
Abstract
Purpose
Multidrug and toxin extrusion proteins (MATEs) are multispecific organic cation transporters mediating the efflux of various cationic drugs into the urine. The present study aimed at identifying endogenous compounds in human plasma and urine specimens as biomarkers to evaluate drug interactions involving MATEs in the kidney without administration of their exogenous probe drugs.
Methods
An untargeted metabolomic analysis was performed using urine and plasma samples from healthy volunteers and mice treated with or without the potent MATE inhibitor, pyrimethamine. Plasma and urinary concentrations of candidate markers were measured using liquid chromatography-mass spectrometry. Transport activities were determined in MATE- or OCT2-expressing HEK293 cells. The deuterium-labeled compounds of candidates were administered to mice for pharmacokinetics study.
Results
Urinary excretion of eleven compounds including thiamine and carnitine was significantly lower in the pyrimethamine-treatment group in humans and mice, whereas no endogenous compound was noticeably accumulated in the plasma. The renal clearance of thiamine and carnitine was decreased by 70%–84% and 90%–94% (p < 0.05), respectively, in human. The specific uptake of thiamine was observed in MATE1-, MATE2-K- or OCT2-expressing HEK293 cells with Km of 3.5 ± 1.0, 3.9 ± 0.8 and 59.9 ± 6.7 μM, respectively. The renal clearance of carnitine-d 3 was decreased by 62% in mice treated with pyrimethamine.
Conclusions
Our findings indicate that MATEs account for the efflux of thiamine and perhaps carnitine as well as drugs into the urine. The urinary excretion of thiamine is useful to detect drug interaction involving MATEs in the kidney.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
INTRODUCTION
Multidrug and toxin extrusion proteins (MATEs) facilitate the exchange of organic cations and H+. Human MATEs are comprised of two isoforms, MATE1/SLC47A1 and MATE2-K/SLC47A2, both of which are expressed on the brush border membrane (BBM) of renal proximal tubules in the kidney (1,2) and exhibit overlapping substrate specificities for organic cations such as tetraethylammonium (TEA) and metformin (3,4). To date, MATEs are considered to account for the secretion of various cationic drugs into the urine from the blood circulation in conjunction with the basolateral uptake transporter, OCT2/SLC22A2. This concept has been supported in an animal study using Mate1 knockout mice which showed a marked delay in the elimination of metformin from the systemic circulation, accompanied by an elevation of the metformin concentration in the kidney (5). Also, the renal secretory clearance of varenicline was reduced by 55% in the Mate1 knockout mice, compared with that in normal mice (6). Furthermore, a genetic disruption in Mate1 potentiated the nephrotoxicity of the anti-cancer platinum agent cisplatin and of the herbicide paraquat by the marked accumulation of these compounds in the kidney (7,8).
We previously reported that, among the drugs in the market, cimetidine and pyrimethamine (PYR) are potent inhibitors of MATE1 and MATE2-K (9–11). PYR completely inhibited the uptake of MATEs substrates such as metformin and N-methylnicotinamide (NMN), by human-kidney BBM vesicles (BBMV), driven by the H+ gradient imposed across the membrane, with Ki values similar to those observed in cDNA transfectants expressing MATE1 and MATE2-K. It is of important to note that a comparison of the Ki values of cimetidine and PYR for MATEs and OCT2 with their maximum unbound plasma concentrations achieved at their usual clinical doses suggested that both drugs can significantly inhibit both MATE1 and MATE2-K without affecting OCT2. Indeed, cimetidine and PYR caused an elevation in the kidney concentrations of Mate substrates in mice (11,12). Cimetidine has been used to investigate the urinary excretion mechanisms of drugs in humans in drug development (12). Recently, we demonstrated that PYR significantly inhibited the renal elimination of metformin in healthy subjects (13). Furthermore, in addition to drugs, we also found that MATEs are involved in the urinary excretion of endogenous metabolites: PYR reduced the renal clearance of creatinine by ~20% (13) and of NMN by ~70% (14). This finding led the concept of prediction of transporter-mediated drug-drug interactions without administration of probe drugs. However, the role of MATEs in the urinary excretion of endogenous compounds needs more investigation.
The purpose of this study was to identify endogenous MATEs substrates in the human, plasma level and/or urinary excretion of which is highly sensitive to the activities of MATEs in the kidney. Untargeted metabolomics is a useful analytical approach for discovering novel endogenous or food-derived substrates (15–17). In this study, metabolomic analyses of plasma and urine specimens from eight healthy male volunteers treated with or without PYR in a crossover fashion were conducted using liquid chromatography-mass spectrometry (LC-MS). Consequently, we identified candidate endogenous substrates for MATEs, the renal clearances of which were markedly decreased by PYR. Our results offer a way of evaluating the inhibitory potency of drugs and drug candidates against MATEs in humans without administration of probe substrates in drug development.
MATERIALS AND METHODS
Materials
PYR was purchased from Wako Pure Chemicals (Osaka, Japan). Thiamine-d 3 hydrochloride was purchased from Toronto Research Chemicals (North York, Canada). Carnitine-d 3, acetylcarnitine-d 3, and 2′-deoxycytidine-15N3 were purchased from Cambridge Isotope Laboratories (Andover, MA). Carnitine-d 9 was purchased from CDN Isotopes (Quebec, Canada). [14C]TEA bromide (3.5 mCi/mmol) and [3H]thiamine hydrochloride (20 Ci/mmol) were purchased from PerkinElmer Life and Analytical Science (Waltham, MA) and American Radiolabeled Chemicals (St. Louis, MO), respectively. All other reagents that were used were commercially available and of analytical grade.
