
Abstract
The use of physiologically based pharmacokinetic (PBPK) modeling to support the drug product quality attributes, also known as physiologically based biopharmaceutics modeling (PBBM) is an evolving field and the interest in using PBBM is increasing. The US-FDA has emphasized on the use of patient centric quality standards and clinically relevant drug product specifications over the years. Establishing an in vitro in vivo link is an important step towards achieving the goal of patient centric quality standard. Such a link can aid in constructing a bioequivalence safe space and establishing clinically relevant drug product specifications. PBBM is an important tool to construct a safe space which can be used during the drug product development and lifecycle management. There are several advantages of using the PBBM approach, though there are also a few challenges, both with in vitro methods and in vivo understanding of drug absorption and disposition, that preclude using this approach and therefore further improvements are needed. In this review we have provided an overview of experience gained so far and the current perspective from regulatory and industry point of view. Collaboration between scientists from regulatory, industry and academic fields can further help to advance this field and deliver on promises that PBBM can offer towards establishing patient centric quality standards.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
INTRODUCTION
The use of modeling in drug product development has made significant progress in last four decades. Though the attempts to model the drug absorption kinetics began in 1950s and 1960s (1, 2); this field made significant progress in the 1980s with the use of very basic mathematical models and considering gastrointestinal tract in a simplified way (3,4,5). Further, in the 1990s, Gordon Amidon, Lawrence Yu and their collaborators (6,7,8,9) did extensive fundamental work in this field, explored various mathematical models and Lawrence Yu and Gordon Amidon laid the foundation of the compartmental absorption and transit (CAT) model (10). The foundation of the CAT model was very significant step in the field of absorption modeling. The development of the CAT model and other fundamental work on mathematical and absorption modeling very quickly resulted in the establishment of a commercial absorption model, using an advanced compartmental absorption and transit (ACAT) model (11). Since then, several other absorption models, like advanced dissolution absorption model (ADAM) (12) have been developed and are commercially used (13, 14) for mechanistic absorption modeling. These advance absorption models are continuing to evolve further and being optimized, as more physiologically relevant information is being obtained; for example, based on work of Mudie et al. (15), recently dynamic fluid volumes were used in the ACAT model for in situ modeling of a weak base (16). In last two decades, the extensive collaboration of academia, pharmaceutical industry, software development companies and regulatory agencies has led to significant progress in the field of PBPK and mechanistic absorption modeling (11, 17,18,19,20,21,22).
Currently, the use of PBPK models to replace human evaluation for predicting drug-drug interactions is widespread in the pharmaceutical industry and accepted by worldwide regulatory agencies (23). However, the application of PBPK models or Physiological Based Biopharmaceutics Models (PBBM) to support drug product quality attributes specification setting, safe space evaluation and bridging of the formulations is relatively recent and only the US FDA has recently published a draft guidance to support this type of simulations (24). PBPK models for biopharmaceutics applications or PBBM are gaining momentum as shown in this review and hold the promise to reduce burdensome human evaluation whilst providing assurance of the product quality for patients.
Different terminologies have been used over the recent years to cover the use of modeling and simulation to support quality aspects of drug products, from mechanistic IVIVC (19), physiological based absorption modeling (19), applications of PBPK in biopharmaceutics (24), or PBBM (20). A literature survey over the last 15 years, reveals that publications on clinically relevant dissolution specifications, PBPK or PBBM applications linked to dissolution specifications, and PBPK or PBBM applications linked to formulation bridging, have started approximately in 2011 and their number is growing exponentially since then (Fig. 1). The articles mentioning PBBM started in 2019 following a Regulatory Education for Industry (REdI) workshop titled Current State and Future Expectations of Translational Modeling Strategies to Support Drug Product Development, Manufacturing Changes and Controls that took place on September 23–25, 2019 at the University of Maryland, which introduced this new term PBBM (20). The field of PBBM includes biopharmaceutics of not only the drug absorbed through GI tract but also the drug which are designed to use locally through oral or non-oral route of administration. As of end December 2021, there was an annual average close to 50 papers published on this topic worldwide (Fig. 1). This survey shows that although more recent, the term PBBM has picked up some pace over the last 3 years and represents the majority of the literature topics in this field.

Annual publications over the last 15 years on various topics relevant to this review (see legend and text).
The purpose of this review is to give an overview of the current literature on these topics and highlight how PBBM can assist in product development through a product’s lifecycle, from early development to post-approval changes. PBBM, as discussed below, is an important tool that can aid in identifying critical attributes and limits beyond which changes in these critical attributes can affect drug bioavailability. PBBM can help in identifying critical drug product quality attributes, specification setting, safe space evaluation and bridging of the drug product formulations etc. While interest and use of PBBM to support product quality is increasing there are several potential areas where development and improvement in these models is needed. We are presenting the current regulatory perspective and industry applications to help identify advantages of using PBBM, advances made so far, current challenges and aspects where further developments are needed which can help to improve these tools to support drug product quality and minimize burdensome in vivo trials.
The authors acknowledge that the PBPK modeling approach is widely used for the Clinical Pharmacology applications in IND/NDA submissions (23) and it is expanding in bioequivalence/biowaiver applications in ANDA submissions (25, 26). However, those areas are beyond the scope of this article. This review article is mainly focusing on the application of PBPK models or more specifically the PBBM models to support quality aspects of the drug product development and the regulatory and industrial perspective and the related applications.
REGULATORY PERSPECTIVE
Role of PBBM in Biopharmaceutics
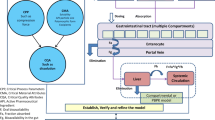
The role and the use of PBPK modeling in biopharmaceutics i.e. PBBM is expanding during the complex multi-phase drug product development, regulatory approval and life cycle management. The use of PBBM focuses on the use of biopharmaceutics modeling for the establishment of an in vitro in vivo link. Once this link is established then the PBBM modeling can facilitate the following: i) improve drug product risk assessment; ii) development of patient-centric quality standards; iii) expand regulatory flexibility and facilitate the drug product approval; iv) improve the drug product life cycle management (24, 27, 28). Figure 2 describes various regulatory areas where PBBM in biopharmaceutics plays important role to support quality aspects of the drug product. Figure 3 further presents the distribution of the regulatory submissions containing PBBM information and the role for Biopharmaceutics Assessment.

Regulatory impact of Biopharmaceutics Modeling.

Distribution of regulatory submission including PBBM for Biopharmaceutics Assessment.
The US-FDA has continuously emphasized on establishing patient-centric quality standards (29). The patients, who use the medication, expects that each dose unit of the drug product that they use is of highest quality and continuously and consistently achieves its therapeutic goals (29). In last several years the OPQ/CDER has stressed on to build in the quality in drug product rather than relying on the end product testing alone (30). Developing clinically relevant dissolution specification (method and acceptance criteria) is critical to achieving the objective of building patient-centric quality standards (31). Clinically relevant dissolution specification is “a specification that takes into consideration the clinical effect of variations in dissolution ensuring a consistent safety and efficacy profile” (24) of the drug product. The expansion of the use of the PBBM approach in drug product development and regulatory decision making is an important step in achieving the patient-centric quality standards.
Applications of PBBM
Identification of Critical Bioavailability Attributes and Quality Risk Assessment
The “Critical Bioavailability Attributes” (CBAs) are the formulation or process attributes those are expected to critically impact the bioavailability (absorption rate and extent) of a drug product (32, 33). The Critical Bioavailability Attributes (CBAs) is relatively a newer term and organically evolved during discussions between the US-FDA and the pharmaceutical industry. The CBAs may include critical material attributes (CMAs),(31) and critical process parameters (CPPs) (31) which can potentially affect bioavailability, for example drug substance particle size, polymorphic form, lubrication, coating levels etc.
Traditionally, a BCS based framework, the prior knowledge and experience is used to understand the possible challenges and risks towards developing the drug product formulations and identifying the possible list of CBAs for oral dosage forms. For example, the BCS framework can be easily used for the highly soluble drugs formulated into the drug products which dissolves rapidly and across physiological pH. However, BCS alone may not provide a clear understanding of all the CBAs for the poorly soluble immediate release drug products and the modified release formulations. In addition, using this approach finding the ranges for the CBAs beyond which they can critically affect bioavailability is challenging. A thorough understanding of the basic biopharmaceutics characteristics of the active substances and the drug product and using the PBPK modeling approach in biopharmaceutics, the CBAs can be clearly identified and the ranges in which they are most critical can be understood and established thus clearly understanding the risk and ranges where the risk is elevated or may be reduced.
Currently, while developing the in vitro dissolution method and assessing the initial biopharmaceutics risk, identification, and detection of the CBAs is considered very important. In cases of the drug products containing poorly soluble drugs and extended-release drug products, if CBAs cannot be clearly identified detected and controlled, for example using a discriminating dissolution specification, then initial biopharmaceutics risk is considered high to very high. In these cases, the use of PBBM approach to understand how the CBAs can affect the clinical performance, establish IVIVR and biopharmaceutics risk mitigation strategy becomes very significant (32, 33). The PBBM approach can also help in demonstrating and establishing the manufacturing design space which is clinically relevant and establishing control strategies to mitigate the biopharmaceutics quality risks and patient centric quality standards (24). For example, Fang et al. reported that in one case, to investigate the impact of dissolution, of two formulations different in the polymorphic form of the drug, on the pharmacokinetics (PK) parameters (Cmax and AUC) a sensitivity analysis on z-factor was performed. Parameter sensitivity analysis identified the range of the z-factor that would not significantly affect the bioavailability of the drug product developed with two different polymorphic forms. Parrott et al. reported using parameter sensitivity analysis to demonstrate that the maximal plasma concentrations were not affected by the change in precipitation and permeability, however, decrease in solubility and increase in particle size, more than 12 µm, has potential to affect the Cmax levels (34). The use of PBPK modeling approach in biopharmaceutics to identify the CBAs and the most optimal range may also help to establish the Quality Target Product Profile (QTPP) and develop the drug product using a Quality by Design (QbD) systemic approach (17).
Establishing an In Vitro-In Vivo Link and Biopredictive Dissolution Method
One of the key focus of the Division of Biopharmaceutics/US-FDA is to establish a link between the drug product quality and its clinical performance (35).. Establishing an in vitro-in vivo link helps in developing clinically relevant drug product specifications and establishing the patient-centric quality standards (24). Traditionally, this link has been established using an in vitro/in vivo correlation (IVIVC) or an in vitro-in vivo relationship (IVIVR) approach. In this approach a mathematical model is developed, without mechanistic description of the absorption or the PK characteristics (36), which can predict the relationship between an in vitro property (usually drug dissolution or release) of the drug product and an in vivo response (for example plasma drug concentration or amount of drug absorbed). However, in general this traditional approach of establishing a link between the in vitro and in vivo has been considered challenging, due to hurdles related to knowledge and resources and ethical considerations about conducting human trials. In addition, it was reported that using this approach the regulatory acceptance rate of the model was low (approximately 40%) (35, 37).
