
Abstract
Purpose
H102, a novel β-sheet breaker peptide, was encapsulated into liposomes to reduce its degradation and increase its brain penetration through intranasal administration for the treatment of Alzheimer’s disease (AD).
Methods
The H102 liposomes were prepared using a modified thin film hydration method, and their transport characteristics were tested on Calu-3 cell monolayers. The pharmacokinetics in rats’ blood and brains were also investigated. Behavioral experiments were performed to evaluate the improvements on AD rats’ spatial memory impairment. The neuroprotective effects were tested by detecting acetylcholinesterase (AchE), choline acetyltransferase (ChAT) and insulin degrading enzyme (IDE) activity and conducting histological assays. The safety was evaluated on rats’ nasal mucosa and cilia.
Results
The liposomes prepared could penetrate Calu-3 cell monolayers consistently. After intranasal administration, H102 could be effectively delivered to the brain, and the AUC of H102 liposomes in the hippocampus was 2.92-fold larger than that of solution group. H102 liposomes could excellently ameliorate spatial memory impairment of AD model rats, increase the activities of ChAT and IDE and inhibit plaque deposition, even in a lower dosage compared with H102 intranasal solution. H102 nasal formulations showed no toxicity on nasal mucosa.
Conclusions
The H102-loaded liposome prepared in this study for nasal administration is stable, effective and safe, which has great potential for AD treatment.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Alzheimer’s disease (AD), a devastating neurodegenerative disease characterized by progressive cognitive impairment, has become one of the most lethal diseases in elderly people, with the incidence rate ranking next to cardiovascular disease, cancer and apoplexy [1]. By the year 2050, the worldwide prevalence of Alzheimer’s will grow to 106.8 million [2], which will have a great influence on society.
The pathological hallmarks of AD are the presence of senile plaques and neurofibrillary tangles in the brains of patients. The current treatment for AD is to address biochemical sequelae of nerve cell loss in certain neuronal populations, but it does not target the underlying pathology [3]. Clinical drugs such as cholinesterase inhibitors or glutamate receptor antagonists can only ameliorate the symptoms instead of curing the disease, and an actual therapeutic strategy for AD is still lacking. Investigations have proven that β-amyloid protein (Aβ) plays a key role in AD, and the pathological folding of Aβ and the formation of Aβ oligomer might trigger AD pathogenesis [4]. As β-sheets of Aβ may be responsible for the accumulation of Aβ oligomers [5, 6] and have been reported to be toxic to various cultured cerebral cells [7], inhibiting the formation of β-sheets may be meaningful for AD treatment.
β-sheet breaker peptides are oligopeptides structurally analogous to specific epitopes of Aβ [8] and can specifically interfere with β-sheets within Aβ [7]. The first β-sheet breaker peptides were reported by Soto et al. [9] in 1996. They designed a series of β-sheet blockers to interact with the central hydrophobic region of Aβ1–40 at residues 17–21 (Aβ17–21), substituting proline instead of valine, which is one of the critical residues in β-sheet formation. Among them, iAβ5 and its end-terminally protected version iAβ5p, had high affinity with Aβ, preventing the folding and deposition of Aβ both in vitro and in vivo. H102 peptide (HKQLPFFEED) is a recently discovered β-sheet breaker designed according to the structure of Aβ [8]. It could specifically bind to the Aβ monomer (Aβ1–42) and stabilize its structure, thus inhibiting the formation of β-sheets and then blocking the early steps of misfolding and aggregation of the soluble Aβ. In addition, it could also increase the viability and relieve the impairment of nerve cells by resolving the existing Aβ fibers. After intracerebroventricular injection, it could improve the spatial memory impairment of Aβ precursor protein (APP) transgenic mice and reduce the quantity of senile plaques and the level of APP and Aβ, as well as enhance the activity of choline acetyltransferase (ChAT) and decrease the activity of acetylcholinesterase (AchE). This indicates that H102 may be a promising drug for AD treatment.
As a peptide drug, the easy degeneration and poor central nervous system (CNS) penetration of H102 limits its clinical application. Therefore, it might be meaningful to find a strategy to improve the stability and enhance the brain delivery of H102 for its use in AD treatment. In recent years, nasal administration has been rapidly developing in brain targeting and has been applied to brain delivery. It has been proven that peptide drugs such as insulin, insulin-like growth factor-one (IGF1), nerve growth factor (NGF) and basic fibroblast growth factor (bFGF) could enter the brain through the olfactory nerve and trigeminal nerve after nasal administration (nose-brain direct pathways), thus avoiding the restriction of the blood–brain barrier (BBB) [10–13]. Unfortunately, factors such as high molecular weights, poor nasal mucosa penetration capacity, easy enzymatic degradation and rapid mucociliary clearance (the residence time of drug formulations in the nasal cavity is approximately 15–20 min), result in the low total amount of peptide drug transport into the brain, which could not reach the therapeutic level [14]. Encapsulating peptides and proteins into nanometer-sized particles could protect them from degradation and facilitate their transport across the mucosal barrier. Among these nano-formulations, liposomes present excellent characteristics, such as good biocompatibility, safety and easy industrialization. For instance, formulations such as Exparel (bupivacaine liposome injectable suspension), Doxorubicin Hydrochloride Liposome Injection and NDA 202497 Marqibo (vincristine sulfate liposomes injection) are approved for use. In summary, liposomes could have great potential as peptide and protein carriers. Accordingly, in this study, we designed and prepared H102 peptide-loaded nasal liposomes with poly ethylene glycol (PEG) on the surface and subsequently determined its second conformation using circular dichroism, the release in vitro and the penetration capacity through Calu-3 cell monolayers. Further, we evaluated whether the liposomes could improve the peptides’ stability and enhance the brain delivery via intranasal administration. The Morris water maze experiment was also conducted to study the improving effect of the liposomes on rat spatial memory impairment compared with H102 solution administered intranasally containing 1% chitosan as an absorption enhancer.