Clinical Samples
The study protocol was approved by the Ethics Review Boards of both the Graduate School of Pharmaceutical Sciences of the University of Tokyo and the Clinical Investigation Center, Kitasato University East Hospital.
Plasma and urine specimens were obtained from healthy subjects participating in a pharmacokinetic interaction study, which was registered with the UMIN Clinical Trials Registry at www.umin.ac.jp/ctr/index.htm (UMIN000002739) (13). No clinically undesirable signs or symptoms possibly attributable to the administration of metformin or pyrimethamine were identified throughout the study, except for a slight increase in the plasma creatinine level that did not result from kidney injury. All the subjects completed the study successfully according to the following protocol: briefly, eight male subjects were enrolled in the study after they had provided their written informed consent. The subjects were physically normal upon clinical examination and routine clinical testing, and none of the subjects had any history of significant medical illness or hypersensitivity to any drugs. The mean ± SD values of the age, body weight and serum creatinine level of the volunteers were 28.0 ± 4.5 years (range 22–34 years), 63.9 ± 6.9 kg (range 54.1–76.6 kg) and 0.82 ± 0.10 mg/dL (range 0.66–0.94 mg/dL), respectively.
The pharmacokinetic interaction study consisted of four periods and was conducted in a cross over fashion. Subjects received a microdose (100 μg) of metformin after an overnight fast during Periods 1 and 2 and received a therapeutic dose (250 mg) of metformin during Periods 3 and 4. During Periods 2 and 4, the volunteers ingested a Daraprim® tablet (50 mg pyrimethamine/tablet) 1 h before the administration of metformin. Blood samples were taken by direct venipuncture (sodium heparin anticoagulant) and were centrifuged to isolate the plasma, which was then stored at −80°C until analysis.
Animal Experiments
All the animal studies were approved by the Taisho Pharmaceutical Animal Care Committee. Male ddY mice were purchased from Japan SLC (Shizuoka, Japan). They were housed in an air-conditioned room set to a 12-h light/dark cycle, given access ad libitum to water and a standard laboratory diet (MF; Oriental Yeast, Tokyo, Japan), and used at the age of 7–8 weeks for experiments. In the PYR-treated group, a bolus dose of PYR (0.5 mg/kg in 0.9% NaCl and 10% ethanol) was administered via the tail vein 30 min before the injection of carnitine-d 3 (0.25 mg/kg). The vehicle was administered in the control group. The animals were anesthetized with isoflurane, and blood and urine samples were collected from the postcava and bladder, respectively, at predose and at 5, 15, 30, 60, and 120 min after the administration of carnitine-d 3. After the animals were sacrificed, their kidneys were quickly removed and weighed, then homogenized in 4 volumes of ice-cold water. The blood sample was collected in a tube containing EDTA as an anticoagulant and was centrifuged to obtain the plasma.
Metabolomic Analysis
Metabolomic analyses were performed using the following samples: human urine and plasma obtained 7 h after PYR dosing during Periods 1 and 2 and mouse urine, plasma and kidney samples obtained 120 min after dosing with carnitine-d 3. Cold solvent (acetonitrile: methanol = 9:1, 200 μL) was added to 50 μL of a biological sample. The mixture was vortexed and centrifuged for 5 min at 10,000 × g, and the supernatant was subjected to an LC-MS analysis. The LC system used was an Agilent 1100 series HPLC (Agilent Technologies, Santa Clara, CA) equipped with an XBridge HILIC column (3.5 μm, 4.6 × 50 mm; Waters, Milford, MA). The mobile phase consisted of 10 mM ammonium acetate (pH 5.0) as solvent A and acetonitrile as solvent B, and was delivered at 1.0 mL/min using 95% solvent B between 0 and 1 min followed by a linear gradient to 70% B at 10 min and 50% B at 11 min, then isocratic to 12 min with 50% B. The MS data were acquired using an LTQ Orbitrap XL mass spectrometer (Thermo Fisher Scientific, Waltham, MA). The samples were analyzed using positive ion-heated electrospray ionization. The MS conditions were set as follows: spray voltage, 2,500 V; scanning range, m/z 100–800; resolving power, 30,000; and normalized collision energy (CID), 35%. The freely available MZmine software (18) was used to align the data in the time domain and to integrate the peak area automatically. The HMDB (19) and KEGG (20) databases were used for structural searching based on accurate masses.
Quantification Using LC-MS/MS
Thiamine-d 3, dCyd-15N3, carnitine-d 9, and acetylcarnitine-d 3 were used as internal standards for the quantification of the corresponding analytes. The sample-pretreatment methods, LC system, and solvents for the mobile phase were the same as those used for metabolomic analysis conditions. The composition of the mobile phase for the quantification of thiamine and 2′-deoxycytidine (dCyd) was 90% solvent B (0–0.5 min), followed by a linear gradient to 80% B at 2.2 min, 60% B at 2.7 min, 50% B at 4.5 min and then isocratic to 5 min with 50% B. The composition of the mobile phase for carnitine and acetylcarnitine was 75% B (0–0.2 min), followed by a linear gradient to 50% B at 2.5 min, and then isocratic to 3 min with 50% B. MS data were acquired using an API4000 tandem mass spectrometer (AB Sciex, Foster City, CA). The analytes were detected by exploiting the transitions of m/z 265 → 122 for thiamine, m/z 228→/112 for dCyd, m/z 268 → 125 for thiamine-d 3, m/z 231 → 115 for dCyd-15 N3, m/z 162 → 103 for carnitine, m/z 204 → 85 for acetylcarnitine, m/z 171 → 103 for carnitine-d 9 and m/z 207 → 85 for acetylcarnitine-d 3. The lower limit of quantification was 1 ng/mL except for dCyd (0.3 ng/mL). The PYR levels were analyzed by exploiting the transitions of m/z 249 → 177 and using propranolol as an internal standard.