One of the most appealing use of the PBBM is that it can be used to link in vitro drug product attributes (for example dissolution and/or particle size) to the in vivo characteristics, (for example PK performance) (21, 38,39,40,41). To achieve this objective, one of the possible approaches is to use in vitro dissolution data as an input to predict drug absorption kinetics and establish a biopredictive dissolution method (28). Though significant progress has been made towards using the biorelevant medium, and mechanistic absorption modeling, the use of in vitro dissolution data to predict the drug absorption quantitatively remains challenging (24, 27). In PBBM approach, the mechanistic gastrointestinal absorption and disposition kinetics understanding, and the simulation of in vivo absorption profiles are used to estimate the in vivo dissolution profiles. Once the estimation of in vivo dissolution profiles is available, using a thorough understanding of the drug substance physiological properties and drug product CBAs, appropriate in vitro dissolution conditions can be developed such than a link can be established between the in vitro dissolution and in vivo dissolution and absorption (21, 24, 42).
Once biopredictive dissolution/drug release testing conditions, capable of predicting the PK profiles, are established then the in vitro test method can be used to predict the systemic exposure after administration of the drug product and establish the clinically relevant dissolution specifications. If in vitro dissolution/drug release is used to establish the in vitro-in vivo link then it is important that the dissolution/drug release method has the discriminating ability with regard to the CBAs; so that different dissolution profiles can be obtained from the drug product variants manufactured by altering the CBAs and clinically relevant drug product specifications can be established (20, 21, 24, 42).
If a biopredictive dissolution method has been developed, then it is expected that the same method is used for the quality control (QC) dissolution testing. Previously, the US-FDA has recommended to use only one dissolution method for QC and other regulatory purposes, for example biowaiver or bridging. However, as per the FDA’s draft Guidance, if a biopredictive dissolution method is too complex and may not be used for routine quality control (QC) dissolution testing then an alternative dissolution method can be used for routine quality control (QC) dissolution testing. This indicates that the US-FDA is open to embracing the possibility of accepting more than one dissolution method for a drug product if this approach encourages the development of more biopredictive dissolution methods. If the drug product development includes development of a biopredictive dissolution method, then parallel dissolution studies using a biopredictive and a potential QC dissolution method can be conducted throughout the drug product development (21, 24). In these scenarios, PBBM can be very useful in linking biopredictive and QC dissolution methods (20, 21, 41).
Challenges in Establishing an In Vitro-In Vivo Link
Although establishing an in vitro in vivo link is highly desirable, it remains a challenge. Limitations exist at both ends, in vitro, finding appropriate methods to generate in vitro dissolution data that is useful to establish a link and in vivo, understanding conditions that influence in vivo release and absorption/disposition characteristics (43).
Many drug candidates, especially several drug products recently developed and currently under development, have low aqueous solubility and understanding CBAs can be challenging for such products (44). Good understanding of drug substance properties and formulation properties is essential to identify CBAs; however, pH dependent solubility of drug substance, particle size distribution (PSD), presence of different polymorphic forms, conversion between such forms during product manufacturing and storage and presence of co-crystals can add to complexity of understanding the CBAs. This can lead to difficulties in identifying appropriate in vitro dissolution method. Factors such as presence of supersaturation, precipitation with change in pH gradient as product moves through gastrointestinal tract, presence of bile acids can affect in vivo dissolution and absorption. Various excipients can affect in vitro and in vivo dissolution e.g., presence of disintegrant, surfactant and its effect on wetting ability and solubilization, complex interplay of drug and excipient as in case of solubility enhancing excipients that cause dissolution, precipitation, and absorption. In case of modified release products in addition to factors mentioned above, characteristics of release controlling polymers such as polymer swelling and erosion through polymer matrix can affect dissolution. An appropriate dissolution method that is discriminating towards CBAs can provide insights in in vivo dissolution and aid in establishing in vitro in vivo correlation (45, 46). Though, the availability of standardized methods that can be used for dissolution testing under such scenarios is limited; however, efforts are currently underway to develop in vitro tools that can aid in understanding in vivo performance (14, 43, 47, 48). Though the use of biorelevant dissolution media is increasing in determination of the biorelevant solubility etc., till date, using the biorelevant media no biopredictive dissolution using IVIVC or PBBM has been approved as a regulatory dissolution method by the US-FDA(27). The authors note that many times standard dissolution testing conditions, like pharmacopeial buffers and methods, for example paddle at 50 rpm or basket at 100 rpm are used to generate in vitro dissolution profiles with an objective to establish an in vivo link and as an input in the PBBM model. These standard dissolution testing conditions may not always provide most appropriate dissolution profiles and use of non-standard dissolution testing conditions may be needed. For example, Kato et al. showed that for a tablet product, using a non-pharmacopeial buffer (tosylate solution, pH 3.0) and using a lower paddle rotational speed of 25 rpm, in vitro dissolution profiles could be obtained and used in a PBPK model to establish clinically relevant drug product specifications (39).
In addition to challenges associated with drug substance and product formulation, gastrointestinal (GI) physiological factors such as gastric emptying, regional pH and regional permeability and changes in permeability along the GI tract can affect absorption. Several in vivo factors affect product disposition and understanding these factors is critical to establishing in vitro in vivo link. Some drug substances may have nonlinear PK, which may be due to factors like solubility limitation, need for a specific transporter, factors affecting metabolism such as saturation of metabolizing enzymes which can affect in vivo absorption and disposition. Limited understanding of such factors creates a challenge towards establishing in vitro and in vivo link (28).
Establishing Bioequivalence Safe Space
A “safe space” or a bioequivalence space is constructed by the ranges of the in vitro specifications of drug product quality attributes, for example dissolution, drug substance particle size etc. (19, 24). Creating a safe space ensures that the variant drug batches manufactured within that safe space can be deemed bioequivalent to each other or the pivotal clinical batch(es) (24). There are several ways in which a “safe space” or a bioequivalence space can be built, for example using classical IVIVC or IVIVR (rank-order relationship) and using exposure –response analysis(40). However, the PBBM based IVIVR/IVIVC approach is most recent approach that is being used for building safe space, both in the NDA and ANDA applications, and obtain regulatory flexibility (24, 28, 32, 40). There can be more than one way of establishing the safe space using PBBM based IVIVR/IVIVC approach. One of the possible methods to establish the safe space has been described by Tycho et al. (28). In this approach having in vivo data (typically PK profiles) from a pivotal clinical /bioequivalent batch and biopredictive dissolution data, appropriate model development and validations are the critical elements. Briefly in this approach of developing safe space, i) initially a baseline PBBM model needs to be developed based on the drug physiochemical properties, drug product characteristics and mechanistic absorption and disposition kinetics of the drug; ii) use pivotal clinical /bioequivalent batch to generate dissolution profiles and develop an appropriate scheme (for example Weibull, Z-factor etc.) to input dissolution data in the PBBM model, the use of biopredictive dissolution method/data is preferable; iii) establish an appropriate criterion for model validation and show that the model can predict the observed PK profiles and perform appropriate validations; iv) if possible, demonstrate the model’s ability to predict the in vivo performance of the non-bioequivalent batches; v) use virtual bioequivalence trials to establish the safe space and widen the drug product quality specifications, if needed (28, 49). To use a PBBM approach for establishing the safe space, developing, verifying and validating a PBBM model is required (21, 28, 38). The validated model then can be used to predict the systemic exposures using virtual BE analysis. Fang et al. reported that virtual BE analysis with parameter sensitivity analysis can be used to establish, verify and widen the proposed ranges for product CBAs (CMAs, CPPs, and critical quality attributes (CQAs) (37, 50) Tycho et al. reported a case of poorly soluble weak base dasatinib, where the dissolution of a lower strength (20 mg) was faster than the dissolution of the bio-strength (100 mg) and therefore in vitro dissolution based biowaiver for the lower strength was denied. However, PPBM model and simulation were used to show that the faster dissolution of the lower strength (20 mg) did not affect the bioequivalence outcome and the Applicant obtained the biowaiver for the lower strength, thus avoiding conducting a BE study (38).
Establishing Clinically Relevant Dissolution Acceptance Criteria
An in vivo and in vitro link can establish that the method is biopredictive; however, establishing the link alone is not sufficient enough to establish the patient centric quality standards. To continuously ensure that the clinical performance of the commercial batches is similar to the clinical batches, drug product specifications like dissolution acceptance criteria are established. Traditionally, the dissolution acceptance criteria are established based on the average dissolution data of the pivotal clinical batches (e.g. ± 10% variation range of an extended release drug product and where Q = 80% dissolution occurs for immediate release drug products); while using this approach, if the relationship between the CBAs, in vitro dissolution and in vivo performance is not clear then the acceptance criteria may be too stringent or too wide and irrelevant to the clinical performance (51). Therefore, using PBBM, setting up the in vitro/vivo link, building safe space and establishing clinically relevant dissolution acceptance criteria, which i) are related to the clinical performance; ii) are not unnecessarily tight or too wide; and iii) can reject the non-bioequivalent batches; is very meaningful. If a biopredictive dissolution is used to establish a safe space, then using the PBBM approach, it is possible that a wider clinically relevant dissolution acceptance criteria can be established than the dissolution acceptance criteria established using the traditional approach(49). It has been reported that PBBM has been used to support a wider acceptance criterion of Q value of 75% instead of 80% at 30 min(37). In another case, the PBBM approach was used to show that the predicted Cmax and AUCs from a drug product batch with slower dissolution and batches with faster dissolution profiles were similar and a wider dissolution acceptance criterion at 45 min could be justified (49).
PBBM approaches have been used to support the Scale-up and Post-Approval Changes (SUPAC) (37), to obtain biowaivers (52) and bridging formulation changes. Tycho et al. reported a case of using PBBM approach to support the biowaiver request for a manufacturing site change of a drug product containing poorly soluble drug. In this case, due to batch-to-batch variability, f2 similarity testing showed that the dissolution profiles of the pre-change and post changes batches were not similar. However, using the PBBM approach a safe space could be established and dissolution profile boundaries between the bioequivalent and non-bioequivalent batches were set up (38). The PBBM based simulation showed that the dissolution profiles of the batches indicated to be not-similar by f2 similarity testing were still within the safe space, therefore assuring bioequivalence of pre-change and post changes batches (38).
Clinically Relevant Drug Product Specifications Other than Dissolution
To establish patient centric drug product quality standards, demonstrating clinically relevant drug product specifications using a link between the CBAs, CQA and PK performance is essential. Clinically relevant drug product specification considers the influence of variations in the CBAs on the in vivo performance of the drug product. Based on the drug product, the CBAs, including CMA (for example drug substance particle size, polymorphic form etc.) and CPP (for example granulation, compression force, release controlling coating levels etc.) needs to be clearly identified. Typically, the effect of the CBAs and changes in CBAs on in vivo performance should be studied using bioavailability (BA)/bioequivalence (BE) study in human subjects. However, conducting these BA/BE studies in humans many times is not practical and is burdensome from not only time, economical but also ethical reasons. Therefore, PBBM approach provides an opportunity where prior knowledge of CBAs and their impact on dissolution can be leveraged and used to make predictions about possible widening of the ranges of the CBAs (24, 37). For example, using a PPBM approach for a weakly acidic drug and parameter sensitive analysis using permeability, solubility, and particle size distribution, Pepin et al. (53) showed that the drug substance particle size (< 100µ), which can impact dissolution rate, may not impact the Cmax levels, however a higher particle size range (> 100µ), has potential to impact the Cmax levels. Further, Pepin et al. reported using a validated PBBM model, and “fitting of a theoretical particle size distribution to dissolution data” predicted the in vivo dissolution of the drug product and showed that a drug product manufactured using the proposed particle size limits would be bioequivalent to the clinical batches. Pepin et al. further reported that the successful use of the PBBM approach led to regulatory flexibility and establishing of clinically meaningful drug product specifications (53). In another case, for a capsule drug product containing poorly soluble and poorly permeable drug, a PBBM approach using a validated model was used to establish the drug substance particle size specifications (54).