Materials and Methods
Materials and Animals
H102 peptide (HKQLPFFEED) was synthesized by GL Biochem (Shanghai) Ltd. (purity 97%). Eptifibatide (internal standard) was synthesized by Chengdu KaiJie Biopharm Co., Ltd. (purity 95%). 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethylene glycol) 2000] (DSPE-PEG2000), egg phosphatidylcholine (EPC, PC-98 T) and cholesterol were purchased from Shanghai Advanced Vehicle Technology L.T.D. Co. Chitosan hydrochloride (Mw ~300 kDa) was a gift of Golden-Shell Biochemical Co. (Nanjing, China). AChE and ChAT activity assay kits were provided by the Jiancheng Bioengineering Institute (Nanjing, China). Anti-insulin degrading enzyme/IDE antibody was obtained from Abcam (USA), and Aβ1–40 was from the Chinese Peptide Company (Hangzhou, China). The human airway serous cell line (Calu-3) was obtained from the Chinese Academy of Sciences Cells Bank (Shanghai, China). Dulbecco’s Modified Eagle Medium Nutrient Mixture F12 (DMEM/F12), fetal bovine serum (FBS) and trypsin-EDTA solutions were purchased from Gibco (CA). All other chemicals used were of analytical or HPLC grade.
Male Sprague–Dawley (SD) rats (200–220 g) were supplied by Shanghai Sino-British Sippr/BK Lab Animial Ltd. (Shanghai, China). Animals were housed in cages under optimum light conditions (12:12 h light–dark cycle), temperature (25 ± 1°C) and humidity (50–60%), with food and water provided ad libitum. The animal studies were performed according to the protocols approved by the ethical committee of Fudan University.
H102 Liposome Preparation
The H102 liposomes were prepared using a modified thin film hydration method. Briefly, the mixture of EPC, DSPE-PEG2000 and cholesterol in the molar ratio of 20:1:5 was dissolved with chloroform in a pear-shaped flask and was subsequently evaporated to a dry film using a rotary evaporator under a vacuum. The lipid film was hydrated with H102 in pH 6.5 PBS buffer (Conc. 0.1%) and agitated for 2 h at 40°C. Then, the suspension was extruded through a 0.22-μm microporous membrane (Millex-GP, SLGP033RS, Millipore) three times. The liposomes were collected by ultrafiltration (molecular cut off 10000, Amicon Ultra-0.5, UFC901096, Millipore) at 6000 rpm for 30 min at 4°C, and lyophilized (Virtis Model Benchtop K, USA) and stored at −20°C in airtight vials.
Liposome Characterization
The liposomes’ morphology was examined using transmission electron microscopy (TEOL2010, JEM) and negative staining with 2% phosphotungstic acid solution. The particle sizes, polydispersity index (PDI) and zeta potential of the liposomes were measured using the light scattering method with a Malvern Zetasizer Nano-ZS (Malvern Instruments, Malvern, UK).
The encapsulation efficiency of H102 in liposomes was determined by the ultrafiltration- centrifugation technique. Briefly, 1 ml liposomal suspension was ultra-filtered at 4°C, 6000 rpm for 30 min. Then, 20 μl ultrafiltrate was injected into the Agilent 1200 HPLC system [15]. The total concentration of H102 in the formulations (100 μl) was determined after destructing the liposomes with 400 μl 0.5% Triton X-100, then mixing with 500 μl of mobile phase for analysis. The associated H102 within the liposomes was calculated from the difference between the total and the free drug concentrations determined in the liposome suspension and in the ultrafiltrate, respectively.
Circular Dichroism (CD) Studies
CD spectra of H102 and liposomes were recorded using a Jasco spectropolarimeter (J-800). Samples with a H102 concentration of 100 μg/ml were prepared and placed in a cuvette of 1 cm path length, respectively. Spectra were recorded between 190 and 240 nm with a scan rate of 50 nm/min. Deconvolution was carried out using a K2D3 web server to estimate the α helix and β strand content of a protein.
Liposome In Vitro Release
In vitro release of H102 from the liposomes was carried out using the dialysis bag. One milliliter of liposomal suspensions (0.0325%) in dialysis tubing was immersed in 20 ml artificial nasal fluid [16] (pH 6.3, containing 0.1% BSA), which was incubated at 37°C with constant shaking at 100 rpm. Samples were taken at 0.5, 1, 2, 4, 6, 8, 10 and 12 h, respectively, and immediately replaced with equal volumes of the medium after each sampling. Released H102 was detected by HPLC and the cumulative release percentage was calculated.
Stability of H102 in Plasma
H102 and H102 liposomes were dispersed in blank rat plasma and then incubated at 37°C. Samples (n = 4) were collected at 0, 0.5, 1 and 2 h, and then analyzed by HPLC/MS as described previously [15]. The changes in H102 concentration before and after incubation were calculated.
Transport Studies of Liposomes Across Calu-3 Monolayers
The human airway serous cell line Calu-3 was cultured in complete DMEM/F12, containing 10% (v/v) FBS, 1% (v/v) nonessential amino acid solution, penicillin (100 U/ml) and streptomycin (100 μg/ml) under standardized conditions (95% relative humidity, 5% CO2, 37°C).
To establish the monolayers, cells were seeded at a density of 1 × 105 cells/cm2 onto 12-well Transwell filters (3.0 μm, Millipore, USA). After 15–18 days, the transendothelial electrical resistance (TEER) was measured using an epithelial voltmeter (MILLICELL®-ERS, Millipore, USA). Only cell monolayers with TEER exceeding 350Ω•cm2 were selected for the following experiment.
Briefly, the monolayers were balanced with pre-warmed DMEM/F12 (37°C) and 1 ml pre-warmed DMEM/F12 was added to the basolateral compartments. Approximately 0.5 ml DMEM/F12 containing H102 loaded liposomes or H102 solution with 1% chitosan were then, respectively added to the apical compartments at a final concentration of 1 mg/ml per well and incubated at 37°C. At 2, 5, 10, 15, 30, 45, 60, 90, 120, 180, 240 and 360 min, 100 μl samples were taken from the basal compartments and the volume was replaced with equal fresh DMEM/F12. The concentrations of H102 in samples were determined by HPLC after the addition of 100 μl 0.5% Triton X-100. The results were averaged from three determinations.
