
ABSTRACT
Cancer cells catabolise nutrients in a different way than healthy cells. Healthy cells mainly rely on oxidative phosphorylation, while cancer cells employ aerobic glycolysis. Glucose is the main nutrient catabolised by healthy cells, while cancer cells often depend on catabolism of both glucose and glutamine. A key organelle involved in this altered metabolism is mitochondria. Mitochondria coordinate the catabolism of glucose and glutamine across the cancer cell. Targeting mitochondrial metabolism in cancer cells has potential for the treatment of this disease. Perhaps the most promising target is the hexokinase-voltage dependent anion channel-adenine nucleotide translocase complex that spans the outer- and inner-mitochondrial membranes. This complex links glycolysis, oxidative phosphorylation and mitochondrial-mediated apoptosis in cancer cells. This review discusses cancer cell mitochondrial metabolism and the small molecule inhibitors of this metabolism that are in pre-clinical or clinical development.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
CANCER CELL METABOLISM
In a healthy cell the vast majority of energy is supplied by oxidative phosphorylation. Glucose is the main source of energy, undergoing glycolysis in the cytosol, before pyruvate enters the mitochondria and is converted to acetyl CoA. The tricarboxylic acid (TCA) cycle moves the carbon backbone around, generating FADH2 and NADH which can then enter oxidative phosphorylation and produce ATP.
Many cancer cells exhibit increased glycolysis, with much of the pyruvate being converted to lactate, in contrast to entry into the mitochondria and conversion to acetyl CoA. This is known as the Warburg effect (1). The increase in glucose metabolism to lactate, though, provides only a fraction of the ATP that is provided by oxidative phosphorylation. The relative contribution of mitochondria to ATP production varies between cancer type (for a review see (2)). Up to 34 molecules of ATP are produced by the complete metabolism of glucose, with glycolysis contributing only two of these molecules. A number of theories have been proposed to explain the apparently inefficient switch in energy supply, beginning with Warburg’s original proposal that mitochondria are defective in cancer (1). It has become clear, though, that cancer cells often have normal levels of oxidative phosphorylation (2–4). The current theory for the Warburg effect is that the increased biosynthesis required by rapidly proliferating cells (anabolism of DNA, lipids and proteins) is more efficiently supplied by aerobic glycolysis than oxidative phosphorylation (5–7).
Rapidly proliferating cancer cells require not only energy, but also building blocks such as amino acids and nucleic acids for cell biosynthesis and mitosis. In that regard, various glycolytic and TCA cycle intermediates are siphoned off, requiring greater amounts of glucose, or an alternative, to supply the greater demand for biosynthetic components. Glutamine catabolism appears to be the alternative for the supply of these components in cancer cells (8–10). As a cell prepares for mitosis, the expression of enzymes involved in nucleotide, carbohydrate and lipid synthesis increases. These processes are supported by precursors from glycolysis, the pentose phosphate pathway, the tricarboxylic acid pathway, oxidative phosphorylation and amino acid metabolism (in particular glutaminolysis).
Tumors are characterized by regions of hypoxia resulting in low levels of nutrients and increased levels of waste (11). One of the key responses to hypoxia within a cell is the expression of hypoxia inducible factor 1 (HIF1). HIF1 shifts the cell’s metabolism from oxygen-dependent energy generation to oxygen-independent mechanisms. In tumor cells, HIF1 expression can be upregulated even under normal oxygen conditions. A number of oncogenes contribute to HIF1 expression in cancer, including Ras, SRC and phosphoinositide 3-kinase, or the loss of tumor suppressor genes such as von Hippel-Lindau or PTEN (12). HIF1 is a transcription factor that activates genes involved in the characteristic shift in metabolism of tumor cells, including enzymes of glycolysis such as hexokinase, phosphofructokinase, lactate dehydrogenase and the glucose transporter GLUT1 (12,13). Pyruvate dehydrogenase kinase is activated by HIF1, which inhibits the entry of pyruvate into the TCA cycle. HIF1 upregulation, therefore, accounts for many of the changes associated with the Warburg effect.
Along with HIF1, the expression of the transcription factors c-Myc and p53 are also altered in many cancer cells. A frequently expressed oncogene, c-Myc is involved in the transactivation of lactate dehydrogenase and the glucose transporter GLUT1 (14,15), and the induction of glutaminolysis (16). HIF1 can cooperate with c-Myc to increase pyruvate dehydrogenase kinase and hexokinase II expression (17). p53 is a tumor suppressor gene that is frequently expressed in a mutated form in tumors, leading to an increase in hexokinase II expression, an increase in glucose uptake, a reduced inhibition of phosphofructokinase-1 (increased activity), and reduced assembly of the cytochrome c oxidase complex (complex IV of the electron transport chain) (18).
Cancers, therefore, often exhibit altered expression of metabolic enzymes. Where cancer cell metabolism differs from healthy cell metabolism provides an opportunity to selectively target this disease. In this review, we focus on the role of mitochondria in cancer cell metabolism. A number of small molecule inhibitors of mitochondrial metabolism that target a number of mitochondrial transporters or enzymes are in pre-clinical or clinical development. We discuss the mechanisms of action and state of development of these inhibitors.
MITOCHONDRIAL METABOLISM IN CANCER CELLS
Despite the shift from oxidative phosphorylation to aerobic glycolysis in cancer cells, mitochondria still play a key role in their survival. In cancer cells, mitochondria supply anywhere from 40 to 75% of the cells ATP requirements (19). This means that disrupting oxidative phosphorylation will significantly interrupt the cancer cell’s energy supply. However, it is not just the mitochondria’s importance in energy supply that makes them a good target for cancer therapy. Mitochondria also play a major role in the synthesis of amino acids, purines, pyrimidines, carbohydrates and fatty acids by supplying intermediates from the TCA cycle.
In the following sections we discuss the role that mitochondria play in cancer cell glycolysis, oxidative phosphorylation, ATP transport and glutaminolysis. Synthetic and natural inhibitors of proteins involved in these respective pathways are discussed and the current state of their development as anti-cancer agents is presented.
Glycolysis
Glycolysis is the catabolic pathway through which glucose is broken down to pyruvate. It involves ten reactions, consuming two and producing four molecules of ATP per glucose molecule. Other monosaccharides can enter glycolysis after being first converted to any one of the intermediates. Intermediates can be siphoned off to fuel other metabolic pathways, for example glucose 6-phosphate is the first substrate of the pentose phosphate pathway. Glycolysis occurs in the cytosol under aerobic or anaerobic conditions. Two phases of glycolysis exist: the first phase consumes ATP whilst converting glucose to two molecules of glyceraldehyde 3-phosphate, while the second phase converts each molecule of glyceraldehyde 3-phosphate to pyruvate producing four ATP in the process.
Hexokinase (HK, EC 2.7.1.1) mediates the first step of glucose catabolism, phosphorylating glucose to produce glucose 6-phosphate. Once phosphorylated, glucose is essentially trapped in the cell, as GLUT, the passive bidirectional transporter of glucose, is unable to transport glucose 6-phosphate out of the cell (20). There are four isoforms of HK, numbered I through IV. Isoforms I and II can interact with the outer-mitochondrial membrane, isoform III is found in a perinuclear compartment, and isoform IV (also known as glucokinase) is cytosolic (21,22). HKI is predominantly expressed in the brain, HKII in insulin sensitive tissue, HKIII has very low expression but can be detected in the lung, and HKIV is found in the liver and the pancreas (22). HKII expression is silenced in normal cells due to methylation of its promoter (23). In malignant cells, HKII is overexpressed (21,24,25) and generally accounts for the increased activity of hexokinases. Hypoxia (26) and the mutated p53 gene (27) are responsible for increased HKII expression in cancer cells.
When tumors arise in organs that predominantly express HKIV, such as the liver and the pancreas, one of the first changes observed is a switch in expression from HKIV to HKII (28,29). The properties of HKII are an important factor in aerobic glycolysis: i) HKI-III have a higher affinity for glucose than HKIV (Km of 0.02 mM for I–III, and 5 mM for IV) (21), ii) the N-terminal hydrophobic domain allows HKI and II to associate with the mitochondrial membrane in contact with the voltage dependant anion channel (VDAC) and, iii) in contrast to HKI, HKII has two active glucokinase-equivalent-domains (21) and is more efficient at phosphorylating glucose. HKII expression in cancer cells, therefore, enhances their ability to catabolise glucose.