In Vitro Transport Study Using cDNA Transfectants
HEK293 cells stably expressing OCT2, MATE1 or MATE2-K were constructed previously (9,21). Cells were seeded 72 h before the transport assay in poly-l-lysine- and poly-l-ornithine-coated 12-well plates at a density of 1.5 × 105 cells per well. The cell culture medium was replaced with culture medium supplemented with 5 mM sodium butyrate 24 h before the transport assay to induce the expression of the transporters. The in vitro transport study was conducted as described previously (22). Briefly, the cells were washed twice and preincubated with Krebs–Henseleit buffer at 37°C for 15 min. The Krebs–Henseleit buffer consisted of 118 mM NaCl, 23.8 mM NaHCO3, 4.8 mM KCl, 1.0 mM KH2PO4, 1.2 mM MgSO4, 12.5 mM HEPES, 5.0 mM glucose, and 1.5 mM CaCl2 and was adjusted to pH 7.4. Uptake was initiated by the addition of [14C]TEA or [3H]thiamine in the presence or absence of TEA (5 mM) at 37°C. For evaluation of MATE-mediated transport, cells were preincubated in the presence of NH4Cl (20 mM) for intracellular acidification. Uptake was terminated at the designated times by the addition of ice-cold Krebs–Henseleit buffer after the removal of the incubation buffer. The cells were solubilized with NaOH overnight at 4°C, then neutralized with HCl. The radioactivity in the aliquots was measured using liquid scintillation counting. The protein concentration was determined using the Lowry method (23).
Kinetic Analyses
Specific uptake was calculated by subtracting the uptake by the vector-transfected cells from the uptake by the cDNA-transfected cells. The kinetic parameters were calculated using the following equations:
where Pdif is the nonsaturable uptake clearance, v is the uptake velocity of the substrate, Vmax is the maximum uptake rate, Km is the Michaelis constant, and S is the substrate concentration in the medium. Fitting was performed using a nonlinear least-squares method using the MULTI program (24).
Pharmacokinetic Analysis
The area under the plasma concentration–time curve (AUC) was calculated using the trapezoidal rule. The renal clearance (CLR) was calculated by dividing the cumulative urinary excretion (Xurine) by the corresponding AUC. The total plasma clearance (CLtot) was calculated by dividing the dose by the corresponding AUC.
Statistical Analysis
Data are presented as the mean ± S.E.M. A Student two-tailed paired t-test was used to identify significant differences between the two sets of data in the clinical study. A Student unpaired t-test for equal variances or the Welch test for unequal variances was used for the animal experiment. A value of P < 0.05 was considered statistically significant. All the statistical analyses were performed using SAS 9 (SAS Institute, Cary, NC).
RESULTS
Untargeted Metabolomic Analysis
An untargeted metabolomic analysis was performed using urine samples obtained from humans and mice treated with or without PYR. An LC-MS analysis detected approximately 2,000 and 3,000 peaks in the human and mice specimens, respectively. Eleven compounds of interest were selected based on the following three criteria: 1) the relative urinary excretion (the product of the peak area and the urine volume) was decreased by more than half of the control value in both humans and mice; 2) the decrease was seen in at least 7 of the 8 volunteers; and 3) the peak intensity was strong enough for obtaining the product ion spectrum. The chromatographic and mass spectral data of these compounds are shown, along with the fold changes, in Table I. Online databases, HMDB (19) and KEGG (20), were searched according to the accurate mass-to-charge ratio (m/z) to suggest structural candidates. The LC/MS data of six compounds agreed with those of authentic standards: NMN, thiamine, dCyd, carnitine and the acyl-derivatives of carnitine, acetylcarnitine and propionylcarnitine. The structures of U146 and U160 were speculated to be 4-N-trimethylaminobutyrate and its CH2-elongated analogue by comparing their spectral data with that of carnitine. The structures of the other three unknown compounds have yet to be identified.
Plasma and kidney samples were also subjected to metabolomic analyses. No fold-changes greater than 2-fold were observed with the exception of PYR (m/z 249.09043) and its metabolites, the oxidized form (m/z 265.08527) and glucuronide (m/z 425.12233). The plasma levels in the PYR-treatment group of the eleven compounds described above were comparable to those in the control group (Table I). Their accumulations in mouse kidney reached a maximum of only 1.7-fold.
Effect of PYR on Plasma Levels and Urinary Excretion of MATE Substrate Candidates
The plasma and urinary concentrations of thiamine, carnitine, acetylcarnitine and dCyd were quantified using the corresponding stable isotopes as internal standards. The plasma concentration–time profiles of thiamine, carnitine and acetylcarnitine are shown in Fig. 1a. Their AUCs and CLR are listed in Table II. The dCyd concentration in the plasma was below the lower limit of quantification (0.3 ng/mL). This result is presumably due to its unstable nature in plasma. The stability of dCyd in human plasma was determined in vitro using cryopreserved human plasma. The residual amount of exogenous dCyd added to plasma (48 ng/mL) was decreased to 2.2% at an ambient temperature, and even to 28% on ice after 2 h. The plasma concentration of thiamine remained constant until 8 h during both the control and the PYR-treatment periods (Fig. 1a). The 12 h-samples from the control showed a slight elevation. The AUC did not differ between the control and the PYR-treatment periods except during the interval of 7–13 h falling within Periods 3 and 4 (Table II). No significant diurnal rhythms in the plasma carnitine level were observed (Fig. 1a). The inter-individual variation in the carnitine level (CV, 20%) was larger than the diurnal rhythm (CV, 6%). The plasma acetylcarnitine level tended to be higher in the morning. The AUCs of plasma carnitine and acetylcarnitine during the PYR-treatment periods were l5% lower than those during the untreated periods (Table II). The Xurine of the four compounds including dCyd is shown in Fig. 1b. The Xurine of all four compounds were markedly lower during the PYR-treatment periods (Periods 2 and 4) compared with that during the untreated periods (Periods 1 and 3); these results agree with the results of the metabolomic analysis. The CLR of thiamine, carnitine and acetylcarnitine were significantly reduced by 70%–84%, 90%–94% and 87%–91%, respectively, during the PYR-treatment periods, compared with the corresponding values during the untreated periods (Table II).