Common Limitation Observed in the PBBM Modeling Approaches Submitted in Regulatory Submissions
In last decade, since 2010, the FDA has noticed a significant increase in the usage of PBBK approach for regulatory decisions making including related to the drug product quality decisions (37). In general, for NDAs, the acceptance rate (approximately 75%) of the PBBM models have been significantly higher as compared to the traditional IVIVC model acceptance rate (approximately 40%) (28, 35, 51). The authors also noticed that the use of PBBM modeling has increased in immediate release drug products, whereas the traditional IVIVC modeling was relatively limited to the extended-release dosage forms. For the review by the US-FDA, the PBBM models have been submitted through the IND submissions as well as the NDA and ANDA applications. It is worth noting that though the submission/applications containing PBBM modeling are increasing there are some general limitations those have been noticed in these submissions (37). These limitations can be broadly categorized into three categories and are summarized below:
i. Limitations related to input data: for example, lack of biorelevant solubilities and biopredictive dissolution data. Inadequate clinical data: model input parameters not justified; unreliable parameters estimation and coefficient values and not considering population variabilities (37).
ii. Limitations related to model development: for example, inadequate justification for parameter selection; improper selection of dissolution model and dosage forms; improper assumptions about the bioavailability, metabolism and elimination. Use of single release rate formulation (37).
iii. Limitations related to model validation: for example, inadequate PK data and lack of information on criteria for method validation and limited information on model predictive performance (37).
The following sections focuses on the industrial perspective and applications of the PBBM:
INDUSTRIAL PERSPECTIVE AND APPLICATIONS
PBBMs, when based on first principles, provide a link between quantitative quality attributes and human exposure to a batch of drug product. They can be used to gain a mechanistic understanding of what limits drug absorption and how a product quality attribute can outside a certain range, start to impact the exposure of the drug in a given human population or for a given human being. These models can then be used to reduce unnecessary human evaluation, improve drug product robustness to a physiological variable, predict the impact of change in certain excipients or predict the outcome of a relative bioavailability or bioequivalence study. PBBMs are an integral part of the predictive science efforts supporting the development, registration and post approval management of drug products in the pharmaceutical industry (Fig. 4).

Role of PBBM in model informed drug product design during development and post approval.
In the field of product quality and over the value chain, PBBM is used for material choices (drug substance polymorph (42) and particle size distribution (34, 55), formulation components (56, 57), drug product design (58))and also impact of manufacturing process (59) or critical process parameters (60) on the drug PK. In addition, as the development progresses and drugs products are being tested in clinical phases, PBBM also help to perform risk evaluation (change in polymorph during storage (41), change in batch dissolution profile (59, 61), change in excipient quality or amount (57)) to support rational decisions to be made, to ensure product robustness for the commercial phase (62). Finally, PBBM support analytical method development and control strategies through the choice of dissolution methods, establishment of their clinical relevance, and justification of critical biopharmaceutical attributes. Post approval, these models are also useful to manage the changes related to formulation composition, excipient grades or suppliers, introduction of new manufacturing sites, provided that these changes can be shown to happen within the safe space established for the drug substance and drug product CBA (53, 63).
Post approval generic formulation development can also benefit from a PBBM approach to understand the key characteristics of the product and which design space for a generic formulation will be successful to achieve bioequivalence with the reference product (64, 65). The assessment of generic products from different sources and estimation of the impact of formulation and dissolution differences on the human PK can be supported by PBBMs (66). The use of PBBM is also reported to support the change of dissolution specifications post approval (67).
These models also serve to combine and federate different types of measurements such as solubility pH profiles, impact of bile salts on drug solubility, precipitation rate of super-saturated drug solutions, drug substance particle size distribution, drug product dissolution rate with the data related to the drug metabolism, distribution and excretion to provide for a comprehensive mechanistic model for systemic or organ exposure prediction.
PBBMs have evolved from PBPK models without losing any of their benefits, but adding the mechanism of drug release, dissolution and absorption (where relevant) from the site of administration. The impact of physiological changes (age, disease (68), stomach pH, prandial state, gastro-intestinal surgery or co-administration of drugs or excipients modifying the transit (69, 70) or the release rate of drug formulations (56)) can be simulated, and their impact on drug dissolution from the dosage form can also be anticipated. The type of dosage form and their transit along the GI tract can be simulated as well, making these tools important in the design phase of a drug product development.
Options to Integrate Drug Product Dissolution in a PBBM
Overall the following options are currently available to integrate drug product in vitro dissolution in a PBBM (71).
-
1.
Direct input
-
2.
Data fitted with Weibull functions (single or multiple phases)
-
3.
Constant Z-factor
-
4.
Z-factor vs pH profile
-
5.
Drug Substance (DS) PSD + Diffusion Layer Model (DLM) scaling factor with mechanistic dissolution model
-
6.
P-PSD with mechanistic dissolution model
These options are not equivalent and they have to be carefully selected since they can influence the outcome of the prediction (72). In addition, some restrictions exist regarding which option to select.
Option 1 (direct input) is the least favored option of all: Unless the dissolution sampling time frequency is high, the linear interpolation between the measured time points will be a source of errors for the simulation. In addition, if the dissolution data is not generated until complete dissolution, extrapolation errors may also occur. Option 2, (Weibull functions), are quite flexible, and can be applied to dissolution profiles showing lag times, single or multiple phases and can cover from sigmoidal to first order types of dissolution rates. Both options 1 and 2 are non-mechanistic and will force the in vivo drug dissolution to happen as a function of time, i.e. disconnected from GI transit and not dependent on solubility and volume in the GI tract. In some PBBM platforms, the Weibull function can be used to control the release of undissolved drug. The drug dissolution can happen as a second step in a mechanistic way based on the drug substance particle size, and local conditions in the GI tract at the time of release.
Option 3 is the z-factor proposed by Takano et al. (73) as described in Eq. (1).
where D (m2.s-1) is the drug diffusion coefficient, \({\rho }_{S}\) the true density (kg.m-3), h the thickness of the unstirred water layer (m) and r0 is the initial dissolving particle radius (m). The z-factor has SI units of m3.kg-1.s-1 and lumps the drug substance and drug product properties in one parameter. The z-factor is an approximation of the real behavior of particles during dissolution which shrink to disappearance in sink conditions, as it considers only the initial particle radius. The z-factor should not be dependent on the drug solubility in the dissolution medium, the drug dose, or the volume of the medium since these three parameters are considered in addition to the z-factor to predict dissolution rate (73). Therefore, a single z-factor is expected to be obtained for any given DS powder or drug product (DP) formulation for all the media tested. Z-factors should not be applied to media comprising micelles, for which the fraction of drug unbound will change as a function of micelle concentration, as the diffusion coefficient and UWL thickness of a micelle bound drug are very different from that of the free drug (74). Hofsäss et al. also cautioned against using the z-factor when the release is incomplete (due to coning for example) or when there is a time needed for disintegration and proposed a methodology to improve the fit of dissolution data in these conditions and reduce/eliminate these bias (75).
Option 4 (z-factor vs pH profile), which is sometimes used in the literature to describe pH dependent dissolution of drug products, should be used with caution and justified with additional information. A variation of z-factor with pH may attest from effects of excipients which can vary with pH or differences in the drug substance wettability with pH (76).
Option 5 and Option 6 rely on mechanistic dissolution equations such as the Wang Flanagan or Johnson equations to predict in vitro or in vivo dissolution (77, 78). Option 5 relies on the assumption that the PSD of a drug substance comprised in a drug product can be predictive of the drug product dissolution by applying a single scaling factor to the drug substance PSD. Although interesting, this approach still needs to be demonstrated as applicable for numerous product batches, but there are several scientific hurdles to the application of Option 5 for solid oral dosage forms as explained by Pepin et al. (79). Option 6 proposes to fit a product particle size (P-PSD) to the DP dissolution and apply this P-PSD as a batch specific parameter to calculate in vitro or in vivo dissolution. This approach was shown to be predictive of human PK for a variety of compounds and is exemplified in Table 1 (supplementary material). One advantage of the mechanistic dissolution approaches is that they can be translated from one population, e.g. healthy adults, to target populations (diseased, pediatric etc.), whilst a PBBM validated in healthy adults with options 1 or 2 for integration of dissolution data cannot be adapted to another population. The P-PSD can be obtained using commercial tools such as SIVA for Simcyp®, DDDPlus® for GastroPlus™, or from non-commercial Excel tools (79). There is currently no comparison of the different models of in vitro dissolution using Option 6, and further work is needed to define dissolution model performance on data obtained under different conditions for the same drug product batch. The 166 dissolution profile database on 18 different drugs used by Pepin et al. (79) could be a starting point.
Over the thirty-two (32) compounds and models reviewed in this article (Table 1), twenty seven (27) were developed for immediate release products and 5 were developed for modified release products. All the 5 models developed for the modified release products used a Weibull function to integrate dissolution in the PBBM. The options selected by modelers for immediate release dissolution integration in the PBBM are shown in Fig. 5 and show a preference of z-factor (either constant or as a function of pH) to integrate dissolution mechanistically. Non-mechanistic integration of dissolution (Weibull and direct input) still represents 40% of the case studies reported. There are only a few research articles which compare the different methods to integrate dissolution in PBBM (28, 53, 72, 80, 81), and further work would be needed to define the best practices.

Options selected for introduction of IR product dissolution in PBBM from 27 examples in the literature (See Table 1, Supplemental material).
Clinical Relevance of a Dissolution Method
Establishing the clinical relevance of a discriminating dissolution method is of importance to ensure that the drug product quality differences can be picked up, and that the specifications established for a particular method will be of relevance for the patients. Clinically relevant dissolution methods can evaluate large or small formulation changes such as the presence of pH-active excipients for example, as was shown by Cámara-Martinez et al. (57) for two non-bioequivalent 200 mg ibuprofen fixed dose combinations with pseudoephedrine. For all oral products, whether immediate or modified release, clinical PK should be measured for formulation and/or process variants relevant to the commercial dose and drug product (28), and relevant to the CBA on which the specification is being established. For illustration a non-exhaustive list of types of variants which may be considered is given below:
-
Immediate release formulations: drug substance particle size, polymorphic form, drug product granule size or density, binder amount, compression force.
-
Modified release matrix formulations: different grades/amounts of matrix forming agents, molecular weight or viscosity of the polymers.
-
Modified release pellet formulations: Coating level/thickness, molecular weight or viscosity of the polymer, pore forming agent content.
-
Eroding tablet formulations: surface to volume ratio, compression force.
-
Formulations containing amorphous drug substance: Surface to volume ratio, crystalline drug substance (spiking of formulations with crystalline material).
If feasible, generation of PK data for an intravenous formulation may be advisable depending on the complexity of drug metabolism. In addition, the inclusion of an oral solution that does not precipitate in a PK study is desired to provide for a reference PK profile. These two latter sets of PK data may be used for PBBM setup or verification. For situations where there is no significant first-pass metabolism, oral solution PK may also be used to inform/verify drug disposition in the model.