HKII binds to the voltage dependent anion channel (VDAC) of the outer mitochondrial membrane, which interacts with adenine nucleotide translocase (ANT) of the inner-mitochondrial membrane (30) (Fig. 1). This has implications for both glycolysis and apoptosis of cancer cells. HKII’s association with VDAC affords it preferential access to ATP produced by oxidative phosphorylation (31), while blunting glucose 6-phosphate feedback inhibition of the enzyme (32). This allows for greater flux of glucose in cancer cells. Moreover, binding of HKII to VDAC has been shown to inhibit apoptosis of the cell (33–35). Binding of HKII to VDAC blocks binding of the pro-apoptotic molecule Bax to VDAC (33). Bax binding to VDAC initiates cytochrome c release from the mitochondria, propagating apoptosis across the cell. The HKII interaction is not specific for a particular VDAC isoform (36).

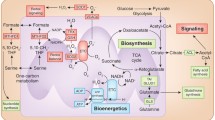
Mitochondrial metabolism in cancer cells. Cancer cells fuel their growth through the catabolism of two main nutrients: glucose and glutamine. The first step in glycolysis, conversion of glucose to glucose-6-phosphate (G-6-P), is catalyzed by hexokinase and cancer cells mostly employ an isoform (HKII) that is bound to mitochondria via interaction with the voltage dependent anion channel (VDAC). This affords HKII preferential access to the mitochondrial ATP, generated by oxidative phosphorylation (complexes I-V), via inner-membrane adenine nucleotide translocase (ANT). The pyruvate (Pyr) generated by glycolysis is converted to lactic acid (Lac) or enters the mitochondrial matrix where it is catabolised to acetyl coenzyme A (AcCoA) by pyruvate dehydrogenase (PDH), which is controlled by pyruvate dehydrogenase kinase (PDHK). The expression of genes required for glutamine uptake and catabolism are increased in cancer cells and mitochondrial metabolism is reprogrammed to depend more on glutamine catabolism for TCA cycle carbon in the form of α-ketoglutarate (αKG). One mechanism of conversion of Glu to αKG is catalysed by glutamate pyruvate transaminase (GPT). Mutations in the TCA cycle enzymes succinate dehydrogenase (SDH), fumarate hydratase (FH) and isocitrate dehydrogenase (IDH) have been linked to tumorigenesis.
The nature of the HKII-VDAC interaction has been probed in bovine brain mitochondria (37,38). There appears to be two types of interaction, one that is dissociable by glucose 6-phosphate and one that is not (38). The HK in this system utilizes predominantly mitochondrial ATP as its co-substrate, rather than cytosolic sources of ATP (38). Binding of HK to VDAC is enhanced when VDAC is associated with ANT (39,40). In addition, binding of HK to VDAC induces channel closure that is reversed by glucose 6-phosphate (41). Interestingly, the HK-VDAC association in cancer cells appears to influence the lipid (cholesterol) distribution of the mitochondrial membrane (42). Changes in membrane cholesterol content have the potential to affect the function of membrane bound enzymes, such as those of the electron transport chain.
It has been proposed that binding of HK to VDAC-ANT in cancer cells helps to maintain the mitochondrial membrane potential by facilitating the reverse reaction catalyzed by HK, that is, conversion of glucose-6-phosphate to glucose (43,44). This then leads to mitochondrial import of ATP, not export. However, this model is controversial as HK has also been reported to inhibit VDAC-mediated import of ATP into mitochondria (45).
Pyruvate dehydrogenase (PDH, EC 1.2.4.1) is the connecting step between glycolysis and the TCA cycle (Fig. 1). Following entry of pyruvate into the mitochondria, by an as yet unknown transporter, PDH catalyzes its conversion into acetyl CoA in an irreversible reaction (46). PDH activity is regulated by phosphorylation, which is controlled by two enzymes PDH kinase (EC 2.7.11.2) and PDH phosphatase (EC 3.1.3.43) (Fig. 1). PDH is inactive in its phosphorylated state. Cancer cells express high levels of PDH kinase (17,47–50) and so maintain PDH in its inactive form, which promotes aerobic glycolysis. Inhibiting PDH kinase permits PDH activity and the channeling of pyruvate into the TCA cycle (51).
The TCA cycle, also known as the citric acid cycle or Krebs cycle, continues the breakdown of glucose linking glycolysis to oxidative phosphorylation (Fig. 1). It consists of a series of reactions transferring hydride ions to NAD and FAD (52). The entry of newly synthesized acetyl CoA is commonly considered as the starting point of the cycle. In the cycle, succinate undergoes oxidation by succinate dehydrogenase (SDH, EC 1.3.99.1, 1.3.5.1) to form fumarate, while fumarate undergoes hydration by fumarate hydratase (FH, EC 4.2.1.2) to form malate (Fig. 1). SDH and FH have been described as tumor suppressors. When either of these genes are mutated it can lead to paragangliomas (53,54) and phaerochromocytomas, and in the case of FH, cutaneous and uterine leiomyomas and renal cell cancer (55–57). Mutations of SDH and FH prevent the degradation of HIF1, as they lead to accumulation of succinate and fumarate that inhibits prolyl hydroxylase (58,59). Prolyl hydroxylase is responsible for triggering HIF1 degradation (58,59). Further, somatic mutations of isocitrate dehydrogenase (IDH, 1.1.1.42) have been found in glioblastoma cells (60). Notably, IDH mutations can result in a novel enzymatic reaction that converts α-ketoglutarate to 2-hydroxyglutarate (61), which may contribute to tumorigenesis (3).
Glycolysis Inhibitors
HKII is a promising cancer drug target as it links cancer cell glycolysis with mitochondrial-mediated apoptosis. Several HKII inhibitors are currently being studied as potential anti-cancer drugs, including analogues for glucose and azole derivatives. 2-Deoxyglucose is a glucose analogue and a competitive inhibitor of HK (Fig. 2). HK phosphorylates 2-deoxyglucose but it is not processed further by glucose 6-phosphate isomerase. As a result, the phosphorylated 2-deoxyglucose accumulates in the cell and competitively inhibits HK by negative feedback. Taxol- and melphalan-resistant cells have been shown to accumulate 2-deoxyglucose-6-phosphate to a greater extent than sensitive cells, which resulted in growth inhibition (62). Phase I clinical trials have determined the safety and pharmacokinetic parameters of 2-deoxyglucose in prostate cancer patients (63) (Table I). 5-Thioglucose and mannoheptulose (64) are two other analogues of glucose that inhibit HK.

Small molecule mitochondrial metabolism inhibitors in pre-clinical or clinical development for the treatment of cancer. 2-Deoxyglucose, 3-bromopyruvate and lonidamine target HKII. Jasmonates and azole derivatives disrupt the association between HKII and VDAC, while the organoarsenicals GSAO and PENAO, retinoic acids and lonidamine target ANT. Betulinic acid and honokiol target the mitochondrial permeability transition pore, of which VDAC and ANT are thought to be components. Dichloroacetate and AZD7545 inhibit PDHK. Rotenone and bullatacin inhibit complex I of the electron transport chain, vitamin E analogues inhibit complex II, benzylisothiocyanate inhibits complex III, and resveratrol inhibits complex V. Aminooxyacetate inhibits GPT, that functions in glutamine catabolism.
Azole derivatives such as clotrimazole or bifonazole are traditionally used as antifungals. These compounds are also thought to displace HK from the mitochondrial membrane (65) (Fig. 2), having a significant effect on cell viability (65–67) and sensitizing glioblastoma cells to radiation (67). Although this class of drugs has a calmodulin antagonistic action, the disassociation of HK from VDAC appears to be independent of this function (68). Clotrimazole has anti-tumor effects in a rat model of glioma (66).
PDH activity is negatively regulated by phosphorylation. The phosphorylation is mediated by PDH kinase and PDH phosphatase. PDH kinase is upregulated in cancer (47–50). Dichloroacetate is a pyruvate mimetic that allosterically inhibits PDH kinase (Fig. 2), allowing pyruvate to be channeled into the TCA cycle (69). Dichloroacetate has cytotoxic effects in a number of cancer cell lines (69–74). Interestingly, cancer cells with defects in the electron transport chain appear to be more sensitive to dichloroacetate (72). This observation suggests that dichloroacetate inhibition of PDH kinase leads to a greater reliance on mitochondrial metabolism, making the cell more susceptible to compounds that disrupt oxidative phosphorylation. Dichloroacetate has modest anti-tumor activity in mice (70,72,73), though a small trial of five patients with glioblastoma showed promising results (75). Unfortunately, the publicity of these results and the ease of access of dichloroacetate has led to self-medication and little motivation from pharmaceutical companies to take dichloroacetate through clinical trials as a cancer therapy (76). AZD7545 is a specific inhibitor of PDH kinase (77,78) (Fig. 2) that has been developed to treat diabetes by improving glucose control (79). It has potential to be used in the treatment of cancer, although it has not been studied as an anti-cancer agent. AZ12 and Nov3r are related compounds to AZD7545 that also inhibit PDH kinase (78).