After transport study, the drug and liposomes were carefully removed. The monolayers were then rinsed with HBSS, supplied with the culture medium and allowed to regenerate for 1 day at the incubated atmosphere. TEER was periodically measured during the recovery period.
Brain Uptake Study of H102 Liposomes
Ninety-six rats were randomly divided into three groups to receive i) intravenous injection (i.v.) of H102 solution (0.2 mg/kg) with 0.1% BSA (to reduce non-specific adsorption); ii) intranasal administration (i.n.) of H102 solution (2 mg/kg) with 1% chitosan and 0.1% BSA; and iii) i.n. H102 liposomes (2 mg/kg). For the i.n. group, 20 μl of each preparation was given into the nostrils of rats by a polyethylene 10 (PE 10) tube sheathed with a microliter syringe (10 μl for each nostril). For the i.v. group, 200 μl of the preparation was injected into the tail vein rapidly.
At the predetermined time, the blood samples were taken from the heart and rats were perfused with saline. Afterwards, brains were harvested and dissected into four parts: cerebrum (CR), cerebellum (CL), hippocampus (HI) and olfactory bulb (OB), which were weighed and homogenized in ice-cold saline using a tissue homogenizer. The supernatant was obtained after centrifugation at 5000 rpm for 10 min at 4°C. Blood samples were centrifuged to collect plasma. Then, 100 μl of the plasma samples or the supernatant of tissue homogenates were mixed with 10 μl of the internal standard (5 μg/ml eptifibatide in tri-distilled water). After the addition of 200 μl methanol, the mixtures were vortexed for 2 min and centrifuged at 12,000 rpm, 4°C for 20 min. Then, 20 μl of supernatant was analyzed by HPLC/MS (Agilent 1100 LC-MS, USA) [15]. Standards and control samples of the plasma and tissues for drug analysis were prepared in the same manner.
The area under the blood or brain tissue concentration of the H102 versus time curve (AUC0→t) was calculated with the trapezoidal method. The variance of AUC0→t was estimated using the method described by Yuan [17].
Pharmacodynamic Study
In Vivo Model of Alzheimer’s Disease
Bilateral injections of Aβ1–40 into the hippocampus has been demonstrated to induce the impairment of memory acquisition in rats, and this animal model could be used to develop and evaluate potential drugs for AD therapy [18]. Aβ1–40 was dissolved in saline at the concentration of 5 mg/ml and incubated at 37°C for 7 days to obtain fibril formation. Male SD rats were anesthetized and fixed in a David Krof stereotaxic apparatus. Two microliters of Aβ1–40 solution was slowly bilaterally injected into the rat hippocampus (±2.0 mm lateral to the midline, 3.5 mm posterior to the bregma and 2.7 mm ventral to the skull surface) over 5 min. The needle was left in place for another 5 min to minimize liquid backflow and to allow an appropriate diffusion. Sham group rats were injected with the same volume of saline. The animals were kept in cages to recover for 1 week.
Drug Treatment and Experimental Design
Rats were divided into seven groups (Table I): Sham group, AD control group, and five test groups were given the preparations according to Table I after 1-week recovery. The Morris water maze test was performed after 7 days of administration.
Morris Water Maze Test
The Morris water maze test was carried out in a circular tank (180 cm in diameter and 30 cm in depth), divided into four equal quadrants and filled with black-dyed water. The water temperature was adjusted to 24 ± 2°C. A black platform (9 cm in diameter) was placed 2 cm under the water level in the center of the southwest quadrant. Several unchanged strategic cues were fixed at distinct positions around the pool. The swimming paths of the rats were recorded using the digital camera.
The procedure of the Morris water maze consisted of a 4-day navigation testing session and a probe trail. Rats were given four trails per day for four consecutive days. H102 formulations or saline were administered 1 h before the first trial of each day. The rats were placed into the water facing the wall and were allowed to swim for 60 s to seek the invisible platform. Rats that found the platform within 60 s were required to stay there for 10 s, while the rats that failed to locate the platform were guided to it and allowed to rest for 10 s.
The probe trial was performed immediately after last training trial. The hidden platform was removed and the rats were introduced into the water from the northeast quadrant and were allowed to swim freely for 60 s. The swimming tracks were acquired by a computerized video tracking system and measures such as the percentage of time and distance in the target quadrant were calculated for each rat.
Determination of Biochemical Indexes
After the Morris water maze test, animals were anesthetized and transcardially perfused with saline. Hippocampal tissues were rapidly harvested and homogenized in ice-cold saline. Supernatant was obtained after centrifugation to determine AChE and ChAT activity with the assay kit. The rest of the brains homogenized in saline were used to determine IDE activity using western blot analysis. The protein concentration was measured by the Quantity Protein assay kit. The activity of the enzymes was expressed as units of enzyme per mg protein.
Histology
The animals were anesthetized and perfused with saline followed by 4% paraformaldehyde after the probe test. Brains were removed and doused in 4% paraformaldehyde overnight. After dehydration in 20% phosphate-buffered sucrose solution, brains were embedded in paraffin and cut into 6 μm sections from the injection site. Sections were stained with Congo red to determine the amyloid plaques. The hippocampal areas were examined under a Leica optical microscope (Leica, Germany) equipped with ImageJ software (v1.41, NIH, USA).
Nasal Ciliotoxicity
Nasal ciliotoxicity was examined using a modified in vivo rat nasal mucosa model described by Jiang et al. [19]. Twelve rats were randomly divided into four groups: i) i.n. saline (negative control); ii) i.n. H102 solution with 1% chitosan (5 mg/ml); iii) i.n. H102 liposomes (5 mg/ml); iv) 1% deoxy-sodium cholate (positive control). The rats received 50 μl of preparations in the left nostril once a day for seven consecutive days. Twenty-four hours after the last administration, rats were sacrificed and the nasal mucosa was peeled off. The mucocilia were examined under a scanning electronic microscope (SEM) (JSM-T300, Japan).