The search for a clinically relevant method can be done using a traditional IVIVC approach (57, 82) or a PBBM approach. For approaches using PBBM to establish an IVIVC, the approach can be “top down”, i.e. using the PBBM to fit the in vivo dissolution to the observed oral PK profile and then comparing the in vivo dissolution to the in vitro dissolution. For diltiazem-HCl MR formulations (83), this approach was used to show that the QC method was not clinically relevant and propose a new method which was clinically relevant. For Basmisanil IR formulations (84), this method was used to demonstrate that the QC method was clinically relevant.
Another option is a “bottom-up” approach with a direct integration of the in vitro dissolution profile in a PBBM again using a Weibull function for example, to predict the impact of dissolution changes on the PK, and establish the clinical relevance of the method. This was shown for Felodipine MR tablets (71), Zolpidem hemitartrate MR tablets (83), Lithium carbonate IR and MR tablets (28), UK-369,003 MR tablets (85), Paracetamol MR tablets (83) and osmotic pump products comprising oxybutin chloride, venlafaxine HCl, carbamazepine, glipizide, nifedipine, paliperidone, metformin-HCl, pseudoephedrine-HCl (86). This approach, although non-mechanistic, relies on the principle that the dissolution method conditions chosen (volume, agitation or pH) will lead to a good representation of the in vivo release or in vivo dissolution. Indeed, without a mechanistic approach, the in vivo release will strictly match the in vitro release. The chances of success for this approach are high predominantly for BCS 1 or BCS 1-like drugs, i.e. when there is no solubility limitation to absorption, since typically the volumes of fluids used in vitro are much higher than the ones in the gastro-intestinal tract (15, 87, 88). In addition, the Weibull approach can be successful when the release rate from the dosage form will limit absorption, which is typically the case for modified release formulations, or immediate release formulations that release slower than the gastric emptying rate in vivo. Despite the lack of mechanistic dissolution, these models are useful since they can still feature mechanistic models for post release processes such as dissolution (if not already governed by the Weibull function), precipitation, influx or efflux transporters or first pass gut extraction. For all these processes, the varying luminal conditions and transit along the GI tract would still apply. For example, the model for Diltiazem-HCl MR formulations, even using a Weibull function to determine in vivo dissolution, was able to reproduce the higher exposure of females compared to males in the clinical study at equivalent body weight due to the prolonged residence time of the dosage form in the large intestine of females compared to males (83, 89). Similarly, the use of a Weibull function for modified release formulations of BCS class 3 drug MK-0941 in combination with absorption windows for the drug along the GI tract, improved the prediction of observed human oral PK data for these formulations compared to a classical IVIVC (90). For osmotic pumps, adjustment of colonic permeability was found needed by Ni et al. for venlafaxine-HCl (BCS 1) and pseudoephedrine-HCl (BCS 3) to match observed PK profiles for the MR formulations whilst for paliperodone (BCS 2), the GI transit time was increased to 24 h and all absorption scaling factors needed to be adjusted along the GI tract to match the observed profile (86). The need to adjust the absorption parameters in the gastro-intestinal model of PBBMs in certain cases to match the absorption of modified release formulations could point to currently inadequate models for drug colonic permeability and a need for improvement (91). Interestingly, for Felodipine ER tablets (71), Zolpidem hemitartrate MR tablets (83, 92), MK-0941 mesylate MR tablets and pellets (90), or BMS-663068 MR tablets (62, 93), the clinical relevant dissolution method that was used in conjunction with a Weibull function to predict in vivo performance, was performed in USP 2(paddle) at 100 rpm. This level of agitation in USP 2(paddle) corresponds to average fluid velocities of around 10-17 cm/s (79, 94). In 8 healthy volunteers, Worsoe et al. measured that the top velocity of magnetic labeled formulations in the upper intestine 2 h after pylorus passage was ranging from 0.3 to 0.6 cm/s and was only reached 3% of the 2 h exploration time. In addition, the average velocity over 2 h exploration was of 0.25 mm/s irrespective of prandial state (95). The fact that clinical relevance is achieved for MR matrix formulation with such large agitation rates in USP 2 apparatus compared the biorelevant fluid velocities, may be related to the different nature of stress generated while using USP 2 in vitro as compared to in vivo. In vivo unilateral compression forces exerted by the gastro-intestinal tract which can be measured by Smartpills® or manometers are ubiquitous along the GI tract (96), and especially strong in the colon at specific times during the day such as wake up and post prandial phase (97). For the case of darifenacin-HBr (83), or felodipine (71) extended release gelling matrix tablets, multiple absorption phases are observed when the dosage form is present in the colon at times coinciding with subject food intake, which could be related to the effect of colonic pressure waves on the drug release from the matrix. The inclusion of data resulting from more complex dissolution tools such as the USP 3 apparatus (98), GastroDuo (99), or TIM-1 model (100, 101), which exert a controlled compression on the dosage form, could improve the prediction of the impact of pressure on drug release from modified release products and explain within and between subject variability observed in the PK for these dosage forms, provided the PBBM can simulate this effect as well.
For immediate release BCS class 2 or 4 drug products, a non-mechanistic way to integrate dissolution in PBBM is likely to not succeed and the use of mechanistic models for dissolution is recommended. If the BCS class 2 drug is formulated as an MR drug, depending on the release mechanism, the use of a Weibull function which controls the release but not the drug dissolution may be advisable. Based on the analysis of the case studies reported in Table 1 for immediate release products and separating them highly or low BCS solubility classes, it appears that modellers have preferred more mechanistic models for low solubility products (Fig. 6).

Options selected for introduction of IR product dissolution in PBBM by BCS solubility class (See Table 1, Supplemental material).
For IR BCS class 2 drugs, which are able to change their ionization or bind to micelles, and therefore increase their solubility when moving from the stomach to the intestine, there may be additional recommendations for model building to consider. For example, a BCS class 2 weak acid, poorly soluble in the stomach may be able to dissolve in the intestine and behave as a BCS 1 like product for its absorption. For weak acid drugs with low stomach solubility, a significant proportion of individual PK profiles will show lag times and multiple peaking, which signal that a portion of the solid drug is retained in the stomach and that the stomach empties in multiple phases (102). In this case, the absorption may be complete but the PK profile will be impacted by the gastric emptying profile as was shown for lesinurad (53), naproxen (63), ibuprofen (102), and other NSAIDs (103). In this situation, a mechanistic model for integrating dissolution in a PBBM would still allow the development of a safe space for product dissolution, provided that the gastric emptying is accounted for in the model. Average PK profiles are not a good representation of a multiphasic gastric emptying (63) and the building of a representative population for which gastric emptying is made of multiple phases is sometimes needed to account for the impact on Cmax (53). Recently, Komasaka et al. have shown a way to integrate acid delayed dissolution of raltegravir potassium tablets in a PBBM, which could be used to run sensitivity analyses on the impact of within and between subject variability in gastric emptying on the in vivo PK (104).
This phenomenon of partial gastric emptying although more pronounced for drugs that are poorly-soluble in the stomach, has also been reported for a variety of compounds including high solubility products like cimetidine (105), zolpidem hemitartrate (106). For BCS class 2 weak basic drugs solubilized in the stomach when its pH is low, gastric emptying and gastric retention may also be a determining factor for Cmax control and presence of lag phases in the PK profiles, as shown for acalabrutinib capsules, were the co-administration of a Smartpill® allowed to link for certain subjects the end of observed lag time in PK profile, to flushing of the stomach following fluid ingestion by the subject (107).
Similarly, modified release formulations which are sensitive to pressure or increased hydrodynamics for drug release, will also show lag times and multiple phases in the absorption profile depending on the localization of the drug product in the stomach (108, 109).
Best Practices and Hurdles Towards Building and Validation of PBBM Models
Some best practices have started to be defined regarding the different stages of model building, verification, validation and use of PBBM but there are several aspects which need to be addressed to allow for a wider and broader utilization of these models (110). The main issues reported so far (and solutions proposed) are around the handling of uncertainty in the PBBM input parameters, the establishing of criteria for model verification and validation, the characterization of within and between subject variability, when and how to optimize an input parameter or a system parameter.
Models can be used in a “bottom up”, “top down” or “middle out” approach using human data as input to PBBM as they become available during development, to refine or scale in vitro inputs during model verification (106, 111, 112).
The type of questions that PBBM can answer in a regulatory submission need to be defined as science and model progresses in the future. The current US-FDA draft guidance (24), provides a framework for the application of PBBM in the field of product quality to support CBA specification setting, safe space definition or changes pre- or post-approval. A scientific discussion is now needed amongst regulators, academia and pharmaceutical industry to understand the requirements on number and design of clinical studies that can support the validation of those models, the type of formulation and process variants which should be tested clinically and to refine acceptance criteria for PBBMs validation and application. To this effect, the IQ consortium (113) provided comments to the FDA on the draft guidance adapting the concept proposed by Kuemmel et al. on model influence and model risk (114). The model influence considers the contribution of the model to the overall decision, with the highest influence when the model is considered alone and a lower influence if supportive clinical or preclinical evidence exist to support the decision. The model risk is ranking the possibility of a negative outcome should the decision be incorrect based on model predictions. Both these grids could be used to proposed adapted acceptance criteria for model validation and model use depending on the question that the model is aimed to inform. In addition, factors such as the within subject variability and the presence of a large therapeutic index, may be used to apply reference-scaled average bioequivalence approach for conducting virtual bioequivalence (115). Finally, the existence of PK-PD and PK-Tox relationships may be used to define a safe space for a product beyond the strict bioequivalence limits. (92); however, a more thorough scientific discussion is needed on all the above stated aspects of the PBBM.
One of the main hurdles to the rapid adoption of PBBM by the pharmaceutical industry, is the need to harmonize the regulatory acceptation of PBBMs in order to waive human evaluation in setting drug product CBA specifications and associated safe spaces. As mentioned previously, only the US FDA has proposed a draft guidance on PBBM as of today, and a quick ICH on this topic is highly desired, as most pharmaceutical companies operate worldwide. The objectives of PBBM are to reduce unnecessary human testing, and to provide a quantitative and mechanistic understanding of what limits drug absorption in humans based on the drug substance properties and drug product performance. If these objectives are to be realized, the adoption of PBBM as a standard tool will need to be generalized and widely accepted. To this effect, an intensive scientific collaboration between regulatory agencies worldwide, software developers, pharmaceutical companies and academics is needed to ensure that the merits of PBBM are recognized and that these models are appropriately used to support product development and patient access to high quality drug products (116).
PBBM as a Game Changer
Drug development is a costly and sometimes unsuccessful, according to Wong et al. over the period 2000-2015, with an overall probability of success of 13.8% from phase 1 to approval, and a probability of success of 59% from phase 3 to approval. (117). Some of the reasons behind the failures are reported to be an improper dose selection, non-optimal assessment schedules, inappropriate efficacy metrics/markers, and more generally the lack of understanding of how the investigational product interacts with the human body. Another aspect to consider in these clinical failures is the application of the old paradigm of “one size fits all” for the clinical evaluation of drug products, which creates variability in exposure, efficacy and toxicity in a given population due to the inherent variability across (and within) subjects in terms of body composition, physiological functions following disease evolution and proteome.
With the advance of medical imaging to quantify body composition and physiology, and liquid biopsies (118) to quantify the expression level of various proteins in the body including enzymes responsible of drug metabolism or the proteins which are the target of pharmacological or toxicological action, PBBMs could be used in the future to set up individual patient virtual twins (119), in order to calculate for each patient, the dose/release rate and scheduling requirements for optimal treatment. These PBBMs could be used in combination with smart manufacturing technologies or smart delivery devices to achieve the potential of model informed precision dosing for each patient (120, 121).