Oxidative Phosphorylation
Oxidative phosphorylation (also referred to as respiration) utilizes the reducing power of NADH and FADH2 produced by the TCA cycle to reduce molecular oxygen to water. This releases a large amount of energy in the form of a proton gradient across the inner-mitochondrial membrane, which is then utilized to synthesize ATP via ATP synthase. The respiratory chain (the electron transport chain) consists of 5 complexes: NADH-ubiquinone reductase (complex I), succinate-Q reductase (complex II), cytochrome c oxidoreductase (complex III), cytochrome c oxidase (complex IV) and ATP synthase (complex V). Complexes I, III and IV combine the reduction of their substrates with the movement of protons into the inter-membrane space, creating the gradient that drives ATP synthase (complex V) (Fig. 1).
Complex I (EC 1.6.5.3) transfers two electrons from NADH to coenzyme Q, and translocates four protons into the inter-membrane space. Coenzyme Q then carries the electrons to Complex III. Premature electron leakage commonly occurs at complex I and is one of the main sites for production of reactive oxygen species (80,81). Complex II (EC 1.3.5.1) transfers two electrons from succinate (from the TCA cycle) to FADH2 to coenzyme Q, then coenzyme Q carries two protons across the membrane. Succinate dehydrogenase, the TCA cycle enzyme, forms part of complex II. Complex III (EC 1.10.2.2) transfers a pair of electrons from coenzyme Q to cytochrome c, and concurrently pumps a further four protons into the inter-membrane space. Complex IV (also known as cytochrome c oxidase, EC 1.9.3.1) catalyzes the oxidation of cytochrome c. This reaction is coupled to the reduction of O2 to two molecules of water and a further two protons are pumped into the inter-membrane space. Subunit four of cytochrome c oxidase is a regulatory subunit of complex IV. Under hypoxic conditions, expression switches from isoform 1 to isoform 2, allowing for optimal activity of cytochrome c oxidase (82). The final step of oxidative phosphorylation is catalyzed by ATP synthase (complex V, EC 3.6.3.6). ATP synthase uses the proton gradient across the inner-mitochondrial membrane to drive the production of ATP from ADP and orthophosphate.
All complexes of the electron transport chain have been implicated in cancer. Decreased expression and activity has been observed for complex I (83–85), complex II (86), complex III (85,86), complex IV (86) and complex V (86–88).
Oxidative Phosphorylation Inhibitors
Rotenone is an inhibitor of complex I (Fig. 2). It is isolated from the stems and roots of several plants and is commonly used as a natural insecticide (89). Rotenone induces apoptosis in a variety of cancer cell lines, including breast cancer, melanoma, leukemia, lymphoma and neuroblastoma (89–91). Studies have not progressed beyond cell culture experiments at this time.
Bullatacin is a member of the annonaceous acetogenins family of proteins, isolated from the fruit of Annona atemoya (92). It inhibits the pumping action of complex I of the electron transport chain (93) (Fig. 2). Bullatacin has cytotoxic activity against multidrug resistant human mammary adenocarcinoma MCF-7/Adr cells (94–96) and is efficacious in ovarian and leukemia murine tumor models (93). Toxicity was observed at doses greater than 1.4 mg/kg in an ovarian tumor model (94).
Vitamin E analogues such as alpha-tocopheryl succinate (alpha-TOS) can induce mitochondrial-mediated apoptosis in tumor cells. Alpha-TOS targets complex II, preventing binding of ubiquinone (97) (Fig. 2). This leads to electron leakage and subsequent generation of reactive oxygen species (97,98). Alpha-TOS is cytotoxic for proliferating endothelial cells (99) and tumor cells (100) and has anti-tumor activity in mouse breast and lung cancer models (101). In addition, dietary vitamin E intake is inversely related to bladder cancer incidence, which suggests a chemo-preventative action (102).
Benzylisothiocyanate has been isolated from cruciferous vegetables. In the breast cancer cell lines, MCF-7 and MDA-MB-231, this compound was shown to inhibit complex III (Fig. 2), generating reactive oxygen species and inducing apoptosis (103). It also has chemo-preventative properties in liver and lung solid tumor models in mice (104,105).
Resveratrol is found in grapes, belonging to the polyphenolic phytoalexins class of compounds (106). It binds to complex V and inhibits ATP synthesis (107,108) (Fig. 2). Resveratrol also inhibits the action of many anti-apoptotic proteins in vitro, including Bcl-xL (109), and has anti-tumor and chemo-preventative actions in skin and neuroblastoma cancer models (106,110). Derivatives of resveratrol bound to the membrane-permeable lipophilic triphenylphosphonium cation were developed and found to enhance resveratrol accumulation in mitochondria and improve selectivity for C-26 mouse colon cancer cells (111). Resveratrol has been studied in both healthy volunteers and colorectal cancer patients to determine suitable dosing levels for its chemo-preventative action (112,113) (Table I).
Mitochondrial ATP Transport
ATP synthase is closely aligned to ANT, the transporter responsible for supplying it with ADP for the synthesis of ATP, in a complex known as the ATP synthasome (114,115). ANT exchanges the newly formed ATP with spent ADP in the cytosol. There are four isoforms of ANT in humans (116). ANT1 is specific to muscle and brain tissue, ANT2 is mostly expressed in proliferative undifferentiated cells, ANT3 is ubiquitous and ANT4 is found in the liver, testis and brain (116,117).
ANT2 expression is upregulated in a variety of cancers, including hormone-sensitive cancers of the cervix, uterus and testis, whilst ANT1 expression is repressed in a number of cancer cell lines (43,44,118–120). The ANTs are also thought to play a role in mitochondrial-mediated apoptosis. ANT1 and ANT3 are pro-apoptotic (118,121,122), while overexpression of ANT4 reduces the sensitivity of cells to apoptotic inducers (123). There is also a link between lonidamine efficacy and ANT2 expression, although the mechanism of this effect is not known (119,120).
ATP is transported across the outer-mitochondrial membrane by VDAC. VDAC is also permeable to small and large ions (124) and functions in mitochondrial-mediated apoptosis. There are three isoforms of VDAC (125). VDAC1 is expressed in many tissues and its over-expression in cells leads to an increase in apoptosis (126). VDAC2 is found in spermatozoa (127), while VDAC3 expression has been observed in the testes (128). VDAC1 expression is increased in a number of cancer cell lines (129), although the reason for this is not apparent.
VDAC and ANT are thought to be components of the mitochondrial permeability transition pore. An increase in matrix calcium levels triggers the formation and opening of the pore, allowing equilibration of molecules with molecular weight lower than 1.5 kDa across the inner-mitochondrial membrane. If left unchecked, this can lead to swelling of the inner membrane, rupture of the outer membrane and cell death. VDAC and ANT do not appear to be essential components of the transition pore (130,131), although the transporters co-purify from cells (132,133) and are thought to interact in the inner-membrane space (134,135).
Mitochondrial ATP Transport Inhibitors
GSAO (4-(N-(S-glutathionylacetyl)amino)phenylarsonous acid) is a tripeptide trivalent arsenical (136,137). It is a pro-drug that is activated by γ-glutamyl transpeptidase at the cell surface to produce GCAO ((4-(N-(S-cysteinylglycylacetyl)amino) phenylarsonous acid)) (138). GCAO enters the cell via an organic ion transporter and is further processed by dipeptidases to CAO (4-(N-(S-cysteinylacetyl)amino) phenylarsonous acid) in the cytosol. CAO enters the mitochondrial matrix, through an unknown transporter, where its arsenical moiety cross-links Cys160 and Cys257 on the matrix face of ANT. This covalent interaction inactivates the transporter, blocking ATP delivery to VDAC-bound HKII (Fig. 2). The perturbation of ANT also leads to partial uncoupling of oxidative phosphorylation, an increase in superoxide production and the arrest of proliferation of the cell (136). CAO preferentially reacts with ANT in proliferating cells, which appears to be a consequence of the higher matrix concentrations of calcium (136). Cytosolic levels of GCAO and CAO are controlled by the multidrug-resistance associated protein isoforms 1 and 2 (139). These transporters are poorly expressed in proliferating endothelial cells which provides a selectivity for angiogenic endothelial cells in vitro and in vivo. GSAO is currently being tested in a Phase I clinical trial in adults with solid tumors refractory to standard therapy (Table I).