Statistical Analysis
All data were expressed as the mean ± S.E.M. or the mean ± S.D. Significant differences were computed with one-way analysis of variance (ANOVA), followed by post hoc Dunnett tests for multi-group comparison. Student’s t-test was used for two-group comparisons and p < 0.05 was considered statistically significant.
Results
Preparation and Characterization of H102 Liposomes
H102 peptide liposomes were successfully prepared using the thin film hydration method. The mean particle size of the liposomes was 112.2 ± 6.4 nm with a narrow particle distribution (PDI = 0.185 ± 0.012), and the surface charge of the liposomes was −2.96 ± 0.38 mV. The observed liposomes presented elliptic and uniform characteristics under the transmission electron microscope (Fig. 1). H102 liposomes had an encapsulation efficiency of 71.35 ± 0.87%.

Transmission electron micrograph of H102 liposomes, bar = 100 nm.
Conformation Analysis
The secondary structure conformation of H102 peptide in solution or liposomes was investigated using CD (Fig. 2(a)). The unencapsulated H102 had approximately 15.46% α-helix and 9.16% β-sheets, but while encapsulated in liposomes, its content percentage changed to 8.15 and 14.08%, respectively. This change might represent the interaction of H102 with liposomes, and the enhanced β-sheets could result from the confinement of the peptide in a small volume that could promote greater intermolecular associations, leading to β-sheet formation [20].

a CD spectrum of H102 and H102-loaded liposomes. b Release of H102 from liposomes in artificial nasal fluid. n = 6.
Liposomes In Vitro Release
The in vitro release of H102 from liposomes in artificial nasal fluid was shown in Fig. 2(b). It was characterized by a burst release during the first 0.5 h (approximately 40%) followed by a step of slower release. Approximately 97% of the drug was released after 12 h. The release profile fit the first-order rate equation: ln(1-Q) = −0.3828 t-0.3214, R2 = 0.9928.
Stability of H102 in Plasma
The stability of free H102 and H102 entrapped in the liposome in plasma were detected using HPLC/MS. For free H102, almost all H102 was degraded, and only 6.5% could be detected after 1 h of incubation in plasma, indicating that H102 was unstable in plasma (Table S1). It also showed a rapid degradation when incubated with brain homogenate (Table S2), only 3.0% could be detected after 2 h. The encapsulation of H102 into liposomes significantly enhanced H102 stability, as more than 63% of H102 remained after 2 h incubation in plasma (Table S1).
Calu-3 Monolayer Transportation of Liposomes
Calu-3 was a well-characterized cell line derived from human bronchial epithelium. It could be grown as confluent sheets at an air-media interface with physical and electrical properties comparable to nasal mucosa [21]. Calu-3 cell line has been used as an in vitro nasal platform to investigate the nanoparticles and polymer gels for protein delivery [21, 22]. Figure 3 shows the cumulative amount of H102 transported across the Calu-3 cell monolayers. In the solution group, H102 transport was quick and reached a platform after 15 min, which might have resulted from the ability of chitosan to open tight junctions between cells [23], demonstrated by the fact that the TEER of Calu-3 monolayers in the solution group decreased to 45.7 ± 1.2% within 30 min. While in the liposome group, the concentration of H102 that permeated across the monolayers increased slowly at initial sampling time points, but was higher than that in the solution group from 30 min and rose consistently. Liposomes had a little influence on monolayers’ integrity, and the TEER declined to 78.0 ± 1.6% ~ 86.0 ± 0.8% of the initial value during the transport period. After removing the drug solution and liposomes, TEER values completely recovered to initial values within 24 h, indicating that the effect of chitosan and liposomes on tight junctions was evidently reversible.

Cumulative amount of H102 transported across the Calu-3 cell monolayers.
Brain Uptake Study of H102 Formulations
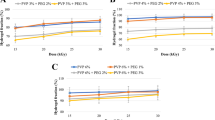
The concentration of H102 in plasma achieved 247.97 ± 39.01 ng/ml 1 min after i.v. injection, and then decreased rapidly below the detection limit at 5 min (Fig. 4). I.n. administration of H102 solution presented a quick absorption, with the peak concentration of 88.20 ± 12.02 ng/ml in plasma at 5 min. The following fast elimination made H102 undetectable 45 min after the administration, and the absolute bioavailability of H102 nasal solution was 11.3%. Unlike i.n. H102 solution, nasal liposomes showed a slower absorption with tmax of 30 min. H102 was found in plasma even 90 min after administration, which was markedly longer than i.n. H102 solution (45 min). The absolute and relative bioavailabilities of i.n. H102 liposomes were 30.2 and 269%, respectively, suggesting that liposome significantly enhanced the nasal absorption of H102.

Concentration–time profiles of H102. a H102 in plasma following intranasal (i.n.) administration of H102 solution, liposomes and intravenous (i.v.) administration of H102 solution to rats. Brain uptake of H102 following intranasal administration of H102 nasal solution and H102 liposomes in the olfactory bulb (b), cerebrum (c), cerebellum (d) and hippocampus (e) (mean ± SD, n = 4).
No drug could be detected in each brain region after intravenous administration, indicating that H102 could not penetrate the BBB. However, two intranasal preparations produced significant absorption in the brain, especially in the olfactory bulb (OB), suggesting that intranasal administration was effective for brain delivery of H102 and that H102 could enter the brain directly through the nose-brain pathway. The AUC0→90min values of H102 in the OB, CR, CL and HI of rats following the nasal application of H102 liposomes were 1.67-, 1.68-, 1.59- and 2.92-fold greater than that of the rats with i.n. H102 solution, respectively (Table II).