FUTURE IMPROVEMENTS OF PBBMS
Average Values or Adequate Variation for System Parameters
As was reported in a previous workshop, agreement on the physiological parameters of the human GI tract is still a topic for discussion. Most of the average system parameters are well described (122), however research is now focused on understanding variability in these parameters within and between subjects (123). In addition, efforts are being made to understand whether the best simulation strategy for physiological parameters is to adopt an average constant value or a time dependent value and some randomization within observed ranges (96, 102, 124).
Since the local transient conditions of pH, osmolarity and bile salt are eminently variable between or within subjects in a given prandial state (123, 125), the analysis of average PK study results for drugs that are sensitive to these parameters may be impaired by the between and within subject variability. Recently an evaluation of NSAID PK profiles has shown that is difficult to calculate in vivo dissolution from PK profiles in the absence of a separate and independent measurement of gastric emptying (103). In this respect, a well-defined PBBM could be used to run sensitivity analyses and identify the key variable of human exposure for a given drug formulation. This may be a physiological value such as pH, bile salt, volume or gastric emptying. In this case, one interesting strategy would be to co-administer the drug with a biomarker of interest which could then be used to inform individual model and reduce the clinical variability to improve the in vitro in vivo correlation. Bermejo et al. have shown how individual level patient data, on luminal pH and gastric emptying, can help to improve predictions of the pharmacokinetics of ibuprofen and predict relevant in vivo dissolution. This work illustrates the importance of pH and gastric emptying phases to control the PK (102). In most situations that level of information is absent for individual subjects in a given clinical trial but the establishment of ranges of variation in gastric emptying and types of gastric emptying profiles in the fasted and fed state, can be re-used to inform how system parameters should vary in a virtual BE for other compounds (53). Clinical studies utilizing biomarkers are therefore invaluable to increase the relevance of PBBM system parameters and run VBEs with the right level of variation within and between subjects.
Volume for Dissolution
A number of authors (53, 59, 61, 71, 83, 88, 102, 112, 126) have proposed a reduction of the default compartment fluid volumes in the GI tract following the observations made by Schiller et al. (87) and confirmed by Mudie et al. (15). In addition, the volume in the intestinal compartments should be a function of the fluid osmolarity (127, 128) and osmotic agent digestibility (129). However, the role of the mucus layer for all dosage forms and nanoparticulates (112) should be clarified, since it could affect the volume available for drug dilution after dissolution or the transit of solid particulates (130). At the moment the Simcyp (from v18) simulator allows to use the unstirred water boundary layer as a space for particulate dissolution. One could question whether this should remain a model option or be the default situation, since mucus can represent, as it is the case in the colon, the majority of the water available for dissolution (112). A systematic evaluation of the impact of mucus on permeation and dissolution should be conducted for drugs and particulates and changes be made if needed to the models.
Gastric Emptying
Current models for fasted gastric emptying predict single phase first or zero order emptying immediately happening or following the IMMC cycle. Individual PK data in the fasted state for a variety of drugs (IR or MR) show lag phases and multiple peaking phenomena which can be related to gastric retention and partial stomach emptying (53, 103, 105, 106, 109, 131). A stomach model which would differentiate the liquid phase emptying from the solid phase emptying (132,133,134) and allow drug products and particles sediment or float according to their density and wettability (135) would improve predictions of gastric retention and gastric emptying and is a pre-requisite to predicting between and within subject variability in gastric emptying. The fed state is believed to be less variable than the fasted state, however recent insights around the presence of the Magenstrasse, through which water dosed after a meal, goes around the viscous chyme and exist the stomach in matter of seconds, can lead, depending on the disintegration rate, density and size of the drug product, to very different mixing situations: the dosage form may sit on top of the stomach within the fat of the meal, mix with the liquid phase of the fed stomach contents or empty very fast with the administration fluid (135,136,137).
Health, Disease, Drugs and Excipient Effects
Patient centric development should be enabled by the use of PBBM and some of the changes to the human physiology brought by a disease, surgical procedures or co-administration with drugs or excipients which may impact the GI physiology should be integrated in the modeling strategy depending on the type of populations who will receive the drug product. To give a few examples, the impact of diabetes on gastric emptying is well described (138, 139), bariatric surgery can be included in the PBBMs; the effect of metoclopramide (140, 141) or proton pump inhibitors (PPIs) (140,141,142) on the gastric emptying, stomach pH and volume are also understood. Polyethylene glycols (143, 144), and low digestible carbohydrates such as fructose mannitol or sorbitol (145), accelerate transit through GIT in a dose dependent way, and PBBM can be used to evaluate their impact and set acceptable limits (70).
Viscosity Effects
The viscosity of the chyme is expected in the fed state to show large variation along the GI tract and would also depend on the type of food. In the stomach, if the drug product mixes with the chyme, the impact of increased viscosity in the fed state would be a reduction of the drug dissolution which should be accounted to predict the impact of food on PK (146, 147).
Tablet Disintegration
Integration of disintegration models in PBBM and the impact of GI fluid composition could also help with the simulation of formulation and food effects (147). Current modeling efforts show that a mechanistic disintegration model could be available soon, where factors such as tablet hardness, porosity, wettability and liquid viscosity can be used together with some excipient information such as type and amount of disintegrants and their swelling capacity. This could help predict the formulation disintegration time, which may in certain cases control the in vivo release of the drug (148).
Integration of Dissolution
Not enough PBBM applications show how they apply in vitro dissolution as a mechanistic input, nor show a validation of the in vitro input prior to integration in the PBBM. Ideally, the integration of dissolution should always be mechanistic for an immediate release formulation, the parameter used (P-PSD, Z-factor or DLM scaling factor) should be batch specific and allow to explain all dissolution data in multiple media. The use of pH dependent Z-factors or ranges of DLM scaling factors depending on the dissolution method chosen for a given batch, should be justified and alternatives explored. It could be that the drug wettability varies with pH (76) or dissolution medium composition (149), or that the surface pH and surface solubility was ignored, leading to a failure of mechanistic models to be good representatives of batch dissolution in all tested conditions. In addition, the mechanistic dissolution models should further evolve to capture the effect of hydrodynamics or sedimentation on dissolution (79). The examples gathered in this review illustrate a single clinically relevant method can be determined and used to assess formulation or process changes in a regulatory context (Table 1).
CONCLUDING REMARKS AND FUTURE OUTLOOK
As shown in this review, the concept of PBBM and its application to support drug product quality is a relatively new concept. However, the understanding of human gut physiology across age, health and disease and how it varies with time, drug or excipient exposure hold the promise to use PBBMs to support more patient centric evaluations in the future and waive more burdensome clinical trials.
From an industrial point of view, the use of PBBM to support product quality applications is likely to gain more importance in near future; and to unlock the benefits of these approaches in terms of defining safe space and reduction of unnecessary human testing, a harmonization of regulatory standards and modeling best practices is needed. There are still a lot of scientific elements to be addressed around these aspects and an intense scientific collaboration between pharmaceutical industry, worldwide regulators, software providers and academia is needed. Education of modelers on best practices and biopharmaceutics is also necessary to support this ambition and deliver high quality PBBMs.
In the future, there is a scope to use PBBM to support drug-drug interactions or special population modeling, blurring the current separation between PBPK and PBBMs. As an example, predicting the impact of pH related DDI or prandial state on drug exposure should be conducted using relevant batch level information and the use of non-mechanistic absorption models should go down. PBBMs built with clinical data could then be used to verify that the range of commercial products that patients will be prescribed will meet the same standards of quality and safety than those used during pivotal clinical trials (59, 61, 150). In addition, PBBM could also integrate PK-PD or PK-Tox models to allow biopharmaceutics, pharmacokinetics and pharmacometrics to join forces.
PBBMs are not only for oral routes and should be developed for other important drug product administration routes. Subcutaneous and lung administration cover a growing number of administration routes for new drug modalities such as peptides, antibody-drug conjugates or anti-sense oligonucleotides. PBBMs could be used in the future to assessment of the impact of excipients, drug concentration, administration vehicle or device to support the quality aspects of these products and drug formulations.
Finally, the mechanistic understanding of product-patient interactions and the level of variability in individual human physiology also can lead the path to precision dosing, where PBBMs could become virtual twins for individual patients to calculate dose, administration schedule or release rate requirements for each one of us based on relevant biomarkers of our physiology and proteome. PBBMs hold many promises for patients, regulators and the pharmaceutical industry. Now is the time to deliver on these !
References
Wagner JG. Biopharmaceutics: Gastrointestinal Absorption Aspects. Antibiot Chemother. 1964;12:53–84.
Wagner JG. Biopharmaceutics: absorption aspects. J Pharm Sci. 1961;50:359–87.
Ho NFH, Merkle HP, Higuchi WI. Quantitative, mechanistic and physiologically realistic approach to the biopharmaceutical design of oral drug delivery systems. Drug Dev Ind Pharm. 1983;9(7):1111–84.
Dressman JB, Fleisher D, Amidon GL. Physicochemical model for dose-dependent drug absorption. J Pharm Sci. 1984;73(9):1274–9.
Dressman JB, Fleisher D. Mixing-tank model for predicting dissolution rate control or oral absorption. J Pharm Sci. 1986;75(2):109–16.
Sinko PJ, Leesman GD, Amidon GL. Predicting fraction dose absorbed in humans using a macroscopic mass balance approach. Pharm Res. 1991;8(8):979–88.
Yu LX, Crison JR, Amidon GL. Compartmental transit and dispersion model analysis of small intestinal transit flow in humans. Int J Pharm. 1996;140(1):111–8.
Yu LX, Lipka E, Crison JR, Amidon GL. Transport approaches to the biopharmaceutical design of oral drug delivery systems: prediction of intestinal absorption. Adv Drug Deliv Rev. 1996;19(3):359–76.
Yu LX, Amidon GL. Characterization of small intestinal transit time distribution in humans. Int J Pharm. 1998;171(2):157–63.
Yu LX, Amidon GL. A compartmental absorption and transit model for estimating oral drug absorption. Int J Pharm. 1999;186(2):119–25.
Agoram B, Woltosz WS, Bolger MB. Predicting the impact of physiological and biochemical processes on oral drug bioavailability. Adv Drug Deliv Rev. 2001;50(Suppl 1):S41-67.
Jamei M. Recent Advances in Development and Application of Physiologically-Based Pharmacokinetic (PBPK) Models: a Transition from Academic Curiosity to Regulatory Acceptance. Curr Pharmacol Rep. 2016;2:161–9.
Stamatis SD, Rose JP. Lilly Absorption Modeling Platform: A Tool for Early Absorption Assessment. Mol Pharm. 2022;19(1):213–26.
Kostewicz ES, Aarons L, Bergstrand M, Bolger MB, Galetin A, Hatley O, et al. PBPK models for the prediction of in vivo performance of oral dosage forms. Eur J Pharm Sci. 2014;57:300–21.
Mudie DM, Murray K, Hoad CL, Pritchard SE, Garnett MC, Amidon GL, et al. Quantification of gastrointestinal liquid volumes and distribution following a 240 mL dose of water in the fasted state. Mol Pharm. 2014;11(9):3039–47.
Hens B, Bolger MB. Application of a Dynamic Fluid and pH Model to Simulate Intraluminal and Systemic Concentrations of a Weak Base in GastroPlus(). J Pharm Sci. 2019;108(1):305–15.