PENAO (4-(N-(S-penicillaminylacetyl)amino)phenylarsonous acid) was designed to bypass the pro-drug processing and metabolism of GSAO (140). PENAO is a cysteine mimetic of CAO. This compound accumulates in cells 85-fold faster than GSAO, which results in a 44-fold increased anti-proliferative activity and a ~20-fold increased anti-tumor efficacy in mice. In contrast to GSAO, PENAO targets both proliferating endothelial and tumor cells. GSAO and PENAO are equally well tolerated in rodents. The cytostatic/cytotoxic and anti-tumor efficacy of GSAO, therefore, has been markedly improved by constructing an analogue of a GSAO metabolite that enters cells faster and is exported slower. The increased residence time of PENAO in the cytosol correlates with its increased mitochondrial toxicity. PENAO will enter a Phase I clinical trial in adults with solid tumors refractory to standard therapy in 2011.
3-Bromopyruvate is a halogenated analogue of pyruvate that alkylates the sulfhydryl groups of HK, interfering with its action (Fig. 2). The compound inhibits the proliferation of melanoma, colon and breast cancer cell lines in culture (141–143) and growth of hepatocellular carcinoma tumors in rats (144). High cytosolic glutathione levels are associated with resistance to 3-bromopyruvate in cell culture (141). 3-Bromopyruvate also alkylates cysteine thiols in glyceraldehyde 3-phosphate dehydrogenase (145) and succinate dehydrogenase (146), so it is not specific for HK (147–149).
Jasmonates belong to a class of compounds that function as plant stress hormones, mediating responses to mechanical and infectious stresses (150). The three most commonly studied natural forms of jasmonates are methyl jasmonate (the most active), jasmonic acid and cis-jasmone. Methyl jasmonate binds to HK and displaces it from VDAC (151) (Fig. 2). Numerous studies in cancer cell lines have been performed (150,152) and selectivity for cancer cells over healthy cells has been observed (153). Synthetic halogenated derivatives of methyl jasmonate are more potent than the parent compound in a variety of cancer cell lines (154,155).
Betulinic acid (also known as bevirimat) is a pentacyclic triterpenoid found in the outer bark of various tree species (156). It triggers apoptosis of cells by disrupting the outer-mitochondrial membrane (157). Bongkrekic acid, which stabilizes the transition pore by binding to ANT (117), inhibits the action of betulinic acid (157,158) (Fig. 2). Bongkrekic acid is also able to prevent mitochondrial permeability transition following the dissociation of HK from VDAC (159). Betulinic acid induces apoptosis of a variety of human cancer cell lines, including doxorubicin-resistant neuroblastoma cells (156,158,160), and has anti-tumor activity in murine melanoma and colorectal cancer models (161,162). The compound was well tolerated in a phase I trial (163) and a phase II trial using betulinic acid as an ointment for the treatment of dysplastic nevi (a mole that has the potential to develop into melanoma) is ongoing (Table I).
Honokiol is a compound from the magnolia tree. It has a range of activities, including antioxidant, antithrombotic, antibacterial and anxiolytic (164). Honokiol has two main cellular mechanisms relevant to cancer. It blocks defective p53 signaling and activated Ras by inhibiting phospholipase D, and it potentiates opening of the mitochondrial permeability transition pore by inducing the expression of cyclophilin D (165) (Fig. 2). It is cytostatic and cytotoxic for a variety of cancer cell lines (164,166–168). The compound is currently being formulated using nanoparticles for intravenous delivery (169).
Lonidamine was first reported as a possible inhibitor of HK in 1981 (170) (Fig. 2). More recent work has suggested that it induces mitochondrial membrane permeabilization by a direct effect on ANT (171,172). Lonidamine has been tested in clinical trials in breast cancer, ovarian cancer, lung cancer, prostate adenoma and glioblastoma (173) (Table I).
Retinoic acids such as all-trans-retinoic acid, 9-cis-retinoic acid and 13-cis-retinoic acid bind to ANT and initiate the mitochondrial membrane permeability transition (174) (Fig. 2). They inhibit leukemia cell growth in vitro and solid tumor growth in vivo (175–178). All-trans retinoic acid is the standard of care in acute promelocytic leukemia (179) (Table I), but provides no benefit in other forms of acute leukemia (180–182).
Glutaminolysis
Glutamine is the most abundant amino acid (59) and is synthesized in the body by glutamine synthetase (EC 6.3.1.2) (8,183). Its catabolism provides precursors for nucleotide and protein biosynthesis and carbon units for the TCA cycle in mitochondria (184). Glutamine is transported across the plasma membrane and the inner-mitochondrial membrane by glutamine transporters. Glutaminases (EC 3.5.1.2) on the inner-mitochondrial membrane start its catabolism. Conversion to α-ketoglutarate by glutamate pyruvate transaminase (EC 2.6.1.15), glutamate dehydrogenase (EC 1.4.1.2) or aspartate aminotransferase (EC 2.6.1.1) provides carbons to the TCA cycle (Fig. 1). Glutamine catabolism in tumor cells is at least 10-fold that of any other amino acid (Fig. 1b) (184,185). In particular, pancreatic carcinoma, glioblastoma, acute myelogenous leukemia and small cell lung cancer cells are sensitive to glutamine starvation (185). Expression of the oncogene c-Myc in glioblastoma cells leads to ‘glutamine addiction’, such that their survival is linked to glutamine catabolism (16,186,187). c-Myc is expressed in more than 80% of gliomas and correlates with the grade of malignancy, with low expression in Grade I and II and high expression in Grade III and IV tumours. c-Myc activates the transcription of genes required for glutamine uptake and catabolism. Glutamine is also thought to supply nitrogen for the synthesis of amino acids in cancer cells when demand outstrips supply (188).
Glutaminolysis Inhibitors
Aminooxyacetate is a transaminase inhibitor that targets glutamate pyruvate transaminase, which converts glutamate to α-ketoglutarate (185) (Fig. 2). In melanoma cell lines, aminooxyacetate sensitizes cells to TRAIL (tumor necrosis factor-related apoptosis inducing-ligand)-induced cell death (189). It inhibits proliferation of MDA-MB-231 breast cancer and SF188 glioblastoma cell lines (16,190) and suppresses growth of MDA-MB-231 xenograft tumors in mice (16,190).
Glutamine analogues such as 6-diazo-5-oxo-L-norleucine, azaserine and acivicin have been explored as possible therapies for cancer. Their development has stalled, though, because of side-effects such as neurotoxicity, gastrointestinal toxicity and myelosuppression (185) (Table I). Inhibiting glutaminolysis at other steps has been explored but none have proved successful to date.
CONCLUSION
Mitochondria play a central role in both healthy and cancer cell metabolism. In cancer cells, this organelle coordinates catabolism of glucose and glutamine across the cytosol. The products of this catabolism fuel the high demand for precursors for protein, DNA and lipid synthesis. Targeting cancer cell mitochondrial metabolism has potential for the treatment of the disease and a number of small-molecule inhibitors are in pre-clinical and clinical development. In our opinion, the most promising mitochondrial target is the HK-VDAC-ANT complex that spans the outer- and inner-mitochondrial membranes. This complex links glycolysis, oxidative phosphorylation and mitochondrial-mediated apoptosis, so its perturbation effects metabolism generally as well as cell viability. Peptide arsenicals that inactivate ANT in proliferating cells show particular promise, as do the inhibitors of HK. The next decade should see great strides in our understanding of mitochondrial metabolism in cancer cells and how we can selectively target this process for cancer treatment.
Abbreviations
- alpha-TOS:
-
alpha-tocopheryl succinate
- ANT:
-
adenine nucleotide translocase
- ATP:
-
adenosine triphosphate
- CAO:
-
4-(N-(S-cysteinylacetyl)amino) phenylarsonous acid
- FADH2 :
-
flavin adenine dinucleotide
- FH:
-
fumarate hydratase
- GCAO:
-
4-(N-(S-cysteinylglycylacetyl)amino) phenylarsonous acid
- G6P:
-
glucose-6-phosphate
- GLUT:
-
glucose transporter
- GPT:
-
glutamate pyruvate transaminase
- GSAO:
-
4-(N-(S-glutathionylacetyl)amino)phenylarsonous acid
- HIF1:
-
hypoxia inducible factor 1
- IDH:
-
isocitrate dehydrogenase
- NADH:
-
nicotinamide adenine dinucleotide (reduced)
- NADPH:
-
nicotinamide adenine dinucleotide phosphate (reduced)
- NSCLC:
-
non-small-cell lung carcinoma
- PENAO:
-
4-(N-(S-penicillaminylacetyl)amino)phenylarsonous acid
- PDH:
-
pyruvate dehydrogenase
- PDHK:
-
pyruvate dehydrogenase kinase
- SDH:
-
succinate dehydrogenase
- TCA:
-
tricarboxylic acid cycle
- VDAC:
-
voltage dependent anion channel
REFERENCES
Warburg O. On the origin of cancer cells. Science. 1956;123(3191):309–14.