Neuroprotection Effects of the H102 Formulations on Alzheimer’s Disease Model Rats
Morris Water Maze Test
Morris water maze was used for spatial learning performance and widely conducted in the study of AD. Moreover, it is a hippocampus-dependent task that is suitable for hippocampus-damaged animal model since the abnormality in hippocampus was highlight in the pathology of AD. In our study, the effect of H102 formulations on the spatial learning and memory deficits of rats was evaluated by the Morris water maze test. The escape latency of all groups of rats displayed a reduction with the 4-day training session (Fig. 5). The latency of the AD control group was significantly longer than that of the sham group, indicating that intrahippocampal injection of Aβ1–40 could successfully induce learning and memory deficits in rats. Intravenous administration of 1000 μg/kg of H102 solution could decrease the escape latency of rats to a certain degree, but with no significance compared to the AD control group. Intranasal administration of H102 solution containing 1% chitosan could ameliorate spatial memory impairment of AD model rats in a dose-dependent manner. H102 solution (50 μg/kg) showed no obvious improvement in the first 2 days, while 500 μg/kg of H102 solution apparently shortened the escape latency on day 1. The escape latency was apparently reduced with H102 liposomes (i.n.) of both 50 and 500 μg/kg/d with the significance observed from the first day of training, presenting a better learning ability.

Mean escape latency in the Morris water maze of rats that received bilateral injection of saline or Aβ1–40 into the hippocampus. The data are shown as the mean ± SEM. (n = 10). * p < 0.05, significantly different from the AD Control group.
In the probe test, the rats in the sham group swam preferentially in the target quadrant, while the AD control group showed no tendency to find the platform (Fig. S1). The animals treated with i.v. H102 solution had little awareness of searching the platform, and spent a very long time finding the platform. The rats treated intranasally with H102 formulations had better knowledge of the platform location, and crossed the platform area approximately 4–5 times, especially in the H102 liposomes (500) group, in which rats had high percentages of swimming distance and time in the target quadrant (46.47 ± 4.70% and 47.85 ± 5.29%, respectively), similar to the sham group (45.40 ± 3.36% and 46.69 ± 3.79%, respectively).
AchE, ChAT and IDE Activity in the Rat Brain
The activity of AChE and ChAT in rat hippocampi was determined to evaluate the neuroprotective effects of H102 formulations. The AChE activity of the AD control group was significantly higher than that of the sham group, while ChAT activity was significantly lower (Fig. 6(a), (b)). As for the IV-H102 group (1000), the activities of AChE and ChAT were similar to the AD control group. After intranasal administration of H102 formulations, the AChE activity obviously decreased to a level with little significance compared to the sham group, while the ChAT activity was significantly higher than that of the AD control group (except the IN-H102 Solution (50) group). Interestingly, the value of ChAT activity approached that of the sham group in the H102 nasal solution (500) and liposome (500) groups, achieving the normal level, which suggested that H102 administered through the nasal cavity could effectively protect central cholinergic nerves from the destruction induced by Aβ1–40.

AChE activity (a), ChAT activity (b) and western blot of insulin degrading enzyme (IDE) expression relative to β-actin (c) in Aβ1–40-treated in rats’ hippocampi. The data are shown as the mean ± SD (n = 6). * p < 0.05, significantly different from the AD Control group, # p < 0.05, significantly different from the Sham group.
Western blot was used to detect the expression level of IDE, a metalloprotease involved in Aβ degradation in the brain, in rat hippocampi. The lowest IDE expression was observed in the AD control group (Fig. 6(c)). Intravenous administration of H102 could not increase IDE concentration, while the IDE activity markedly increased when rats were given intranasal H102 solution and liposomes. The best improvement was achieved in the IN-H102 liposome (500) group, and the IDE expression in the rat brain had no significant difference from the sham group, which indicated that this formulation might prevent amyloids from deposition. Additionally, it was noteworthy that IN-H102 liposomes (50) could achieve similar effects to IN-H102 solution (500) in increasing the IDE expression.
Histology
The formation of Aβ plaques is a pathologic hallmark of AD, and Aβ plaques are stained by Congo red. Obvious orange sediments were easily found in the AD control group (Fig. 7), and the amount of Aβ plaques exhibited no significant change in IV-H102 (1000) group. After nasal administration of H102 solution or liposomes, Aβ plaques were decreased both in quantity and size. Only slightly stained Aβ plaques were detected in the IN-H102 liposome (500) group, which was close to that of the sham group. It is noteworthy that Aβ plaques in liposome groups were fewer and smaller than those in solution groups at the same dosage, indicating that liposomes could have a better effect to prevent the formation of Aβ plaques than solution, which might result from the ability of liposomes to deliver more H102 to the brain.

Representative images of Aβ staining. a AD control; b Sham; c IV-H102; d IN-H102 Solution (50); e IN-H102 Solution (500); f IN-H102 Liposome (50); g IN-H102 Liposome (500). Arrows indicate deposits of β-amyloid. Bar = 100 μm. (H) Quantification of Aβ plaque load in rat hippocampi. The data were shown as the mean ± SD (n = 3). * p < 0.05, significantly different from the AD control group.
Nasal Ciliotoxicity
The nasal ciliotoxicity of H102 formulations was evaluated using an in vivo rat nasal mucosa model. The rats’ nasal cilia were intact with some adhesion or lodging with 1-week treatment of H102 solution (Fig. 8). After treatment with H102 liposomes, the cilia were still intact, dense and arranged in an orderly manner, which showed no significant difference from the saline control group. These results suggested that H102 solution had a slight influence on the rat nasal cilia, and the nasal ciliotoxicity of H102 liposomes was negligible.

Scanning electronic microscope (SEM) images of nasal mucocilia. a negative control; b positive control; c H102 solution; and d liposomes via intranasal administration for 1 week (n = 5). Cilia were indicated by arrows. Bar = 100 μm.