Zhang X, Lionberger RA, Davit BM, Yu LX. Utility of physiologically based absorption modeling in implementing Quality by Design in drug development. AAPS J. 2011;13(1):59–71.
Willmann S, Schmitt W, Keldenich J, Lippert J, Dressman JB. A physiological model for the estimation of the fraction dose absorbed in humans. J Med Chem. 2004;47(16):4022–31.
Abend A, Heimbach T, Cohen M, Kesisoglou F, Pepin X, Suarez-Sharp S. Dissolution and Translational Modeling Strategies Enabling Patient-Centric Drug Product Development: the M-CERSI Workshop Summary Report. The AAPS journal. 2018;20(3).
Pepin XJH, Parrott N, Dressman J, Delvadia P, Mitra A, Zhang X, et al. Current State and Future Expectations of Translational Modeling Strategies to Support Drug Product Development, Manufacturing Changes and Controls: A Workshop Summary Report. J Pharm Sci. 2021;110:555–66.
Mitra A, Suarez-Sharp S, Pepin XJH, Flanagan T, Zhao Y, Kotzagiorgis E, et al. Applications of Physiologically Based Biopharmaceutics Modeling (PBBM) to Support Drug Product Quality: A Workshop Summary Report. J Pharm Sci. 2021;110(2):594–609.
Hens B, Sinko PD, Job N, Dean M, Al-Gousous J, Salehi N, et al. Formulation predictive dissolution (fPD) testing to advance oral drug product development: An introduction to the US FDA funded '21st Century BA/BE' project. Int J Pharm. 2018;548(1):120–7.
Zhao P, Zhang L, Grillo JA, Liu Q, Bullock JM, Moon YJ, et al. Applications of physiologically based pharmacokinetic (PBPK) modeling and simulation during regulatory review. Clin Pharmacol Ther. 2011;89(2):259–67.
U. S. Food and Drug Administration. Draft Guidance for Industry: The Use of Physiologically Based Pharmacokinetic Analyses — Biopharmaceutics Applications for Oral Drug Product Development, Manufacturing Changes, and Controls. 2020. Available from: https://www.fda.gov/media/142500/download. Accessed 15 Dec 2021.
Tsakalozou E, Alam K, Babiskin A, Zhao L. Physiologically-Based Pharmacokinetic Modeling to Support Determination of Bioequivalence for Dermatological Drug Products: Scientific and Regulatory Considerations. Clin Pharmacol Ther. 2021.
The Center for Research on Complex Generics (CRCG): The University of Maryland, Baltimore and University of Michigan. 2021. Available from: http://www.complexgenerics.org/PBPK2021/.
Suarez-Sharp S, Cohen M, Kesisoglou F, Abend A, Marroum P, Delvadia P, et al. Applications of Clinically Relevant Dissolution Testing: Workshop Summary Report. AAPS J. 2018;20(6):93.
Heimbach T, Suarez-Sharp S, Kakhi M, Holmstock N, Olivares-Morales A, Pepin X, et al. Dissolution and Translational Modeling Strategies Toward Establishing an In Vitro-In Vivo Link—a Workshop Summary Report. The AAPS Journal. 2019;21(2).
U.S FDA CDER. CDER patient-focused drug development. [Available from: https://www.fda.gov/drugs/development-approval-process-drugs/cder-patient-focused-drug-development].
J W. The concept of pharmaceutical quality. American Pharmaceutical Review. 2004;7(6):10–5.
Yu LX, Amidon G, Khan MA, Hoag SW, Polli J, Raju GK, et al. Understanding pharmaceutical quality by design. AAPS J. 2014;16(4):771–83.
Anand O. Clinically Relevant Dissolution Specifications a Biopharmaceutics’ Risk Based Approach: an FDA perspective: The Academy of Pharmaceutical Sciences; 2021 [Available from: https://www.apsgb.co.uk/wp-content/uploads/2021/05/Clinically-Relevant-Dissolution-Specifications-an-FDA-Perspective-__Om-Anand.pdf.
Raines K. PBPK Biopharmaceutics Guidance and progress on Risk Assessment 2021 [Available from: http://www.complexgenerics.org/media/SOP/complexgenerics/pdf/Conference-Slides/D2-04%20Kimberly%20Raines_PBPKGuidanceRiskAssessment.pdf
Parrott N, Hainzl D, Scheubel E, Krimmer S, Boetsch C, Guerini E, et al. Physiologically Based Absorption Modelling to Predict the Impact of Drug Properties on Pharmacokinetics of Bitopertin. AAPS J. 2014;16(5):1077–84.
Paul S. Challenges and strategies in establishing an in-vitro invivo link, in Dissolution and translational modeling strategies enabling patient centric product development. 2017.
U. S. Food and Drug Administration. Guidance for Industry: Extended Release Oral Dosage Forms: Development, Evaluation, and Application of In Vitro/In Vivo Correlations. 1997. Available at: https://www.fda.gov/media/70939/download. Accessed 15 Dec 2021.
Wu F, Shah H, Li M, Duan P, Zhao P, Suarez S, et al. Biopharmaceutics Applications of Physiologically Based Pharmacokinetic Absorption Modeling and Simulation in Regulatory Submissions to the US Food and Drug Administration for New Drugs. AAPS J. 2021;23(2):1–14.
Heimbach T, Kesisoglou F, Novakovic J, Tistaert C, Mueller-Zsigmondy M, Kollipara S, et al. Establishing the Bioequivalence Safe Space for Immediate-Release Oral Dosage Forms using Physiologically Based Biopharmaceutics Modeling (PBBM): Case Studies. J Pharm Sci. 2021;110(12):3896–906.
Kato T, Nakagawa H, Mikkaichi T, Miyano T, Matsumoto Y, Ando S. Establishment of a clinically relevant specification for dissolution testing using physiologically based pharmacokinetic (PBPK) modeling approaches. Eur J Pharm Biopharm. 2020;151:45–52.
Zhao Y SS. FDA expectations in building a safe space to gain regulatory flexibility based on PBBM. 2019. Available from: https://cersi.umd.edu/sites/cersi.umd.edu/files/Day%203-1%20Zhao%20Suarez%20LM.pdf.
Tistaert C. Case Study: Bridging physiology-based dissolution testing to quality control testing using PBBM University of Maryland, College Park, MD2019 [Available from: https://cersi.umd.edu/file/day-3-5-christophe-tistaert-finalpdf.
Fan J, Zhang X, Zhao L. Utility of Physiologically Based Pharmacokinetic Absorption Modeling to Predict the Impact of Salt-to-Base Conversion on Prasugrel HCl Product Bioequivalence in the Presence of Proton Pump Inhibitors. AAPS J. 2017;19(5):1479–86.
Butler J, Hens B, Vertzoni M, Brouwers J, Berben P, Dressman J, et al. In vitro models for the prediction of in vivo performance of oral dosage forms: Recent progress from partnership through the IMI OrBiTo collaboration. Eur J Pharm Biopharm. 2019;136:70–83.
Taylor LS, Zhang GGZ. Physical chemistry of supersaturated solutions and implications for oral absorption. Adv Drug Deliv Rev. 2016;101:122–42.
Heimbach T, Kesisoglou F, Novakovic J, Tistaert C, Mueller-Zsigmondy M, Kollipara S, et al. Establishing the Bioequivalence Safe Space for Immediate-Release Oral Dosage Forms using Physiologically Based Biopharmaceutics Modeling (PBBM): Case Studies. Journal of Pharmaceutical Sciences. 2021.
Parrott N, Suarez-Sharp S, Kesisoglou F, Pathak SM, Good D, Wagner C, et al. Best Practices in the Development and Validation of Physiologically Based Biopharmaceutics Modeling. A Workshop Summary Report. J Pharm Sci. 2021;110(2):584–93.
Delvadia PR, Barr WH, Karnes HT. A biorelevant in vitro release/permeation system for oral transmucosal dosage forms. Int J Pharm. 2012;430(1–2):104–13.
Willmann S, Thelen K, Lippert J. Integration of dissolution into physiologically-based pharmacokinetic models III: PK-Sim(R). J Pharm Pharmacol. 2012;64(7):997–1007.
Elagolix Tablets, NDA 210450 Food and Drug Administration. 2018. [Available from: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2018/210450Orig1s000ChemR.pdf].
Miao L, Mousa YM, Zhao L, Raines K, Seo P, Wu F. Using a Physiologically Based Pharmacokinetic Absorption Model to Establish Dissolution Bioequivalence Safe Space for Oseltamivir in Adult and Pediatric Populations. AAPS J. 2020;22(5):107.
Suarez-Sharp S, Li M, Duan J, Shah H, Seo P. Regulatory Experience with In Vivo In Vitro Correlations (IVIVC) in New Drug Applications. AAPS J. 2016;18(6):1379–90.
Triclabendazole Tablets, NDA 208711, Food and Drug Administration. 2018. [Available from: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2018/208711Orig1s000ChemR.pdf].
Pepin XJ, Flanagan TR, Holt DJ, Eidelman A, Treacy D, Rowlings CE. Justification of Drug Product Dissolution Rate and Drug Substance Particle Size Specifications Based on Absorption PBPK Modeling for Lesinurad Immediate Release Tablets. Mol Pharm. 2016;13(9):3256–69.
Duvelisib Capsules, NDA211155, Food and Drug Administration. 2018. [Available from: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2018/211155Orig1Orig2s000ChemR.pdf].
Kesisoglou F, Wang M, Galipeau K, Harmon P, Okoh G, Xu W. Effect of Amorphous Nanoparticle Size on Bioavailability of Anacetrapib in Dogs. J Pharm Sci. 2019;108(9):2917–25.
Đuranović M, Madžarević M, Ivković B, Ibrić S, Cvijić S. The evaluation of the effect of different superdisintegrants on the drug release from FDM 3D printed tablets through different applied strategies: In vitro-in silico assessment. Int J Pharm. 2021;610.
Cámara-Martinez I, Blechar JA, Ruiz-Picazo A, Garcia-Arieta A, Calandria C, Merino-Sanjuan V, et al. Level A IVIVC for immediate release tablets confirms in vivo predictive dissolution testing for ibuprofen. Int J Pharm. 2022;614.
Kesisoglou F, Mitra A. Application of Absorption Modeling in Rational Design of Drug Product Under Quality-by-Design Paradigm. AAPS J. 2015;17(5):1224–36.
Pepin XJH, Moir AJ, Mann JC, Sanderson NJ, Barker R, Meehan E, et al. Bridging in vitro dissolution and in vivo exposure for acalabrutinib. Part II. A mechanistic PBPK model for IR formulation comparison, proton pump inhibitor drug interactions, and administration with acidic juices. European journal of pharmaceutics and biopharmaceutics. 2019;142:435–48.
Dickinson PA, Lee WW, Stott PW, Townsend AI, Smart JP, Ghahramani P, et al. Clinical Relevance of Dissolution Testing in Quality by Design. AAPS J. 2008;10(2):380–90.
Jereb R, Kristl A, Mitra A. Prediction of fasted and fed bioequivalence for immediate release drug products using physiologically based biopharmaceutics modeling (PBBM). Eur J Pharm Sci. 2020;155:105554.
Stillhart C, Pepin X, Tistaert C, Good D, Van Den Bergh A, Parrott N, et al. PBPK Absorption Modeling: Establishing the In Vitro–In Vivo Link—Industry Perspective. The AAPS Journal. 2019;21(2).
