biosynthesis of vitamin C in plant cell suspensions. J Exp Bot 56:2527–2538
Yamazaki M, Gong Z, Fukuchi-Mizutani M, Fukui Y, Tanaka Y, Kusumi T, Saito K (1999) Molecular cloning and biochemical characterization of a novel anthocyanin 5-O-glucosyltransferase by mRNA differential display for plant forms regarding anthocyanin. J Biol Chem 274:7405–7411
Yamazaki M, Saito K (2002) Differential display analysis of gene expression in plants. Cell Mol Life Sci 59:1246–1255
Yamazaki M, Nakajima J, Yamanashi M, Sugiyama M, Makita Y, Springob K, Awazuhara M, Saito K (2003) Metabolomics and differential gene expression in anthocyanin chemo-varietal forms of Perilla frutescens. Phytochemistry 62:987–995
Ziegler J, Diaz-Chávez ML, Kramell R, Ammer C, Kutchan TM (2005) Comparative macroarray analysis of morphine-containing Papaver somniferum and eight morphine-free Papaver species identifies an O-methyltransferase involved in benzylisoquinoline biosynthesis. Planta 222:458–471
 tek A, Azmi A, Goossens A, Inzé D, Van Onckelen H, Roef L (2005) Identification of four adenosine kinase isoforms in tobacco BY-2 cells and their putative role in the cell cycle-regulated cytokinin metabolism. J Biol Chem 280:17512–17519
tek A, Azmi A, Goossens A, Inzé D, Van Onckelen H, Roef L (2005) Identification of four adenosine kinase isoforms in tobacco BY-2 cells and their putative role in the cell cycle-regulated cytokinin metabolism. J Biol Chem 280:17512–17519Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Goossens, A., Rischer, H. Implementation of functional genomics for gene discovery in alkaloid producing plants. Phytochem Rev 6, 35–49 (2007). https://doi.org/10.1007/s11101-006-9018-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11101-006-9018-0