Moreno-Sánchez R, Rodríguez-Enríquez S, Saavedra E, Marín-Hernández A, Gallardo-Pérez JC. The bioenergetics of cancer: is glycolysis the main ATP supplier in all tumor cells? Biofactors. 2009;35(2):209–25.
Cairns RA, Harris IS, Mak TW. Regulation of cancer cell metabolism. Nat Rev Cancer. 2011;11(2):85–95.
Moreno-Sánchez R, Rodríguez-Enríquez S, Marín-Hernández A, Saavedra E. Energy metabolism in tumor cells. FEBS J. 2007;274(6):1393–418.
Solaini G, Sgarbi G, Baracca A. Oxidative phosphorylation in cancer cells. Biochim Biophys Acta Bioenerg. 2011;1807(6):534–42.
Vander Heiden MG, Cantley LC, Thompson CB. Understanding the warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324(5930):1029–33.
Pedersen PL. Warburg, me and Hexokinase 2: multiple discoveries of key molecular events underlying one of cancers’ most common phenotypes, the “Warburg Effect”, i.e., elevated glycolysis in the presence of oxygen. J Bioenerg Biomembr. 2007;39(3):211–22.
Matés JM, Segura JA, Campos-Sandoval JA, Lobo C, Alonso L, Alonso FJ, et al. Glutamine homeostasis and mitochondrial dynamics. Int J Biochem Cell Biol. 2009;41(10):2051–61.
Medina MA, Sánchez-Jiménez F, Márquez J, Rodríguez Quesada A, Núñez I. Relevance of glutamine metabolism to tumor cell growth. Mol Cell Biochem. 1992;113(1):1–15.
Medina MA, De Castro IN. Glutaminolysis and glycolysis interactions in proliferant cells. Int J Biochem. 1990;22(7):681–3.
Dasu A, Toma-Dasu I, Karlsson M. Theoretical simulation of tumour oxygenation and results from acute and chronic hypoxia. Phys Med Biol. 2003;48(17):2829–42.
Denko NC. Hypoxia, HIF1 and glucose metabolism in the solid tumour. Nat Rev Cancer. 2008;8(9):705–13.
Yeung S, Pan J, Lee MH. Roles of p53, Myc and HIF-1 in regulating glycolysis—the seventh hallmark of cancer. Cell Mol Life Sci. 2008;65(24):3981–99.
Osthus RC, Shim H, Kim S, Li Q, Reddy R, Mukherjee M, et al. Deregulation of glucose transporter 1 and glycolytic gene expression by c-Myc. J Biol Chem. 2000;275(29):21797–800.
Shim H, Dolde C, Lewis BC, Wu C-S, Dang G, Jungmann RA, et al. c-Myc transactivation of LDH-A: implications for tumor metabolism and growth. Proc Natl Acad Sci USA. 1997;94(13):6658–63.
Wise DR, DeBerardinis RJ, Mancuso A, Sayed N, Zhang XY, Pfeiffer HK, et al. Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc Natl Acad Sci USA. 2008 Dec 2;105(48):18782–7.
Kim J-W, Gao P, Liu Y-C, Semenza GL, Dang CV. Hypoxia-inducible factor 1 and dysregulated c-Myc cooperatively induce vascular endothelial growth factor and metabolic switches hexokinase 2 and pyruvate dehydrogenase kinase 1. Mol Cell Biol. 2007;27(21):7381–93.
Cuezva JM, Ortega ÁD, Willers I, Sánchez-Cenizo L, Aldea M, Sánchez-Aragó M. The tumor suppressor function of mitochondria: Translation into the clinics. Biochimica et Biophysica Acta—Molecular Basis of Disease. 2009;1792(12):1145–58.
Mathupala SP, Ko YH, Pedersen PL. The pivotal roles of mitochondria in cancer: Warburg and beyond and encouraging prospects for effective therapies. Biochim Biophys Acta. 2010:1225–30.
Wasserman DH, Kang L, Ayala JE, Fueger PT, Lee-Young RS. The physiological regulation of glucose flux into muscle in vivo. J Exp Biol. 2011;214(2):254–62.
Mathupala SP, Ko YH, Pedersen PL. Hexokinase-2 bound to mitochondria: cancer’s stygian link to the “Warburg effect” and a pivotal target for effective therapy. Semin Cancer Biol. 2009;19(1):17–24.
Pastorino JG, Hoek JB. Hexokinase II: the integration of energy metabolism and control of apoptosis. Curr Med Chem. 2003;10(16):1535–51.
Goel A, Mathupala SP, Pedersen PL. Glucose metabolism in cancer: evidence that demethylation events play a role in activating type II hexokinase gene expression. J Biol Chem. 2003;278(17):15333–40.
Rempel A, Mathupala SP, Griffin CA, Hawkins AL, Pedersen PL. Glucose catabolism in cancer cells: amplification of the gene encoding type II hexokinase. Cancer Res. 1996;56(11):2468–71.
Smith TAD. Mammalian hexokinases and their abnormal expression in cancer. Br J Biomed Sci. 2000;57(2):170–8.
Mathupala SP, Rempel A, Pedersen PL. Glucose catabolism in cancer cells: identification and characterization of a marked activation response of the type II hexokinase gene to hypoxic conditions. J Biol Chem. 2001;276(46):43407–12.
Mathupala SP, Heese C, Pedersen PL. Glucose catabolism in cancer cells. The type II hexokinase promoter contains functionally active response elements for the tumor suppressor p53. J Biol Chem. 1997;272(36):22776–80.
Mathupala SP, Ko YH, Pedersen PL. Hexokinase II: cancer’s double-edged sword acting as both facilitator and gatekeeper of malignancy when bound to mitochondria. Oncogene. 2006;25(34):4777–86.
Rempel A, Bannasch P, Mayer D. Differences in expression and intracellular distribution of hexokinase isoenzymes in rat liver cells of different transformation stages. Biochim Biophys Acta Gene Struct Expr. 1994;1219(3):660–8.
Nakashima RA, Mangan PS, Colombini M, Pedersen PL. Hexokinase receptor complex in hepatoma mitochondria: evidence from N, N-dicyclohexylcarbodiimide-labeling studies for the involvement of the pore-forming protein VDAC. Biochemistry. 1986;25(5):1015–21.
Arora KK, Pedersen PL. Functional significance of mitochondrial bound hexokinase in tumor cell metabolism. Evidence for preferential phosphorylation of glucose by intramitochondrially generated ATP. J Biol Chem. 1988;263(33):17422–8.
Bustamante E, Pedersen PL. High aerobic glycolysis of rat hepatoma cells in culture: role of mitochondrial hexokinase. Proc Natl Acad Sci USA. 1977;74(9):3735–9.
Pastorino JG, Shulga N, Hoek JB. Mitochondrial binding of hexokinase II inhibits Bax-induced cytochrome c release and apoptosis. J Biol Chem. 2002;277(9):7610–8.
Arzoine L, Zilberberg N, Ben-Romano R, Shoshan-Barmatz V. Voltage-dependent anion channel 1-based peptides interact with hexokinase to prevent its anti-apoptotic activity. J Biol Chem. 2009;284(6):3946–55.
Pastorino JG, Hoek JB. Regulation of hexokinase binding to VDAC. J Bioenerg Biomembr. 2008;40(3):171–82.
Poleti MD, Tesch AC, Crepaldi CR, Souza GHMF, Eberlin MN, De Cerqueira César M. Relationship between expression of voltage-dependent anion channel (VDAC) isoforms and type of hexokinase binding sites on brain mitochondria. J Mol Neurosci. 2010;41(1):48–54.
Golestani A, Nemat-Gorgani M. Hexokinase ‘binding sites’ of normal and tumoral human brain mitochondria. Mol Cell Biochem. 2000;215(1–2):115–21.
de Cerqueira Cesar M, Wilson JE. Functional characteristics of hexokinase bound to the Type A and Type B sites of bovine brain mitochondria. Arch Biochem Biophys. 2002;397(1):106–12.