Discussion
H102 is a 10-amino-acid β-sheet breaker peptide designed according to the structure and the aggregation mechanism of Aβ [8]. Former studies demonstrated that H102 could specifically bind to the Aβ monomer (Aβ1–42) and lead to the disruption of the β-sheet conformation. Furthermore, it could also resolve the existing Aβ fibers, increase neuronal survival and decrease brain inflammation. After intracerebroventricular injection, H102 could achieve excellent improvement of the spatial learning and memory deficits of APP transgenic rats, enhance the activity of ChAT and decrease the activity of AchE.
Regretfully, H102 peptide was unstable in vivo and was cleared rapidly after intravenous injection with a plasma half-time less than 2 min (Fig. 4 and Table S3). It was also observed in brain homogenate that H102 decrease to 45.73% at 0.5 h (Table S2), which might due to the enzymatic degradation such as aminopeptidases, endopeptidases and carboxypeptidases since the degradation progress could be greatly inhibited at −20°C. To keep the activity of H102 and enhance its brain delivery, liposomes were prepared to protect H102 from the enzymatic degradation, and intranasal administration was adopted for brain targeting in this study. DSPE-PEG2000 was appended to enhance the liposome stability to avoid the in vivo membrane fusion of liposomes, which may lead to drug leakage. The average size of the prepared liposomes was approximately 110 nm, which was suitable for intranasal administration [24].
In our study, H102 liposomes presented a certain sustained release in artificial nasal fluid after a 30-min burst release, which suggested that some H102 peptides were encapsulated into the inner phase of liposomes and protected from the inactivating effect of external conditions (more than 63% of H102 remained after 2 h incubation in plasma). This was also demonstrated by a CD study. The secondary structure conformation obtained from CD suggested an interaction between H102 and liposomes instead of a simple mixture.
Previous studies showed the poor nasal mucosa penetration ability of free H102 (data not shown), and chitosan was demonstrated to have the ability to enhance the nasal absorption of peptides with little toxicity [25]. Therefore, 1% chitosan was added as an absorption enhancer in the nasal solution to provide a comparison to liposomes.
In the Calu-3 cell monolayer transport experiment, H102 solution rapidly crossed the monolayers with the help of chitosan, while liposomes showed slow and sustained transport. One explanation for the phenomenon might be that the endocytosis transport of liposomes would be slower than the passive diffusion of H102 solution [26]. However, H102 was encapsulated in liposomes that could protect H102 from degradation in cells, thus making it easier to deliver more H102 across the monolayers. This also appeared in pharmacokinetics experiments. Administered through the nasal cavity, H102 solution rapidly reached the peak concentration in blood, and the absolute bioavailability was 11.3%. While intranasal H102 liposomes resulted in a slower initial absorption (tmax = 30 min) and longer duration in blood, the AUC value was 2.69 times higher than that of H102 solution. Meanwhile, H102 liposomes could also be easily transported into the brain. The drug amount in the hippocampus of groups that received liposomes was 2.92 times that of the given solution. These values were meaningful for an Aβ-targeted peptide drug to be enriched in the hippocampus, which was among the first affected and critical areas in the pathologic process of AD [27]. These results suggested that intranasal H102 liposomes could facilitate AD therapy with effective drug delivery to the hippocampus of rats.
There were two direct pathways to deliver solutes from the nasal cavity to the brain. One was olfactory pathway including olfactory nerve pathway and olfactory epithelial pathway, which allowed solutes to enter the olfactory bulb and then diffuse to the brain. Another one was trigeminal nerve pathway that propelled solutes to the brain through the pons [28]. Similarly, H102 could be deliver to the brain along these two direct pathways following intranasal administration of solution or liposome [29, 30], although the detailed mechanism still need to be clarified. Compared with IV-H102 group, the increased H102 concentrations in the hippocampus in IN-H102 solution and IN-H102 liposome groups corresponded to the improved memory function reflected in pharmacodynamics studies (water maze experiment, biochemical indexes assay and tissue histology). The rats’ performance was obviously improved in a dose-dependent manner when exposed to different dosages of H102 formulations. H102 liposomes could achieve better ameliorative effects than H102 solution at the same dosage after intranasal administration.
The cholinergic system was partially responsible for the cognitive dysfunction. Disturbances of the acetylcholine metabolism were found in the AD brains with increased levels of AChE (decomposition of acetylcholine) and decreased activity of ChAT (the synthesis enzyme of acetylcholine) [31]. In this study, both H102 nasal solution and liposomes could protect central cholinergic neurons to some degree because they could reduce the AchE, increase the ChAT activity and ameliorate the cholinergic nerve impairment.
The expression level of IDE was also determined to evaluate the neuroprotective effects of H102 formulations. IDE is a metalloprotease that has been involved in Aβ degradation in the brain. Recent evidence indicates that the expression of neuronal IDE was decreased in the brains of severe AD patients [32, 33]. The present study showed that intranasal H102 solution and liposomes could favor to the recovery of this protein, especially the high dosage of H102 liposomes (500), which increased the expression of IDE to almost the normal level, suggesting that this formulation could help reduce Aβ deposition in AD model rats. Similar observations were found in the Congo red staining. Obvious orange plaques were found in the AD control group and the IV-H102 (1000) group, while the plaque number and area were significantly decreased after H102 solution and liposome treatment. The best recoveries were also noticed in the rats of the high dose of liposome group, with few plaques in the hippocampus similar to the normal ones, showing good inhibition of Aβ deposition.
In the last several decades, transgenic mice have enabled dramatic advances in the understanding of the pathogenic mechanisms in AD and of potential therapeutic approaches to the disease [34]. However, the production and the breeding of transgenic mice are time consuming, laborious, inefficient and expensive [35]. For this reason, non-transgenic animal models are still widely used in study of AD [36, 37]. Injection of Aβ oligomers (Aβ25–35,1–40,1–42) into the hippocampus could provoke acute pathological changes, thus inducing brain dysfunction evidenced by neurodegeneration and the impairment of learning and memory [38, 39], which were similar with the symptoms of AD. This type of AD model rat was very suitable for our investigation, as it could be built in a short time and was effective in the evaluation of the neuroprotective effects of our formulations. Further confirmation of the results was conducted using the AD transgenic mice.