Effect of PYR on plasma concentration and cumulative urinary excretion of thiamine, carnitine, acetylcarnitine, and dCyd in healthy subjects. Metformin was administered orally at a dose of 0.1 mg (Periods 1 and 2) or 250 mg (Periods 3 and 4) 1 h after the oral administration of PYR. (a) The plasma concentrations and (b) urinary excretion amount (Xurine) in subjects in the control groups (Periods 1 and 3; open circles and open bars) and the PYR-treatment groups (Periods 2 and 4; closed circles and closed bars) were measured using LC-MS/MS. Each point and vertical bar represent the mean ± S.E.M. (n = 8). * P < 0.05; ** P < 0.01 versus control.
Uptake of Thiamine by HEK293 Cells Expressing MATE1 or MATE2-K
The uptakes of [3H]thiamine (10 nM) and [14C]TEA (a prototypical MATE substrate; 36 μM) in MATE1- or MATE2-K-expressing cells were significantly greater than those in vector-transfected control cells (Fig. 2a). These uptakes were decreased or saturated in the presence of an excess amount of non-labeled TEA. The uptake of thiamine by MATE1- or MATE2-K-expressing cells consisted of a saturable uptake and nonsaturable uptake (Fig. 2b and c). The kinetic parameters obtained by fitting were as follows: Km 3.54 ± 1.02 and 3.94 ± 0.79 μM; Vmax 105 ± 25 and 50.7 ± 9.3 pmol/2 min/mg protein; and Pdif 3.87 ± 0.66 and 3.55 ± 0.27 μL/2 min/mg protein for MATE1 and MATE2-K. [3H]Carnitine uptake by MATE1- and MATE2-K-expressing cells was similar to that by mock-vector transfected cells (data not shown). Because of high uptake of dCyd in mock-vector transfected cells, there was no significant difference in the uptake of dCyd between mock-vector transfected and MATE expressing cells.

Uptake of TEA and thiamine by HEK293 cells expressing MATE1 and MATE2-K. (a) HEK293 cells expressing MATE1 or MATE2-K were incubated with [14C]TEA (36 μM) or [3H]thiamine (10 nM) at 37°C for 10 min in the presence (closed bars) or absence (open bars) of TEA (5 mM) after intracellular acidification by preincubation in the presence of NH4Cl (20 mM). The concentration dependence of the transporter activities in HEK293 cells expressing (b) MATE1 or (c) MATE2-K is shown. The solid lines represent the fitted line obtained by nonlinear regression analysis based on Eq. 1 as described under the “Materials and Methods” section. Data are expressed as the mean ± S.E.M. (n = 3).
Uptake of Thiamine by HEK293 Cells Expressing OCT2
The uptakes of [3H]thiamine (10 nM) in OCT2-expressing cells were significantly greater than those in vector-transfected control cells (Fig. 3a). The uptake of [3H]thiamine by OCT2-expressing cells was markedly decreased in the presence of an excess amount of non-labeled TEA (5 mM). The uptake of thiamine by OCT2 was saturable (Fig. 3b), and the Km and Vmax values were 59.9 ± 6.7 μM and 1.09 ± 0.01 nmol/2 min/mg protein, respectively.

Uptake of thiamine by HEK293 cells expressing OCT2. (a) HEK293 cells expressing OCT2 were incubated with [3H]thiamine (10 nM) at 37°C for 2 min in the presence (closed bars) or absence (open bars) of TEA (5 mM). (b) The concentration dependence of the uptake by OCT2 is shown. The solid lines represent the fitted line obtained by nonlinear regression analysis based on Eq. 1 as described under the “Materials and Methods” section. Data are expressed as the mean ± S.E.M. (n = 3).
Effect of PYR on Disposition of Carnitine in Mice
PYR (0.5 mg/kg) was administered to mice in the PYR-treatment group 30 min before dosing with deuterium-labeled carnitine (carnitine-d 3). The unbound PYR concentration in the kidney was comparable to the Ki value for mouse Mate1 (145 nmol/L) (11). The plasma concentration of carnitine-d 3 did not differ significantly between the control group and the PYR-treatment group except at 120 min after carnitine-d 3 dosing (Fig. 4a). The CLtot were 17.0 and 15.5 mL/min/kg in the control group and the PYR-treatment group, respectively. The cumulative Xurine of carnitine-d 3 was significantly reduced by the PYR-treatment (Fig. 4b). The CLR was reduced by 62% in the PYR-treatment group (0.28 mL/min/kg), compared with that in the control group (0.74 mL/min/kg). Acyl forms derived from carnitine-d 3 in the samples collected at 120 min were detected by the metabolomic analysis. The relative urinary excretions of acetylcarnitine-d 3, propionylcarnitine-d 3, and butyrylcarnitine-d 3 in the PYR-treatment group were reduced by more than half of the value in the control, in addition to the exertion of free carnitine-d 3 (Fig. 4c). On the other hand, the relative plasma and kidney levels of acylcarnitines-d 3 were slightly higher in the PYR-treatment group or were similar in both groups (Figs. 4d and e). Thiamine-d 3 was also administered to the mice as a continuous infusion at rate of 1.86 nmol/kg/min, but its urinary excretion level was below the lower limit of quantification. PYR did not affect the plasma-concentration time profiles of thiamine-d 3 (data not shown).