Loisios-Konstantinidis I, Cristofoletti R, Fotaki N, Turner DB, Dressman J. Establishing virtual bioequivalence and clinically relevant specifications using in vitro biorelevant dissolution testing and physiologically-based population pharmacokinetic modeling. case example: Naproxen. European Journal of Pharmaceutical Sciences. 2020;143:105170.
Macwan JS, Fraczkiewicz G, Bertolino M, Krüger P, Peters S-A. Application of physiologically based biopharmaceutics modeling to understand the impact of dissolution differences on in vivo performance of immediate release products: The case of bisoprolol. 2021;10(6):622-32.
Farhan N, Cristofoletti R, Basu S, Kim S, Lingineni K, Jiang S, et al. Physiologically-based pharmacokinetics modeling to investigate formulation factors influencing the generic substitution of dabigatran etexilate. 2021;10(3):199-210
García MA, Bolger MB, Suarez-Sharp S, Langguth P. Predicting pharmacokinetics of multisource acyclovir oral products through physiologically based biopharmaceutics modeling. Journal of Pharmaceutical Sciences. 2021.
Jaiswal S, Ahmed T, Kollipara S, Bhargava M, Chachad S. Development, validation and application of Physiologically Based Biopharmaceutics Model to justify the change in dissolution specifications for DRL ABC Extended release tablets. Drug Development and Industrial Pharmacy. 2021(just-accepted):1–31.
Wu X, Zhang X, Xu R, Shaik IH, Venkataramanan R. Physiologically based pharmacokinetic modelling of treprostinil after intravenous injection and extended‐release oral tablet administration in healthy volunteers: An extrapolation to other patient populations including patients with hepatic impairment. British Journal of Clinical Pharmacology. 2021.
Matsui K, Takeuchi S, Haruna Y, Yamane M, Shimizu T, Hatsuma Y, et al. Transverse comparison of mannitol content in marketed drug products: Implication for no-effect dose of sugar alcohols on oral drug absorption. Journal of Drug Delivery Science and Technology. 2020;57:101728.
Yamane M, Matsui K, Sugihara M, Tokunaga Y. The Provisional No-Effect Threshold of Sugar Alcohols on Oral Drug Absorption Estimated by Physiologically Based Biopharmaceutics Model. J Pharm Sci. 2021;110(1):467–77.
Jamei M, Abrahamsson B, Brown J, Bevernage J, Bolger MB, Heimbach T, et al. Current status and future opportunities for incorporation of dissolution data in PBPK modeling for pharmaceutical development and regulatory applications: OrBiTo consortium commentary. Eur J Pharm Biopharm. 2020;155:55–68.
Klumpp L, Dressman J. Physiologically based pharmacokinetic model outputs depend on dissolution data and their input: Case examples glibenclamide and dipyridamole. European journal of pharmaceutical sciences : official journal of the European Federation for Pharmaceutical Sciences. 2020;151:105380.
Takano R, Sugano K, Higashida A, Hayashi Y, Machida M, Aso Y, et al. Oral absorption of poorly water-soluble drugs: computer simulation of fraction absorbed in humans from a miniscale dissolution test. Pharm Res. 2006;23(6):1144–56.
Pohl P, Saparov SM, Antonenko YN. The Size of the Unstirred Layer as a Function of the Solute Diffusion Coefficient. Biophys J. 1998;75(3):1403–9.
Hofsäss MA, Dressman J. Suitability of the z-Factor for Dissolution Simulation of Solid Oral Dosage Forms: Potential Pitfalls and Refinements. J Pharm Sci. 2020;109(9):2735–45.
Pepin X, Blanchon S, Couarraze G. Powder dynamic contact angle data in the pharmaceutical industry. Pharm Sci Technolo Today. 1999;2(3):111–8.
Pathak SM, Ruff A, Kostewicz ES, Patel N, Turner DB, Jamei M. Model-Based Analysis of Biopharmaceutic Experiments To Improve Mechanistic Oral Absorption Modeling: An Integrated in Vitro in Vivo Extrapolation Perspective Using Ketoconazole as a Model Drug. Mol Pharm. 2017;14(12):4305–20.
Pepin XJH, Sanderson NJ, Blanazs A, Grover S, Ingallinera TG, Mann JC. Bridging in vitro dissolution and in vivo exposure for acalabrutinib. Part I Mechanistic modelling of drug product dissolution to derive a P-PSD for PBPK model input. European Journal of Pharmaceutics and Biopharmaceutics. 2019;142:421–34.
Pepin X, Goetschy M, Abrahmsén-Alami S. Mechanistic models for USP2 dissolution apparatus, including fluid hydrodynamics and sedimentation. Journal of Pharmaceutical Sciences. 2021.
Laisney M, Heimbach T, Mueller-Zsigmondy M, Blumenstein L, Costa R, Ji Y. Physiologically Based Biopharmaceutics Modeling to Demonstrate Virtual Bioequivalence and Bioequivalence Safe-space for Ribociclib which has Permeation Rate-controlled Absorption. Journal of Pharmaceutical Sciences. 2021.
Kesisoglou F, Chung J, van Asperen J, Heimbach T. Physiologically Based Absorption Modeling to Impact Biopharmaceutics and Formulation Strategies in Drug Development-Industry Case Studies. J Pharm Sci. 2016;105(9):2723–34.
Duan JZ. A Biopharmetrics Approach for Drug Product Quality Control with Clinical Relevance. J Pharm Sci. 2021;110(1):478–88.
Pepin X, editor Use of IVIVc and IVIVe to support formulation development - Industrial case studies. AAPS meeting - 4th November 2014; 2014; San Diego, USA.
Stillhart C, Parrott NJ, Lindenberg M, Chalus P, Bentley D, Szepes A. Characterising Drug Release from Immediate-Release Formulations of a Poorly Soluble Compound, Basmisanil, Through Absorption Modelling and Dissolution Testing. AAPS J. 2017;19(3):827–36.
Watson KJ, Davis J, Jones HM. Application of Physiologically Based Pharmacokinetic Modeling to Understanding the Clinical Pharmacokinetics of UK-369,003. Drug Metab Dispos. 2011;39(7):1203–13.
Ni Z, Talattof A, Fan J, Tsakalozou E, Sharan S, Sun D, et al. Physiologically Based Pharmacokinetic and Absorption Modeling for Osmotic Pump Products. AAPS J. 2017;19(4):1045–53.
Schiller C, Frohlich CP, Giessmann T, Siegmund W, Monnikes H, Hosten N, et al. Intestinal fluid volumes and transit of dosage forms as assessed by magnetic resonance imaging. Aliment Pharmacol Ther. 2005;22(10):971–9.
Sutton SC. Role of physiological intestinal water in oral absorption. AAPS J. 2009;11(2):277–85.
Valentin J. Basic anatomical and physiological data for use in radiological protection: reference values. Ann ICRP. 2002;32(3–4):1–277.
Kesisoglou F, Xia B, Agrawal NGB. Comparison of Deconvolution-Based and Absorption Modeling IVIVC for Extended Release Formulations of a BCS III Drug Development Candidate. AAPS J. 2015;17(6):1492–500.
Eckernäs E, Tannergren C. Physiologically Based Biopharmaceutics Modeling of Regional and Colon Absorption in Dogs. Mol Pharm. 2021;18(4):1699–710.
Paraiso RL, Rose RH, Fotaki N, McAllister M, Dressman JB. The use of PBPK/PD to establish clinically relevant dissolution specifications for zolpidem immediate release tablets. Eur J Pharm Sci. 2020;155:105534.
Brown J, Chien C, Timmins P, Dennis A, Doll W, Sandefer E, et al. Compartmental absorption modeling and site of absorption studies to determine feasibility of an extended-release formulation of an HIV-1 attachment inhibitor phosphate ester prodrug. J Pharm Sci. 2013;102(6):1742–51.
Salehi N, Al-Gousous J, Mudie DM, Amidon GL, Ziff RM, Amidon GE. Hierarchical Mass Transfer Analysis of Drug Particle Dissolution, Highlighting the Hydrodynamics, pH, Particle Size, and Buffer Effects for the Dissolution of Ionizable and Nonionizable Drugs in a Compendial Dissolution Vessel. Mol Pharm. 2020;17(10):3870–84.
Worsoe J, Fynne L, Gregersen T, Schlageter V, Christensen LA, Dahlerup JF, et al. Gastric transit and small intestinal transit time and motility assessed by a magnet tracking system. BMC Gastroenterol. 2011;11:145.
Koziolek M, Grimm M, Schneider F, Jedamzik P, Sager M, Kühn JP, et al. Navigating the human gastrointestinal tract for oral drug delivery: Uncharted waters and new frontiers. Adv Drug Deliv Rev. 2016;101:75–88.
Rao SSC, Sadeghi P, Beaty J, Kavlock R. Ambulatory 24-Hour Colonic Manometry in Slow-Transit Constipation. Am J Gastroenterol. 2004;99(12):2405–16.
Mendes TC, Simon A, Menezes JCV, Pinto EC, Cabral LM, de Sousa VP. Development of USP Apparatus 3 Dissolution Method with IVIVC for Extended Release Tablets of Metformin Hydrochloride and Development of a Generic Formulation. Chem Pharm Bull (Tokyo). 2019;67(1):23–31.
Schick P, Sager M, Wegner F, Wiedmann M, Schapperer E, Weitschies W, et al. Application of the GastroDuo as an in Vitro Dissolution Tool To Simulate the Gastric Emptying of the Postprandial Stomach. Mol Pharm. 2019;16(11):4651–60.
Hopgood M, Reynolds G, Barker R. Using Computational Fluid Dynamics to Compare Shear Rate and Turbulence in the TIM-Automated Gastric Compartment With USP Apparatus II. J Pharm Sci. 2018;107(7):1911–9.
Barker R, Abrahamsson B, Kruusmägi M. Application and Validation of an Advanced Gastrointestinal In Vitro Model for the Evaluation of Drug Product Performance in Pharmaceutical Development. J Pharm Sci. 2014;103(11):3704–12.
Bermejo M, Hens B, Dickens J, Mudie D, Paixão P, Tsume Y, et al. A Mechanistic Physiologically-Based Biopharmaceutics Modeling (PBBM) Approach to Assess the In Vivo Performance of an Orally Administered Drug Product: From IVIVC to IVIVP. Pharmaceutics. 2020;12(1).
Li M, Zhang X, Wu D, Anand O, Chen H, Raines K, et al. Understanding In Vivo Dissolution of Immediate Release (IR) Solid Oral Drug Products Containing Weak Acid BCS Class 2 (BCS Class 2a) Drugs. AAPS J. 2021;23(6):113.
Komasaka T, Dressman J. Simulation of oral absorption from non-bioequivalent dosage forms of the salt of raltegravir, a poorly soluble acidic drug, using a physiologically based biopharmaceutical modeling (PBBM) approach. Eur J Pharm Sci. 2021;157:105630.
Langguth P, Lee KM, Spahn-Langguth H, Amidon GL. Variable gastric emptying and discontinuities in drug absorption profiles: Dependence of rates and extent of cimetidine absorption on motility phase and pH. Biopharm Drug Dispos. 1994;15(9):719–46.
Andreas CJ, Pepin X, Markopoulos C, Vertzoni M, Reppas C, Dressman J. Mechanistic investigation of the negative food effect of modified release zolpidem. Eur J Pharm Sci. 2017;102:284–98.