Vyssokikh MY, Zorova L, Zorov D, Heimlich G, Jürgensmeier JM, Brdiczka D. Bax releases cytochrome c preferentially from a complex between porin and adenine nucleotide translocator. Hexokinase activity suppresses this effect. Mol Biol Rep. 2002;29(1–2):93–6.
Vyssokikh M, Zorova L, Zorov D, Heimlich G, Jürgensmeier J, Schreiner D, et al. The intra-mitochondrial cytochrome c distribution varies correlated to the formation of a complex between VDAC and the adenine nucleotide translocase: this affects Bax-dependent cytochrome c release. Biochim Biophys Acta Mol Cell Res. 2004;1644(1):27–36.
Azoulay-Zohar H, Israelson A, Abu-Hamad S, Shoshan-Barmatz V. In self-defence: hexokinase promotes voltage-dependent anion channel closure and prevents mitochondria-mediated apoptotic cell death. Biochem J. 2004;377(2):347–55.
Campbell AM, Chan SHP. The voltage dependent anion channel affects mitochondrial cholesterol distribution and function. Arch Biochem Biophys. 2007;466(2):203–10.
Chevrollier A, Leiseau D, Stepien G. What is the specific role of ANT2 in cancer cells? Medicine/Sciences. 2005;21(2):156–61.
Chevrollier A, Loiseau D, Chabi B, Renier G, Douay O, Malthiery Y, et al. ANT2 isoform required for cancer cell glycolysis. J Bioenerg Biomembr. 2005;37(5):307–16.
Perevoshchikova IV, Zorov SD, Kotova EA, Zorov DB, Antonenko YN. Hexokinase inhibits flux of fluorescently labeled ATP through mitochondrial outer membrane porin. FEBS Lett. 2010;584(11):2397–402.
Berg JM, Tymoczko JL, Stryer L. Biochemistry. 5th ed. New York Basingstoke: W. H. Freeman and Co.; Palgrave distributor; 2001.
Koukourakis MI, Giatromanolaki A, Sivridis E, Gatter KC, Harris AL. Pyruvate dehydrogenase and pyruvate dehydrogenase kinase expression in non small cell lung cancer and tumor-associated stroma. Neoplasia. 2005;7(1):1–6.
Koukourakis MI, Giatromanolaki A, Bougioukas G, Sivridis E. Lung cancer: a comparative study of metabolism related protein expression in cancer cells and tumor associated stroma. Cancer Biol Ther. 2007;6(9):1476–9.
McFate T, Mohyeldin A, Lu H, Thakar J, Henriques J, Halim ND, et al. Pyruvate dehydrogenase complex activity controls metabolic and malignant phenotype in cancer cells. J Biol Chem. 2008;283(33):22700–8.
Wigfield SM, Winter SC, Giatromanolaki A, Taylor J, Koukourakis ML, Harris AL. PDK-1 regulates lactate production in hypoxia and is associated with poor prognosis in head and neck squamous cancer. Br J Cancer. 2008;98(12):1975–84.
Christofk HR, Vander Heiden MG, Harris MH, Ramanathan A, Gerszten RE, Wei R, et al. The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth. Nature. 2008;452(7184):230–3.
Pathania D, Millard M, Neamati N. Opportunities in discovery and delivery of anticancer drugs targeting mitochondria and cancer cell metabolism. Adv Drug Deliv Rev. 2009;61(14):1250–75.
Hao H-X, Khalimonchuk O, Schraders M, Dephoure N, Bayley J-P, Kunst H, et al. SDH5, a gene required for flavination of succinate dehydrogenase, is mutated in paraganglioma. Science. 2009;325(5944):1139–42.
Baysal BE, Ferrell RE, Willett-Brozick JE, Lawrence EC, Myssiorek D, Bosch A, et al. Mutations in SDHD, a mitochondrial complex II gene, in hereditary paraganglioma. Science. 2000;287(5454):848–51.
Bayley J-P, Devilee P. Warburg tumours and the mechanisms of mitochondrial tumour suppressor genes. Barking up the right tree? Curr Opin Genet Dev. 2010;20(3):324–9.
Rutter J, Winge DR, Schiffman JD. Succinate dehydrogenase—assembly, regulation and role in human disease. Mitochondrion. 2010;10(4):393–401.
Yang Y, Valera VA, Padilla-Nash HM, Sourbier C, Vocke CD, Vira MA, et al. UOK 262 cell line, fumarate hydratase deficient (FH-/FH-) hereditary leiomyomatosis renal cell carcinoma: in vitro and in vivo model of an aberrant energy metabolic pathway in human cancer. Cancer Genet Cytogenet. 2010;196(1):45–55.
Furuta E, Okuda H, Kobayashi A, Watabe K. Metabolic genes in cancer: their roles in tumor progression and clinical implications. Biochim Biophys Acta Rev Canc. 2010;1805(2):141–52.
DeBerardinis RJ, Lum JJ, Hatzivassiliou G, Thompson CB. The biology of cancer: metabolic reprogramming fuels cell growth and proliferation. Cell Metab. 2008;7(1):11–20.
Yan H, Parsons DW, Jin G, McLendon R, Rasheed BA, Yuan W, et al. IDH1 and IDH2 mutations in gliomas. N Engl J Med. 2009;360(8):765–73.
Dang L, White DW, Gross S, Bennett BD, Bittinger MA, Driggers EM, et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature. 2009;462(7274):739–44.
Keenan J, Liang Y, Clynes M. Two-deoxyglucose as an anti-metabolite in human carcinoma cell line RPMI-2650 and drug-resistant variants. Anticancer Res. 2004;24(2A):433–40.
Stein M, Lin H, Jeyamohan C, Dvorzhinski D, Gounder M, Bray K, et al. Targeting tumor metabolism with 2-deoxyglucose in patients with castrate-resistant prostate cancer and advanced malignancies. Prostate. 2010;70(13):1388–94.
Board M, Colquhoun A, Newsholme EA. High Km glucose-phosphorylating (glucokinase) activities in a range of tumor cell lines and inhibition of rates of tumor growth by the specific enzyme inhibitor mannoheptulose. Cancer Res. 1995;55(15):3278–85.
Penso J, Beitner R. Clotrimazole and bifonazole detach hexokinase from mitochondria of melanoma cells. Eur J Pharmacol. 1998;342(1):113–7.
Khalid MH, Tokunaga Y, Caputy AJ, Walters E. Inhibition of tumor growth and prolonged survival of rats with intracranial gliomas following administration of clotrimazole. J Neurosurg. 2005;103(1):79–86.
Liu H, Li Y, Raisch KP. Clotrimazole induces a late G1 cell cycle arrest and sensitizes glioblastoma cells to radiation in vitro. Anticancer Drugs. 2010;21(9):841–9.
Hegemann L, Toso SM, Lahijani KI, Webster GF, Uitto J. Direct interaction of antifungal azole-derivatives with calmodulin: a possible mechanism for their therapeutic activity. J Invest Dermatol. 1993;100(3):343–6.
Papandreou I, Goliasova T, Denko NC. Anticancer drugs that target metabolism: is dichloroacetate the new paradigm? Int J Cancer. 2011;128(5):1001–8.
Bonnet S, Archer SL, Allalunis-Turner J, Haromy A, Beaulieu C, Thompson R, et al. A mitochondria-K+ channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth. Cancer Cell. 2007;11(1):37–51.
Cao W, Yacoub S, Shiverick KT, Namiki K, Sakai Y, Porvasnik S, et al. Dichloroacetate (DCA) sensitizes both wild-type and over expressing bcl-2 prostate cancer cells in vitro to radiation. Prostate. 2008;68(11):1223–31.
Stockwin LH, Yu SX, Borgel S, Hancock C, Wolfe TL, Phillips LR, et al. Sodium dichloroacetate selectively targets cells with defects in the mitochondrial ETC. Int J Cancer. 2010;127(11):2510–9.
Sun R, Fadia M, Dahlstrom J, Parish C, Board P, Blackburn A. Reversal of the glycolytic phenotype by dichloroacetate inhibits metastatic breast cancer cell growth in vitro and in vivo. Breast Cancer Res Treat. 2010;120(1):253–60.
Wong JYY, Huggins GS, Debidda M, Munshi NC, De Vivo I. Dichloroacetate induces apoptosis in endometrial cancer cells. Gynecol Oncol. 2008;109(3):394–402.