Effect of PYR on the disposition of carnitine-d 3 in mice after dosing with carnitine-d 3. PYR (0.5 mg/kg) was administered to mouse intravenously 30 min before a bolus injection of carnitine-d 3 (0.25 mg/kg). (a) The plasma concentrations and (b) urinary excretion amount (Xurine) of carnitine-d 3 were measured using LC-MS/MS in the control group (open circles) and the PYR-treatment group (closed circles) at designated time points. The bladder urine (c), plasma (d), and kidney (e) samples obtained 120 min after dosing with carnitine-d 3 were subjected to metabolomic analysis. The bladder urine (c), plasma (d) and kidney (e) samples obtained 120 min after dosing with carnitine-d 3 were subjected to the metabolomic analysis. (c) The relative Xurine for carnitine-d 3 and its acyl forms represents the product of the urinary volume (mL) and each peak area expressed in an arbitrary unit. (d) The relative plasma concentration (Cplasma) and (e) relative kidney concentration (Ckidney) represent each peak area. Data are expressed as the mean ± S.E.M. of three or four mice. * P < 0.05; ** P < 0.01 versus control.
DISCUSSION
There is growing concern on the probe substrate and in vivo inhibitors for drug transporters since the U S Food and Drug Administration has just released draft guidance on drug interaction studies which proposes to conduct pharmacokinetic interaction studies for drug candidates in humans with in vivo inhibitors and probe drugs to evaluate the magnitude of the interaction during drug discovery and development when the risk is suggested by preclinical studies (25). The present study was designed to elucidate the endogenous substrates of MATEs using metabolomic analyses of clinical samples obtained from pharmacokinetic interaction studies of metformin with PYR in healthy subjects, where CLR of metformin was decreased by ~34% (13).
Untargeted metabolomic analyses of urine specimens from healthy subjects and mice treated with or without PYR picked out 11 compounds as candidates for MATE substrates; PYR treatment reduced the Xurine of each of these compounds by 50% or more (Table I). We were able to identify six of these compounds as NMN, thiamine, carnitine, acetylcarnitine, propionylcarnitine and dCyd. Thiamine is a vitamin (vitamin B1) that is essential for carbohydrate metabolism and neural function (26), and carnitine is indispensable in the transport of long chain fatty acids into mitochondria for the production of cellular energy by β-oxidation (27). Carnitine exists in the body as its free form or as acylcarnitines, which are conjugates produced from carnitine and acyl-CoA by carnitine acyltransferase. dCyd, a pyrimidine deoxyribonucleoside, is recycled in nucleotide synthesis via a salvage pathway or is biotransformed into deoxyuridine, followed by catabolism (28).
The plasma concentration-time profiles and the Xurine of thiamine, carnitine, acetylcarnitine, and dCyd were quantified (Fig. 1). The plasma concentration and CLR obtained in the control groups were comparable to previously reported values (29–31). The plasma concentration of dCyd was undetectable presumably because of its rapid conversion by cytidine deaminase in plasma (32). The plasma concentrations of thiamine and acetylcarnitine showed weak rhythmic patterns, increasing or decreasing along with time, respectively, while that of carnitine was constant at least during the first 12 h. The Xurine of these compounds, including dCyd, was markedly lower in the PYR treatment group. Since the plasma concentrations of thiamine, carnitine and acetylcarnitine were slightly lower in the subjects treated with PYR, the reduced urinary loss may partly result from lower plasma concentrations. However, the fact that PYR-treatment reduced the CLR of thiamine, carnitine and acetylcarnitine by approximately 80%, 90% and 90%, respectively, indicates that PYR inhibits the urinary excretion of these compounds (Table II).
Thiamine was reported to be a substrate of both MATE1 and MATE2-K in an in vitro transport study that used MATE1- and MATE2-K-expressing cells (3). This finding was reproduced in the present study (Fig. 2). In another study that used the BBMV of the rat kidney, thiamine/H+ antiport was observed (33). These in vitro studies also showed that thiamine can be used as a biomarker for evaluating the drug interactions in the urinary excretion that involves MATEs. Conversely, whether carnitine, acylcarnitine, and dCyd are MATE biomarkers remains controversial. The specific uptake of carnitine was not observed in either MATE1- or MATE2-K-expressing cells (data not shown), which agrees with the results of a previous study (3). However, consistent with the clinical observation, the CLR of deuterium-labeled carnitine was reduced by PYR, and the amount of acetylcarnitine excreted into the urine was also decreased by PYR, whereas PYR had no effect on the plasma acetylcarnitine level and on the ratio of acetylcarnitine to free carnitine in mice (Fig. 4). Therefore, it is most likely that the reduced CLR of carnitine and acetylcarnitine is attributable to the effect of PYR on Mate1 in the mouse, but not on the biosynthesis of carnitine and its interconversion between carnitine and acetylcarnitine in mouse. Previously, we reported that the specific uptake of cephalexin was not observed in cDNA-transfected cells; however, both PYR and cimetidine significantly elevated the kidney concentration of cephalexin (12). A marked delay in the urinary excretion of cephalexin was observed in a Mate1 knockout mouse, which supports the predominant role of Mate1 in the urinary excretion of cephalexin (34). That is, the absence of specific uptake of carnitine in the cDNA-transfected cells does not necessarily rule out the possibility that MATEs mediate its efflux into the urine. As for dCyd, there are no additional data supporting its application as a MATE biomarker because of its instability.