Additionally, AD is a chronic disease that requires prolonged care and long-term medication. Therefore, besides being effective, the best formulation for AD treatment should exhibit biocompatibility, little toxicity and safety, which would favor its long-term administration. In our study, H102 solution had a slight influence on the rat nasal cilia and showed weak nasal ciliotoxicity after 1 week of treatment. These might have resulted from the added chitosan, a positively charged polysaccharose, which has a binding affinity on cellular membranes and would result in different degrees of cytotoxicity [25]. Therefore, prolonged exposure to H102 solution might induce irritation in the nasal mucosa. On the other hand, liposomes formed from natural phospholipids, cholesterol and DSPE-PEG2000 have similar structures to the cell membrane, showing good biocompatibility and biodegradation [40–42]. These observations were consistent with the negligible nasal ciliotoxicity observed in H102 liposomes, suggesting that nasal liposomes prepared for the brain delivery of H102 peptide could increase the safety of the preparation and the patients’ compliance. Allergenicity and chronic toxicity testing will be conducted for the further evaluation of nasal liposomes.
Conclusion
The H102 peptide-loaded liposomes prepared in this study for nasal administration could effectively deliver H102 into the brain. Liposomes achieved higher brain absorption, especially in the hippocampus, compared with nasal H102 solution, and could effectively ameliorate spatial memory impairment of AD model rats both at a low dose and a high dose. H102 nasal formulations showed little toxicity on the nasal mucosa. These results demonstrated that the H102 liposomes were safe, effective and stable and might be a good choice for AD therapy.
Abbreviations
- AChE:
-
Acetylcholinesterase
- AD:
-
Alzheimer’s disease
- Aβ:
-
β-amyloid protein
- BBB:
-
Blood–brain barrier
- CD:
-
Circular dichroism
- ChAT:
-
Choline acetyltransferase
- CNS:
-
Central nervous system
- CL:
-
Cerebellum
- CR:
-
Cerebrum
- EPC:
-
Egg phosphatidylcholine
- HI:
-
Hippocampus
- IDE:
-
Insulin degrading enzyme
- OB:
-
Olfactory bulb
- PEG:
-
Poly ethylene glycol
- TEER:
-
Transendothelial electrical resistance
References
Bolukbasi HF, Hatip-Al-Khatib I. Effects of beta-sheet breaker peptides on altered responses of thoracic aorta in rats’ Alzheimer’s disease model induced by intraamygdaloid Aβ40. Life Sci. 2013;92(3):228–36.
Brookmeyer R, Johnson E, Ziegler-Graham K, Arrighi HM. Forecasting the global burden of Alzheimer’s disease. Alzheimers Dement. 2007;3(3):186–91.
Kurz A, Perneczky R. Novel insights for the treatment of Alzheimer’s disease. Prog Neuro-Psychopharmacol Biol Psychiatry. 2011;35(2):373–9.
Liu Y, Hua Q, Lei H, Li P. Effect of Tong Luo Jiu Nao on Aβ-degrading enzymes in AD rat brains. J Ethnopharmacol. 2011;137(2):1035–46.
Chacon MA, Barria MI, Soto C, Inestrosa NC. β-sheet breaker peptide prevents Aβ-induced spatial memory impairments with partial reduction of amyloid deposits. Mol Psychiatry. 2004;9(10):953–61.
Walsh DM, Klyubin I, Fadeeva JV, et al. Naturally secreted oligomers of amyloid β protein potently inhibit hippocampal long-term potentiation in vivo. Nature. 2002;416(6880):535–9.
Bruinsma IB, Karawajczyk A, Schaftenaar G, de Waal RM, Verbeek MM, van Delft FL. A rational design to create hybrid β-sheet breaker peptides to inhibit aggregation and toxicity of amyloid-β. Med Chem Commun. 2011;2(1):60–4.
Lin LX, Bo XY, Tan YZ, Sun FX, Song M, Zhao J, et al. Feasibility of β-sheet breaker peptide-H102 treatment for Alzheimer’s disease based on β-amyloid hypothesis. PLoS One. 2014;9(11), e112052.
Soto C, Kindy MS, Baumann M, Frangione B. Inhibition of Alzheimer’s amyloidosis by peptides that prevent β-sheet conformation. Biochem Biophys Res Commun. 1996;226(3):672–80.
Benedict C, Hallschmid M, Schultes B, Born J, Kern W. Intranasal insulin to improve memory function in humans. Neuroendocrinology. 2007;86(2):136–42.
Vaka SR, Sammeta SM, Day LB, Murthy SN. Delivery of nerve growth factor to brain via intranasal administration and enhancement of brain uptake. J Pharm Sci. 2009;98(10):3640–6.
Ma YP, Ma MM, Cheng SM, et al. Intranasal bFGF-induced progenitor cell proliferation and neuroprotection after transient focal cerebral ischemia. Neurosci Lett. 2008;437(2):93–7.
Feng C, Zhang C, Shao X, et al. Enhancement of nose-to-brain delivery of basic fibroblast growth factor for improving rat memory impairments induced by co-injection of beta-amyloid and ibotenic acid into the bilateral hippocampus. Int J Pharm. 2012;423(2):226–34.
Illum L. Transport of drugs from the nasal cavity to the central nervous system. Eur J Pharm Sci. 2000;11(1):1–18.
Zhang C, Zheng X, Wan X, et al. The potential use of H102 peptide-loaded dual-functional nanoparticles in the treatment of Alzheimer’s disease. J Control Release. 2014;192:317–24.
Cao S, Ren X, Zhang Q, et al. In situ gel based on gellan gum as new carrier for nasal administration of mometasone furoate. Int J Pharm. 2009;365(1):109–15.
Yuan J. Estimation of variance for AUC in animal studies. J Pharm Sci. 1993;82(7):761–3.
Takeda S, Sato N, Niisato K, et al. Validation of Aβ1-40 administration into mouse cerebroventricles as an animal model for Alzheimer disease. Brain Res. 2009;1280:137–47.