Michelakis ED, Sutendra G, Dromparis P, Webster L, Haromy A, Niven E, et al. Metabolic modulation of glioblastoma with dichloroacetate. Sci Translat Med. 2010;2(31).
Pearson H. Cancer patients opt for unapproved drug. Nature. 2007;446(7135):474–5.
Morrell JA, Orme J, Butlin RJ, Roche TE, Mayers RM, Kilgour E. AZD7545 is a selective inhibitor of pyruvate dehydrogenase kinase 2. Biochem Soc Trans. 2003;31(6):1168–70.
Kato M, Li J, Chuang JL, Chuang DT. Distinct structural mechanisms for inhibition of pyruvate dehydrogenase kinase isoforms by AZD7545, dichloroacetate, and radicicol. Structure. 2007;15(8):992–1004.
Mayers RM, Butlin RJ, Kilgour E, Leighton B, Martin D, Myatt J, et al. AZD7545, a novel inhibitor of pyruvate dehydrogenase kinase 2 (PDHK2), activates pyruvate dehydrogenase in vivo and improves blood glucose control in obese (fa/fa) Zucker rats. Biochem Soc Trans. 2003;31(6):1165–7.
Fulda S, Galluzzi L, Kroemer G. Targeting mitochondria for cancer therapy. Nat Rev Drug Discov. 2010;9(6):447–64.
Ishii N. Role of oxidative stress from mitochondria on aging and cancer. Cornea. 2007;26 Suppl 1:S3–9.
Fukuda R, Zhang H, Kim J-W, Shimoda L, Dang CV, Semenza Gregg L. HIF-1 regulates cytochrome oxidase subunits to optimize efficiency of respiration in hypoxic cells. Cell. 2007;129(1):111–22.
Boitier E, Merad-Boudia M, Guguen-Guillouzo C, Defer N, Ceballos-Picot I, Leroux JP, et al. Impairment of the mitochondrial respiratory chain activity in diethylnitrosamine-induced rat hepatomas: possible involvement of oxygen free radicals. Cancer Res. 1995;55(14):3028–35.
Simonnet H, Demont J, Pfeiffer K, Guenaneche L, Bouvier R, Brandt U, et al. Mitochondrial complex I is deficient in renal oncocytomas. Carcinogenesis. 2003;24(9):1461–6.
Bonora E, Porcelli AM, Gasparre G, Biondi A, Ghelli A, Carelli V, et al. Defective oxidative phosphorylation in thyroid oncocytic carcinoma is associated with pathogenic mitochondrial DNA mutations affecting complexes I and III. Cancer Res. 2006;66(12):6087–96.
Simonnet H, Alazard N, Pfeiffer K, Gallou C, Béroud C, Demont J, et al. Low mitochondrial respiratory chain content correlates with tumor aggressiveness in renal cell carcinoma. Carcinogenesis. 2002;23(5):759–68.
Cuezva JM, Krajewska M, De Heredia ML, Krajewski S, Santamaría G, Kim H, et al. The bioenergetic signature of cancer: a marker of tumor progression. Cancer Res. 2002;62(22):6674–81.
Cuezva JM, Chen G, Alonso AM, Isidoro A, Misek DE, Hanash SM, et al. The bioenergetic signature of lung adenocarcinomas is a molecular marker of cancer diagnosis and prognosis. Carcinogenesis. 2004;25(7):1157–63.
Deng Y, Huang H, Lin J. Rotenone induces apoptosis in MCF-7 human breast cancer cell-mediated ROS through JNK and p38 signaling. Mol Carcinog. 2010;49(2):141–51.
Wolvetang EJ, Johnson KL, Krauer K, Ralph SJ, Linnane AW. Mitochondrial respiratory chain inhibitors induce apoptosis. FEBS Lett. 1994;339(1–2):40–4.
Chung WG, Miranda CL, Maier CS. Epigallocatechin gallate (EGCG) potentiates the cytotoxicity of rotenone in neuroblastoma SH-SY5Y cells. Brain Res. 2007;1176(1):133–42.
Liaw C-C, Wu T-Y, Chang F-R, Wu Y-C. Historic perspectives on annonaceous acetogenins from the chemical bench to preclinical trials. Planta Med. 2010;76(13):1390–404.
Ahammadsahib KI, Hollingworth RM, McGovren JP, Hui YH, McLaughlin JL. Mode of action of bullatacin: A potent antitumor and pesticidal Annonaceous acetogenin. Life Sci. 1993;53(14):1113–20.
Holschneider CH, Johnson MT, Knox RM, Rezai A, Ryan WJ, Montz FJ. Bullatacin—In vivo and in vitro experience in an ovarian cancer model. Cancer Chemother Pharmacol. 1994;34(2):166–70.
Chih H-W, Chiu H-F, Tang K-S, Chang F-R, Wu Y-C. Bullatacin, a potent antitumor annonaceous acetogenin, inhibits proliferation of human hepatocarcinoma cell line 2.2.15 by apoptosis induction. Life Sci. 2001;69(11):1321–31.
Oberlies NH, Croy VL, Harrison ML, McLaughlin JL. The Annonaceous acetogenin bullatacin is cytotoxic against multidrug-resistant human mammary adenocarcinoma cells. Cancer Lett. 1997;115(1):73–9.
Dong LF, Low P, Dyason JC, Wang XF, Prochazka L, Witting PK, et al. α-tocopheryl succinate induces apoptosis by targeting ubiquinone-binding sites in mitochondrial respiratory complex II. Oncogene. 2008;27(31):4324–35.
Constantinou C, Papas A, Constantinou AI. Vitamin E and cancer: an insight into the anticancer activities of vitamin E isomers and analogs. Int J Cancer. 2008;123(4):739–52.
Dong LF, Swettenham E, Eliasson J, Wang XF, Gold M, Medunic Y, et al. Vitamin E analogues inhibit angiogenesis by selective induction of apoptosis in proliferating endothelial cells: the role of oxidative stress. Cancer Res. 2007;67(24):11906–13.
Neuzil J, Weber T, Schrӧder A, Lu M, Ostermann G, Gellert N, et al. Induction of cancer cell apoptosis by α-tocopheryl succinate: molecular pathways and structural requirements. FASEB J. 2001;15(2):403–15.
Zhao Y, Neuzil J, Wu K. Vitamin E analogues as mitochondria-targeting compounds: from the bench to the bedside? Mol Nutr Food Res. 2009;53(1):129–39.
Brinkman MT, Karagas MR, Zens MS, Schned A, Reulen RC, Zeegers MP. Minerals and vitamins and the risk of bladder cancer: results from the New Hampshire Study. Cancer Causes Control. 2010;21(4):609–19.
Xiao D, Powolny AA, Singh SV. Benzyl isothiocyanate targets mitochondrial respiratory chain to trigger reactive oxygen species-dependent apoptosis in human breast cancer cells. J Biol Chem. 2008;283(44):30151–63.
Lin JM, Amin S, Trushin N, Hecht SS. Effects of isothiocyanates on tumorigenesis by benzo[a]pyrene in murine tumor models. Cancer Lett. 1993;74(3):151–9.
Sugie S, Okumura A, Tanaka T, Mori H. Inhibitory effects of benzyl isothiocyanate and benzyl thiocyanate on diethylnitrosamine-induced hepatocarcinogenesis in rats. Jpn J Cancer Res. 1993;84(8):865–70.
Athar M, Back JH, Tang X, Kim KH, Kopelovich L, Bickers DR, et al. Resveratrol: a review of preclinical studies for human cancer prevention. Toxicol Appl Pharmacol. 2007;224(3):274–83.
Gledhill JR, Montgomery MG, Leslie AGW, Walker JE. Mechanism of inhibition of bovine F1-ATPase by resveratrol and related polyphenols. Proc Natl Acad Sci. USA. 2007;104(34):13632–7.
Zheng J, Ramirez VD. Inhibition of mitochondrial proton F0F1-ATPase/ATP synthase by polyphenolic phytochemicals. Br J Pharmacol. 2000;130(5):1115–23.
Fulda S. Modulation of apoptosis by natural products for cancer therapy. Planta Med. 2010;76(11):1075–9.
Baur JA, Sinclair DA. Therapeutic potential of resveratrol: the in vivo evidence. Nat Rev Drug Discov. 2006;5(6):493–506.
Biasutto L, Mattarei A, Marotta E, Bradaschia A, Sassi N, Garbisa S, et al. Development of mitochondria-targeted derivatives of resveratrol. Bioorg Med Chem Lett. 2008;18(20):5594–7.