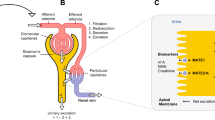
The kidney plays an important role in regulating the body homeostasis of thiamine, carnitine, and acetylcarnitine by extensive reabsorption from the urine after filtration in glomeruli. In fact, the CLR of thiamine, carnitine, and acetylcarnitine (~50, ~3, and ~6 mL/min, respectively) was below the glomerular filtration rate (GFR), given that the GFR is 120 mL/min. The transporters involved in the process of reabsorption of thiamine and carnitine/acetylcarnitine are the thiamine transporters 1 and 2 (THTR1/SLC19A2 and THTR2/SLC19A3) (35) and a high-affinity Na+/l-carnitine cotransporter, OCTN2/SLC22A5 (36), respectively; these transporters are expressed in the BBM of the proximal tubules. Similarly, an equilibrative nucleoside transporter (ENT1/SLC29A1) and concentrative nucleoside transporters (CNT1/SLC28A1, CNT2/SLC28A2, and CNT3/SLC28A3) (37) may mediate the reabsorption of dCyd for nucleoside salvage in the human BBM. Given that PYR completely inhibits the efflux across the BBM in the proximal tubules, but does not affect the reabsorption process, and that biosynthesis and metabolism in the kidney are negligible, the fractions of reabsorption can be estimated by comparing the renal clearance when PYR is given with GFR (Fig. 5a). Based on the estimated fraction of reabsorption, it is suggested that the clearance of thiamine via tubular secretion is 4-fold greater than the GFR. In fact, together with the increase in its plasma concentration, the CLR of thiamine increases and becomes close to the renal plasma flow rate in humans because of the saturation of its reabsorption (38). Of note, it has been reported that specific uptake of thiamine by OCT2 was not observed in cDNA-transfected HeLa cells (39); in contrast, we demonstrated that thiamine is a substrate of OCT2 (Fig. 3). The reason for this discrepancy remains unknown. However, this finding supports the efficient tubular secretion of thiamine upon saturation of reabsorption reported previously. We propose that the removal of thiamine from the systemic circulation when it is present in the blood circulation at high concentrations, to saturate its reabsorption, is one of the physiological roles of OCT2 and MATE in the kidney. This concept needs further verification by examining the effect of PYR in individuals receiving thiamine supplementation. Similarly, carnitine was estimated to be secreted at 9-fold the GFR (Fig. 5b). However, unlike what was observed for thiamine, there is no clinical evidence that carnitine undergoes tubular secretion in the kidney. Even when a high dose of carnitine is administered, the renal carnitine clearance is below the GFR (40). Therefore, another hypothetical diagram assuming negligible uptake from the basolateral side (Fig. 5c) may be appropriate for carnitine. In addition, taking the biosynthesis and metabolism of carnitine in the kidney into consideration will improve the estimates of clearance in the diagram. Further investigations are needed to propose a kinetic model of carnitine disposition in the kidney.

Proposed schematic diagrams showing the disposition of thiamine and carnitine in the kidney in humans. The renal clearance (CLR) of (a) thiamine and (b) carnitine was defined as follows: CLR = (GFR + CLsec) · (1–FR), where GFR, CLsec, and FR represent the glomerular filtration rate, tubular secretion clearance, and fraction of reabsorption from the urine to the blood, respectively. For carnitine (c), another hypothetical diagram was also designed assuming negligible uptake of the compound from the basolateral side. The diagram includes rough estimates of the contribution of the glomerular filtration (black arrows), tubular secretion (gray arrows), and reabsorption (dotted line) under the following assumptions: PYR does not affect GFR and reabsorption [FR · (GFR + CLsec)]. The units of GFR and CLsec are mL/min.
Evaluating the DDI risk involving MATEs is an important issue in the avoidance of severe DDIs since the inhibition of MATEs causes an elevation of the tissue concentrations of the victim drugs, and consequently potentiates its effect in the kidney. Several endogenous compounds with different kinetic characteristics are now available for this purpose. NMN and creatinine are secretion type, whereas thiamine is reabsorption type. The plasma levels of NMN show circadian rhythm for 12 h (14), whereas that of thiamine showed weak circadian rhythm. The fact that the CLR of NMN and thiamine is more susceptible to the changes in the activities of MATEs, which showed a 70–80% reduction, than is metformin (34%, (13)) supports their usefulness as MATE biomarkers. It is obvious that use of endogenous compounds as probe can save time and the expense of additional clinical studies to evaluate the risk of DDIs during drug development. Further studies are necessary to establish such endogenous substrates for other drug transporters.
CONCLUSION
Our findings suggest that MATEs account for the efflux of thiamine, and perhaps carnitine, into the urine. Urinary excretion of thiamine is a useful biomarker for assessing the inhibitory potential of investigational drugs toward MATEs in preclinical and early clinical studies of drug development.