Jiang XG, Cui JB, Fang XL, Wei Y, Xi NZ. Toxicity of drugs on nasal mucocilia and the method of its evaluation. Yao Xue Xue Bao. 1995;30(11):848–53.
Kumaraswamy P, Sethuramanand S, Krishnan UM. Development of a dual nanocarrier system as a potential stratagem against amyloid-induced toxicity. Expert Opin Drug Deliv. 2014;11(8):1131–47.
Driton V, Ruth E, Angeles H, Luca C, Martin G, Lisbeth I, et al. Tight junction modulation by chitosan nanoparticles: comparison with chitosan solution. Int J Pharm. 2010;400(1–2):183–93.
Zheng C, Guo Q, Wu Z, Sun L, Zhang Z, Li C, et al. Amphiphilic glycopolymer nanoparticles as vehicles for nasal delivery of peptides and proteins. Eur J Pharm Sci. 2013;49(4):474–82.
Vllasaliu D, Casettari L, Fowler R, Exposito-Harris R, Garnett M, Illum L, et al. Absorption-promoting effects of chitosan in airway and intestinal cell lines: a comparative study. Int J Pharm. 2012;430(1):151–60.
Illum L. Nanoparticulate systems for nasal delivery of drugs: a real improvement over simple systems? J Pharm Sci. 2007;96(3):473–83.
Kean T, Thanou M. Biodegradation, biodistribution and toxicity of chitosan. Adv Drug Deliv Rev. 2010;62(1):3–11.
Abhay U, Renee R, Wyatt N, Don L, Peter W. Microfluidic preparation of liposomes to determine particle size influence on cellular uptake mechanisms. Pharm Res. 2014;31(2):401–13.
Lotjonen J, Wolz R, Koikkalainen J, Julkunen V, Thurfjell L, Lundqvist R, et al. Fast and robust extraction of hippocampus from MR images for diagnostics of Alzheimer’s disease. NeuroImage. 2011;56(1):185–96.
Liu Q, Shen Y, Chen J, Gao X, Feng C, Wang L, et al. Nose-to-brain transport pathways of wheat germ agglutinin conjugated PEG-PLA nanoparticles. Pharm Res. 2012;29(2):546–58.
Dhuria SV, Hanson LR, Frey 2nd WH. Intranasal delivery to the central nervous system: mechanisms and experimental considerations. J Pharm Sci. 2010;99(4):1654–73.
Liu QF, Shen YH, Chen J, Gao XL, Feng CC, Wang L, et al. Nose-to-brain transport pathways of wheat germ agglutinin conjugated PEG-PLA nanoparticles. Pharm Res. 2012;29(2):546–58.
Carvajal FJ, Inestrosa NC. Interactions of AChE with Aβ aggregates in Alzheimer’s brain: therapeutic relevance of IDN 5706. Front Mol Neurosci. 2011;4:19.
Miners JS, Baig S, Palmer J, Palmer LE, Kehoe PG, Love S. Aβ-degrading enzymes in Alzheimer’s disease. Brain Pathol. 2008;18(2):240–52.
Funalot B, Ouimet T, Claperon A, et al. Endothelin-converting enzyme-1 is expressed in human cerebral cortex and protects against Alzheimer’s disease. Mol Psychiatry. 2004;9(12):1122–8. 1058.
McGowan E, Eriksen J, Hutton M. A decade of modeling Alzheimer’s disease in transgenic mice. Trends Genet. 2006;22(5):281–9.
Chen F, David D, Ferrari A, Gotz J. Posttranslational modifications of tau-role in human tauopathies and modeling in transgenic animals. Curr Drug Targets. 2004;5(6):503–15.
Johann M, Vanessa V, Laurent G, Tangui M. The γ-secretase inhibitor 2-[(1R)-1-[(4-chlorophenyl)sulfonyl](2,5-difluorophenyl) amino]ethyl-5-fluorobenzenebutanoic acid (BMS-299897) alleviates Aβ1–42 seeding and short-term memory deficits in the Aβ25–35 mouse model of Alzheimer’s disease. Eur J Pharmacol. 2013;698(1–3):193–9.
Capurro V, Busquet P, Lopes J, Bertorelli R, Tarozzo G, Bolognesi M, et al. Pharmacological characterization of memoquin, a multi-target compound for the treatment of Alzheimer’s disease. PLoS ONE. 2013;8(2), e56870.
Van DD, De Deyn PP. Drug discovery in dementia: the role of rodent models. Nat Rev Drug Discov. 2006;5(11):956–70.
Nomura I, Takechi H, Kato N. Intraneuronally injected amyloid beta inhibits long-term potentiation in rat hippocampal slices. J Neurophysiol. 2012;107(9):2526–31.
Lian T, Ho RJ. Trends and developments in liposome drug delivery systems. J Pharm Sci. 2001;90(6):667–80.
Samad A, Sultana Y, Aqil M. Liposomal drug delivery systems: an update review. Curr Drug Deliv. 2007;4(4):297–305.
Pujol I, Serracant A, Cano M, Ampudia RM, Rodriguez S, Sanchez A. Use of autoantigen-loaded phosphatidylserine-liposomes to arrest autoimmunity in type 1 diabetes. PLoS ONE. 2015;10(6), e0127057.
ACKNOWLEDGMENTS AND DISCLOSURES
This work was supported by grants from the National Science and Technology Major Project 2009ZX09103-029 and The Open Project Program of Key Lab of Smart Drug Delivery (Fudan University), Ministry of Education, China.
Author information
Authors and Affiliations
Corresponding author
Additional information
Xiaoyao Zheng and Xiayan Shao contributed equally to this work.
Rights and permissions
About this article

Cite this article
Zheng, X., Shao, X., Zhang, C. et al. Intranasal H102 Peptide-Loaded Liposomes for Brain Delivery to Treat Alzheimer’s Disease. Pharm Res 32, 3837–3849 (2015). https://doi.org/10.1007/s11095-015-1744-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11095-015-1744-9