Pepin X. The use of PBBM and biomarkers to provide detailed understanding of in vivo dissolution and absorption for Acalabrutinib University of Maryland, College Park, MD2019 [Available from: https://cersi.umd.edu/sites/cersi.umd.edu/files/Day%203%20-%206%20Xavier%20Pepin%20Final.pdf.
Weitschies W, Friedrich C, Wedemeyer RS, Schmidtmann M, Kosch O, Kinzig M, et al. Bioavailability of amoxicillin and clavulanic acid from extended release tablets depends on intragastric tablet deposition and gastric emptying. Eur J Pharm Biopharm. 2008;70(2):641–8.
Weitschies W, Wedemeyer RS, Kosch O, Fach K, Nagel S, Soderlind E, et al. Impact of the intragastric location of extended release tablets on food interactions. J Control Release. 2005;108(2–3):375–85.
Parrott N, Suarez-Sharp S, Kesisoglou F, Pathak SM, Good D, Wagner C, et al. Best Practices in the Development and Validation of Physiologically Based Biopharmaceutics Modeling. A Workshop Summary Report. Journal of Pharmaceutical Sciences. 2021;110:584–93.
Tsamandouras N, Rostami-Hodjegan A, Aarons L. Combining the ‘bottom up’ and ‘top down’ approaches in pharmacokinetic modelling: fitting PBPK models to observed clinical data. Br J Clin Pharmacol. 2013;79(1):48–55.
Pepin X, Huckle JE, Alluri RV, Basu S, Dodd S, Parrott N, et al. Understanding mechanisms of food effect and developing reliable PBPK models using a middle-out approach. AAPS J. 2021;23.
The International Consortium for Innovation and Quality in Pharmaceutical Development (IQ); https://iqconsortium.org/
Kuemmel C, Yang Y, Zhang X, Florian J, Zhu H, Tegenge M, et al. Consideration of a Credibility Assessment Framework in Model-Informed Drug Development: Potential Application to Physiologically-Based Pharmacokinetic Modeling and Simulation. CPT: Pharmacometrics & Systems Pharmacology. 2020;9(1):21–8.
Davit B, Chen M-L, Conner D, Haidar S, Kim S, Lee C, et al. Implementation of a Reference-Scaled Average Bioequivalence Approach for Highly Variable Generic Drug Products by the US Food and Drug Administration. AAPS J. 2012;14(4):915–24.
Mitra A, Suarez-Sharp S, Pepin XJH, Flanagan T, Zhao Y, Kotzagiorgis E, et al. Applications of Physiologically Based Biopharmaceutics Modeling (PBBM) to support Drug Product Quality: A Workshop Summary Report. J Pharm Sci. 2021;110:594–609.
Wong Chi H, Siah Kien WEI, Lo AW. Estimation of clinical trial success rates and related parameters. Biostatistics. 2019;20(2):273–86.
Achour B, Al-Majdoub ZM, Grybos-Gajniak A, Lea K, Kilford P, Zhang M, et al. Liquid Biopsy Enables Quantification of the Abundance and Interindividual Variability of Hepatic Enzymes and Transporters. Clin Pharmacol Ther. 2021;109(1):222–32.
Polasek TM, Rostami-Hodjegan A. Virtual Twins: Understanding the Data Required for Model-Informed Precision Dosing. Clin Pharmacol Ther. 2020;107(4):742–5.
Darwich AS, Polasek TM, Aronson JK, Ogungbenro K, Wright DFB, Achour B, et al. Model-Informed Precision Dosing: Background, Requirements, Validation, Implementation, and Forward Trajectory of Individualizing Drug Therapy. Annu Rev Pharmacol Toxicol. 2021;61(1):225–45.
Ferreira A, Lapa R, Vale N. PBPK Modeling and Simulation and Therapeutic Drug Monitoring: Possible Ways for Antibiotic Dose Adjustment. Processes. 2021;9(11):2087.
Jereb R, Opara J, Bajc A, Petek B. Evaluating the Impact of Physiological Properties of the Gastrointestinal Tract On Drug In Vivo Performance Using Physiologically Based Biopharmaceutics Modeling and Virtual Clinical Trials. J Pharm Sci. 2021;110(8):3069–81.
Riethorst D, Mols R, Duchateau G, Tack J, Brouwers J, Augustijns P. Characterization of Human Duodenal Fluids in Fasted and Fed State Conditions. J Pharm Sci. 2016;105(2):673–81.
Guiastrennec BS DP, Hansen M, Bagger JL, Lund A, Rehfeld JF, Alskar O, Vilsbøll T, Knop FK, Bergstrand M. Mechanism‐Based Modeling of Gastric Emptying Rate and Gallbladder Emptying in Response to Caloric Intake. CPT: Pharmacomet Syst Pharmacol. 2016;5(12):692–700.
Rabbie SC, Flanagan T, Martin PD, Basit AW. Inter-subject variability in intestinal drug solubility. Int J Pharm. 2015;485(1–2):229–34.
Hofsäss MA, Dressman J. Evaluation of Differences in Dosage Form Performance of Generics Using BCS-Based Biowaiver Specifications and Biopharmaceutical Modeling-Case Examples Amoxicillin and Doxycycline. J Pharm Sci. 2020;109(8):2437–53.
Gisolfi CV, Summers RW, Lambert GP, Xia T. Effect of beverage osmolality on intestinal fluid absorption during exercise. Journal of applied physiology (Bethesda, Md : 1985). 1998;85(5):1941–8.
Lambert PGL, S., Welch R, Shi, X. Combined effects of glucose and fructose on fluid absorption from hypertonic carbohydrate-electrolyte beverages. Journal of Exercise Physiology. 2008;11(2):46–55.
Grimm M, Koziolek M, Saleh M, Schneider F, Garbacz G, Kühn J-P, et al. Gastric Emptying and Small Bowel Water Content after Administration of Grapefruit Juice Compared to Water and Isocaloric Solutions of Glucose and Fructose: A Four-Way Crossover MRI Pilot Study in Healthy Subjects. Mol Pharm. 2018;15(2):548–59.
Arbós P. Influence of the surface characteristics of PVM/MA nanoparticles on their bioadhesive properties. J Control Release. 2003;89(1):19–30.
Lipka E, Lee ID, Langguth P, Spahn-Langguth H, Mutschler E, Amidon GL. Celiprolol double-peak occurrence and gastric motility: nonlinear mixed effects modeling of bioavailability data obtained in dogs. J Pharmacokinet Biopharm. 1995;23(3):267–86.
Collins PJ, Houghton LA, Read NW, Horowitz M, Chatterton BE, Heddle R, et al. Role of the proximal and distal stomach in mixed solid and liquid meal emptying. Gut. 1991;32(6):615–9.
Anjiki H, Sanaka M, Kuyama Y. Dual effects of rabeprazole on solid-phase gastric emptying assessed by the 13C-octanoate breath test. Digestion. 2005;72(2–3):189–94.
Sager M, Jedamzik P, Merdivan S, Grimm M, Schneider F, Kromrey M-L, et al. Low dose caffeine as a salivary tracer for the determination of gastric water emptying in fed and fasted state: A MRI validation study. Eur J Pharm Biopharm. 2018;127:443–52.
Meyer JH, Dressman J, Fink A, Amidon G. Effect of size and density on canine gastric emptying of nondigestible solids. Gastroenterology. 1985;89(4):805–13.
Grimm M, Scholz E, Koziolek M, Kuhn JP, Weitschies W. Gastric Water Emptying under Fed State Clinical Trial Conditions Is as Fast as under Fasted Conditions. Mol Pharm. 2017;14(12):4262–71.
Locatelli I, Mrhar A, Bogataj M. Gastric Emptying of Pellets under Fasting Conditions: A Mathematical Model. Pharm Res. 2009;26(7):1607–17.
Alskar OB J, Røge RM, Knop FK, Karlsson MO, Vilsbøll T, Kjellsson MC. Semimechanistic model describing gastric emptying and glucose absorption in healthy subjects and patients with type 2 diabetes. J Clin Pharmacol. 2016;56(3):340–8.
Cucchiara S, Franzese A, Salvia G, Alfonsi L, Iula VD, Montisci A, et al. Gastric Emptying Delay and Gastric Electrical Derangement in IDDM. Diabetes Care. 1998;21(3):438–43.
Regårdh CG, Lundborg P, Persson BA. The effect of antacid, metoclopramide, and propantheline on the bioavailability of metoprolol and atenolol. Biopharm Drug Dispos. 1981;2(1):79–87.
Marathe PH, Wen Y, Norton J, Greene DS, Barbhaiya RH, Wilding IR. Effect of altered gastric emptying and gastrointestinal motility on metformin absorption. Br J Clin Pharmacol. 2000;50(4):325–32.
de Waal T, Rubbens J, Grimm M, Vandecaveye V, Tack J, Weitschies W, et al. Exploring the Effect of Esomeprazole on Gastric and Duodenal Fluid Volumes and Absorption of Ritonavir. Pharmaceutics. 2020;12(7).
Schulze JDR, Waddington WA, Ell PJ, Parsons GE, Coffin MD, Basit AW. Concentration-Dependent Effects of Polyethylene Glycol 400 on Gastrointestinal Transit and Drug Absorption. Pharm Res. 2003;20(12):1984–8.
Basit AW, Newton JM, Short MD, Waddington WA, Ell PJ, Lacey LF. The Effect of Polyethylene Glycol 400 on Gastrointestinal Transit: Implications for the Formulation of Poorly-Water Soluble Drugs. Pharm Res. 2001;18(8):1146–50.
Adkin D, Davis S, Sparrow R, Huckle P, Phillips A, Wilding I. The Effect of Different Concentrations of Mannitol in Solution on Small Intestinal Transit: Implications for Drug Absorption. Pharm Res. 1995;12(3):393–6.
Cvijić S, Parojčić J, Langguth P. Viscosity-mediated negative food effect on oral absorption of poorly-permeable drugs with an absorption window in the proximal intestine: In vitro experimental simulation and computational verification. Eur J Pharm Sci. 2014;61:40–53.
Radwan A, Amidon GL, Langguth P. Mechanistic investigation of food effect on disintegration and dissolution of BCS class III compound solid formulations: the importance of viscosity. Biopharm Drug Dispos. 2012;33(7):403–16.
Markl D, Zeitler JA. A Review of Disintegration Mechanisms and Measurement Techniques. Pharm Res. 2017;34(5):890–917.
Hörter D, Dressman JB. Influence of physicochemical properties on dissolution of drugs in the gastrointestinal tract. Adv Drug Deliv Rev. 1997;25:3–14.
Mitra A, Parrott N, Miller N, Lloyd R, Tistaert C, Heimbach T, et al. Prediction of pH-dependent drug-drug interactions for basic drugs using physiologically based biopharmaceutics modeling: industry case studies. J Pharm Sci. 2020;109(3):1380–94.
ACKNOWLEDGMENTS AND DISCLOSURES
The authors have no conflicts to declare.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Disclaimer
This article reflects the views of the authors and should not be construed to represent FDA’s views or policies.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Anand, O., Pepin, X.J.H., Kolhatkar, V. et al. The Use of Physiologically Based Pharmacokinetic Analyses—in Biopharmaceutics Applications -Regulatory and Industry Perspectives. Pharm Res 39, 1681–1700 (2022). https://doi.org/10.1007/s11095-022-03280-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11095-022-03280-4