Abbreviations
- AUC:
-
area under the plasma concentration–time curve
- BBM:
-
brush border membrane
- BBMV:
-
brush border membrane vesicles
- CLR :
-
renal clearance
- CLtot :
-
total plasma clearance
- dCyd:
-
2′-deoxycytidine
- GFR:
-
glomerular filtration rate
- LC-MS:
-
liquid chromatography-mass spectrometry
- MATE:
-
multidrug and toxin extrusion protein
- NMN:
-
N-methylnicotinamide
- OCT:
-
organic cation transporter
- PYR:
-
pyrimethamine
- TEA:
-
tetraethylammonium
- Xurine :
-
urinary excretion amount
REFERENCES
Yonezawa A, Inui K. Importance of the multidrug and toxin extrusion MATE/SLC47A family to pharmacokinetics, pharmacodynamics/toxicodynamics and pharmacogenomics. Br J Pharmacol. 2011;164(7):1817–25.
Moriyama Y, Hiasa M, Matsumoto T, Omote H. Multidrug and toxic compound extrusion (MATE)-type proteins as anchor transporters for the excretion of metabolic waste products and xenobiotics. Xenobiotica. 2008;38(7–8):1107–18.
Tanihara Y, Masuda S, Sato T, Katsura T, Ogawa O, Inui K. Substrate specificity of MATE1 and MATE2-K, human multidrug and toxin extrusions/H(+)-organic cation antiporters. Biochem Pharmacol. 2007;74(2):359–71.
Damme K, Nies AT, Schaeffeler E, Schwab M. Mammalian MATE (SLC47A) transport proteins: impact on efflux of endogenous substrates and xenobiotics. Drug Metab Rev. 2011;43(4):499–523.
Tsuda M, Terada T, Mizuno T, Katsura T, Shimakura J, Inui K. Targeted disruption of the multidrug and toxin extrusion 1 (Mate1) gene in mice reduces renal secretion of metformin. Mol Pharmacol. 2009;75(6):1280–6.
Kajiwara M, Masuda S, Watanabe S, Terada T, Katsura T, Inui K. Renal tubular secretion of varenicline by multidrug and toxin extrusion (MATE) transporters. Drug Metab Pharmacokinet. 2012;27(6):563–9.
Nakamura T, Yonezawa A, Hashimoto S, Katsura T, Inui K. Disruption of multidrug and toxin extrusion MATE1 potentiates cisplatin-induced nephrotoxicity. Biochem Pharmacol. 2010;80(11):1762–7.
Li Q, Peng X, Yang H, Wang H, Shu Y. Deficiency of multidrug and toxin extrusion 1 enhances renal accumulation of paraquat and deteriorates kidney injury in mice. Mol Pharm. 2011;8(6):2476–83.
Matsushima S, Maeda K, Inoue K, Ohta KY, Yuasa H, Kondo T, et al. The inhibition of human multidrug and toxin extrusion 1 is involved in the drug-drug interaction caused by cimetidine. Drug Metab Dispos. 2009;37(3):555–9.
Tsuda M, Terada T, Ueba M, Sato T, Masuda S, Katsura T, et al. Involvement of human multidrug and toxin extrusion 1 in the drug interaction between cimetidine and metformin in renal epithelial cells. J Pharmacol Exp Ther. 2009;329(1):185–91.
Ito S, Kusuhara H, Kuroiwa Y, Wu C, Moriyama Y, Inoue K, et al. Potent and specific inhibition of mMate1-mediated efflux of type I organic cations in the liver and kidney by pyrimethamine. J Pharmacol Exp Ther. 2010;333(1):341–50.
Ito S, Kusuhara H, Yokochi M, Toyoshima J, Inoue K, Yuasa H, et al. Competitive inhibition of the luminal efflux by multidrug and toxin extrusions, but not basolateral uptake by organic cation transporter 2, is the likely mechanism underlying the pharmacokinetic drug-drug interactions caused by cimetidine in the kidney. J Pharmacol Exp Ther. 2012;340(2):393–403.
Kusuhara H, Ito S, Kumagai Y, Jiang M, Shiroshita T, Moriyama Y, et al. Effects of a MATE protein inhibitor, pyrimethamine, on the renal elimination of metformin at oral microdose and at therapeutic dose in healthy subjects. Clin Pharmacol Ther. 2011;89(6):837–44.
Ito S, Kusuhara H, Kumagai Y, Moriyama Y, Inoue K, Kondo T, et al. N-Methylnicotinamide is an endogenous probe for evaluation of drug-drug interactions involving multidrug and toxin extrusions (MATE1 and MATE2-K). Clin Pharmacol Ther. 2012;92(5):635–41.
Wikoff WR, Nagle MA, Kouznetsova VL, Tsigelny IF, Nigam SK. Untargeted metabolomics identifies enterobiome metabolites and putative uremic toxins as substrates of organic anion transporter 1 (Oat1). J Proteome Res. 2011;10(6):2842–51.
Krumpochova P, Sapthu S, Brouwers JF, de Haas M, de Vos R, Borst P, et al. Transportomics: screening for substrates of ABC transporters in body fluids using vesicular transport assays. FASEB J. 2012;26(2):738–47.
Kato K, Kusuhara H, Kumagai Y, Ieiri I, Mori H, Ito S, et al. Association of multidrug resistance-associated protein 2 single nucleotide polymorphism rs12762549 with the basal plasma levels of phase II metabolites of isoflavonoids in healthy Japanese individuals. Pharmacogenet Genomics. 2012;22(5):344–54.
Pluskal T, Castillo S, Villar-Briones A, Oresic M. MZmine 2: modular framework for processing, visualizing, and analyzing mass spectrometry-based molecular profile data. BMC Bioinforma. 2010;11:395.
Wishart DS, Knox C, Guo AC, Eisner R, Young N, Gautam B, et al. HMDB: a knowledgebase for the human metabolome. Nucleic Acids Res. 2009;D603-10.
Kanehisa M, Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28(1):27–30.
Busch AE, Karbach U, Miska D, Gorboulev V, Akhoundova A, Volk C, et al. Human neurons express the polyspecific cation transporter hOCT2, which translocates monoamine neurotransmitters, amantadine, and memantine. Mol Pharmacol. 1998;54(2):342–52.
Hirano M, Maeda K, Shitara Y, Sugiyama Y. Contribution of OATP2 (OATP1B1) and OATP8 (OATP1B3) to the hepatic uptake of pitavastatin in humans. J Pharmacol Exp Ther. 2004;311(1):139–46.
Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. Protein measurement with the Folin phenol reagent. J Biol Chem. 1951;193(1):265–75.
Yamaoka K, Tanigawara Y, Nakagawa T, Uno T. A pharmacokinetic analysis program (multi) for microcomputer. J Pharmacobiodyn. 1981;4(11):879–85.
US Food and Drug Administration. Guidance for Industry: drug interaction studies—study design, data analysis, implications for dosing, and labeling recommendations (Draft guidance) (http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM292362.pdf) (14 September 2012).
Finglas PM. Thiamin. Int J Vitam Nutr Res. 1993;63(4):270–4.
Steiber A, Kerner J, Hoppel CL. Carnitine: a nutritional, biosynthetic, and functional perspective. Mol Aspects Med. 2004;25(5–6):455–73.
Moyer JD, Malinowski N, Ayers O. Salvage of circulating pyrimidine nucleosides by tissues of the mouse. J Biol Chem. 1985;260(5):2812–8.
Lynch PL, Young IS. Determination of thiamine by high-performance liquid chromatography. J Chromatogr A. 2000;881(1–2):267–84.
Bain MA, Milne RW, Evans AM. Disposition and metabolite kinetics of oral L-carnitine in humans. J Clin Pharmacol. 2006;46(10):1163–70.
Cao Y, Wang YX, Liu CJ, Wang LX, Han ZW, Wang CB. Comparison of pharmacokinetics of L-carnitine, acetyl-L-carnitine and propionyl-L-carnitine after single oral administration of L-carnitine in healthy volunteers. Clin Invest Med. 2009;32(1):E13–9.
Hale JT, Bigelow JC, Mathews LA, McCormack JJ. Analytical and pharmacokinetic studies with 5-chloro-2′-deoxycytidine. Biochem Pharmacol. 2002;64(10):1493–502.
Gastaldi G, Cova E, Verri A, Laforenza U, Faelli A, Rindi G. Transport of thiamin in rat renal brush border membrane vesicles. Kidney Int. 2000;57(5):2043–54.
Watanabe S, Tsuda M, Terada T, Katsura T, Inui K. Reduced renal clearance of a zwitterionic substrate cephalexin in MATE1-deficient mice. J Pharmacol Exp Ther. 2010;334(2):651–6.
Ashokkumar B, Vaziri ND, Said HM. Thiamin uptake by the human-derived renal epithelial (HEK-293) cells: cellular and molecular mechanisms. Am J Physiol Renal Physiol. 2006;291(4):F796–805.
Nezu J, Tamai I, Oku A, Ohashi R, Yabuuchi H, Hashimoto N, et al. Primary systemic carnitine deficiency is caused by mutations in a gene encoding sodium ion-dependent carnitine transporter. Nat Genet. 1999;21(1):91–4.
Elwi AN, Damaraju VL, Baldwin SA, Young JD, Sawyer MB, Cass CE. Renal nucleoside transporters: physiological and clinical implications. Biochem Cell Biol. 2006;84(6):844–58.
Weber W, Nitz M, Looby M. Nonlinear kinetics of the thiamine cation in humans: saturation of nonrenal clearance and tubular reabsorption. J Pharmacokinet Biopharm. 1990;18(6):501–23.
Dutta B, Huang W, Molero M, Kekuda R, Leibach FH, Devoe LD, et al. Cloning of the human thiamine transporter, a member of the folate transporter family. J Biol Chem. 1999;274(45):31925–9.
Evans AM, Fornasini G. Pharmacokinetics of L-carnitine. Clin Pharmacokinet. 2003;42(11):941–67.
ACKNOWLEDGMENTS AND DISCLOSURE
We thank K. Taguchi, N. Hagima, S. Kamigaso, and K. Iwata of the Taisho Pharmaceutical Company for their skilled and expert technical assistance.
The clinical study was conducted as the NEDO MicroDose-PJ, sponsored by the New Energy and Industrial Technology Development Organization (NEDO), Japan. This study was supported by a Grant-in-Aid for Scientific Research (S) [Grant 24229002], for Scientific Research (B) [Grant 23390034] and for Challenging Exploratory Research [24659071] from Japan Society for the Promotion of Science, Japan, and Scientific Research on Innovative Areas HD-Physiology [Grant 23136101] from the Ministry of Education, Science, and Culture of Japan. It was also supported by a Grant-in-Aid from The Nakatomi Foundation.
K. Kato and H. Mori are full-time employees of Taisho Pharmaceutical Company. The authors have no conflicts of interest that are directly relevant to this study.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Kato, K., Mori, H., Kito, T. et al. Investigation of Endogenous Compounds for Assessing the Drug Interactions in the Urinary Excretion Involving Multidrug and Toxin Extrusion Proteins. Pharm Res 31, 136–147 (2014). https://doi.org/10.1007/s11095-013-1144-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11095-013-1144-y