Brown VA, Patel KR, Viskaduraki M, Crowell JA, Perloff M, Booth TD, et al. Repeat dose study of the cancer chemopreventive agent resveratrol in healthy volunteers: safety, pharmacokinetics, and effect on the insulin-like growth factor axis. Cancer Res. 2010;70(22):9003–11.
Patel KR, Brown VA, Jones DJ, Britton RG, Hemingway D, Miller AS, et al. Clinical pharmacology of resveratrol and its metabolites in colorectal cancer patients. Cancer Res. 2010;70(19):7392–9.
Ko YH, Delannoy M, Hullihen J, Chiu W, Pedersen PL. Mitochondrial ATP synthasome: cristae-enriched membranes and a multiwell detergent screening assay yield dispersed single complexes containing the ATP synthase and carriers for Pi and ADP/ATP. J Biol Chem. 2003;278(14):12305–9.
Chen C, Ko Y, Delannoy M, Ludtke SJ, Chiu W, Pedersen PL. Mitochondrial ATP synthasome. J Biol Chem. 2004;279(30):31761–8.
Dolce V, Scarcia P, Iacopetta D, Palmieri F. A fourth ADP/ATP carrier isoform in man: identification, bacterial expression, functional characterization and tissue distribution. FEBS Lett. 2005;579(3):633–7.
Chevrollier A, Loiseau D, Reynier P, Stepien G. Adenine nucleotide translocase 2 is a key mitochondrial protein in cancer metabolism. Biochim Biophys Acta Bioenerg. 2011;1807(6):562–7.
Jang JY, Choi Y, Jeon YK, Aung KCY, Kim CW. Over-expression of adenine nucleotide translocase 1 (ANT1) induces apoptosis and tumor regression in vivo. BMC Cancer. 2008;8.
Jang JY, Choi Y, Jeon YK, Kim CW. Suppression of adenine nucleotide translocase-2 by vector-based siRNA in human breast cancer cells induces apoptosis and inhibits tumor growth in vitro and in vivo. Breast Canc Res. 2008;10(1).
Le Bras M, Borgne-Sanchez A, Touat Z, El Dein OS, Deniaud A, Maillier E, et al. Chemosensitization by knockdown of adenine nucleotide translocase-2. Cancer Res. 2006;66(18):9143–52.
Bauer MKA, Schubert A, Rocks O, Grimm S. Adenine nucleotide translocase-1, a component of the permeability transition pore, can dominantly induce apoptosis. J Cell Biol. 1999;147(7):1493–501.
Zamora M, Granell M, Mampel T, Viñas O. Adenine nucleotide translocase 3 (ANT3) overexpression induces apoptosis in cultured cells. FEBS Lett. 2004;563(1–3):155–60.
Gallerne C, Touat Z, Chen ZX, Martel C, Mayola E, Sharaf el dein O, et al. The fourth isoform of the adenine nucleotide translocator inhibits mitochondrial apoptosis in cancer cells. Int J Biochem Cell Biol. 2010;42(5):623–9.
Shoshan-Barmatz V, De Pinto V, Zweckstetter M, Raviv Z, Keinan N, Arbel N. VDAC, a multi-functional mitochondrial protein regulating cell life and death. Mol Aspects Med. 2010;31(3):227–85.
De Pinto V, Guarino F, Guarnera A, Messina A, Reina S, Tomasello FM, et al. Characterization of human VDAC isoforms: a peculiar function for VDAC3? Biochim Biophys Acta Bioenerg. 2010;1797(6–7):1268–75.
Zaid H, Abu-Hamad S, Israelson A, Nathan I, Shoshan-Barmatz V. The voltage-dependent anion channel-1 modulates apoptotic cell death. Cell Death Differ. 2005;12(7):751–60.
Menzel VA, Cassará MC, Benz R, De Pinto V, Messina A, Cunsolo V, et al. Molecular and functional characterization of VDAC2 purified from mammal spermatozoa. Biosci Rep. 2009;29(6):351–62.
Liu B, Wang Z, Zhang W, Wang X. Expression and localization of voltage-dependent anion channels (VDAC) in human spermatozoa. Biochem Biophys Res Commun. 2009;378(3):366–70.
Simamura E, Shimada H, Ishigaki Y, Hatta T, Higashi N, Hirai KI. Bioreductive activation of quinone antitumor drugs by mitochondrial voltage-dependent anion channel 1. Anat Sci Int. 2008;83(4):261–6.
Baines CP, Kaiser RA, Sheiko T, Craigen WJ, Molkentin JD. Voltage-dependent anion channels are dispensable for mitochondrial-dependent cell death. Nat Cell Biol. 2007;9(5):550–5.
Kokoszka JE, Waymire KG, Levy SE, Sligh JE, Cai J, Jones DP, et al. The ADP/ATP translocator is not essential for the mitochondrial permeability transition pore. Nature. 2004;427(6973):461–5.
McEnery MW, Snowman AM, Trifiletti RR, Snyder SH. Isolation of the mitochondrial benzodiazepine receptor: association with the voltage-dependent anion channel and the adenine nucleotide carrier. Proc Natl Acad Sci USA. 1992;89(8):3170–4.
Verrier F, Deniaud A, LeBras M, Métivier D, Kroemer G, Mignotte B, et al. Dynamic evolution of the adenine nucleotide translocase interactome during chemotherapy-induced apoptosis. Oncogene. 2004;23(49):8049–64.
Crompton M, Barksby E, Johnson N, Capano M. Mitochondrial intermembrane junctional complexes and their involvement in cell death. Biochimie. 2002;84(2–3):143–52.
Crompton M. Mitochondrial intermembrane junctional complexes and their role in cell death. J Physiol (Lond). 2000;529(1):11–21.
Don AS, Kisker O, Dilda P, Donoghue N, Zhao X, Decollogne S, et al. A peptide trivalent arsenical inhibits tumor angiogenesis by perturbing mitochondrial function in angiogenic endothelial cells. Cancer Cell. 2003;3(5):497–509.
Park D, Dilda PJ. Mitochondria as targets in angiogenesis inhibition. Mol Aspects Med. 2010;31(1):113–31.
Dilda PJ, Ramsay EE, Corti A, Pompella A, Hogg PJ. Metabolism of the tumor angiogenesis inhibitor 4-(N-(S-Glutathionylacetyl)amino)phenylarsonous acid. J Biol Chem. 2008;283(51):35428–34.
Dilda PJ, Don AS, Tanabe KM, Higgins VJ, Allen JD, Dawes IW, et al. Mechanism of selectivity of an angiogenesis inhibitor from screening a genome-wide set of Saccharomyces cerevisiae deletion strains. J Natl Cancer Inst. 2005;97(20):1539–47.
Dilda PJ, Decollogne S, Weerakoon L, Norris MD, Haber M, Allen JD, et al. Optimization of the antitumor efficacy of a synthetic mitochondrial toxin by increasing the residence time in the cytosol. J Med Chem. 2009;52(20):6209–16.
Qin JZ, Xin H, Nickoloff BJ. 3-Bromopyruvate induces necrotic cell death in sensitive melanoma cell lines. Biochem Biophys Res Commun. 2010;396(2):495–500.
Ihrlund LS, Hernlund E, Khan O, Shoshan MC. 3-Bromopyruvate as inhibitor of tumour cell energy metabolism and chemopotentiator of platinum drugs. Mol Oncol. 2008;2(1):94–101.
Liu XH, Zheng XF, Wang YL. Inhibitive effect of 3-bromopyruvic acid on human breast cancer MCF-7 cells involves cell cycle arrest and apoptotic induction. Chin Med J (Engl). 2009;122(14):1681–5.
Ko YH, Smith BL, Wang Y, Pomper MG, Rini DA, Torbenson MS, et al. Advanced cancers: eradication in all cases using 3-bromopyruvate therapy to deplete ATP. Biochem Biophys Res Commun. 2004;324(1):269–75.
Ganapathy-Kanniappan S, Geschwind JFH, Kunjithapatham R, Buijs M, Vossen JA, Tchernyshyov I, et al. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) is pyruvylated during 3-bromopyruvate mediated cancer cell death. Anticancer Res. 2009;29(12):4909–18.
Pereira da Silva AP, El-Bacha T, Kyaw N, dos Santos RS, Da-Silva WS, Almeida FCL, et al. Inhibition of energy-producing pathways of HepG2 cells by 3-bromopyruvate. Biochem J. 2009;417(3):717–26.
Scatena R, Bottoni P, Pontoglio A, Mastrototaro L, Giardina B. Glycolytic enzyme inhibitors in cancer treatment. Expert Opin Investig Drugs. 2008;17(10):1533–45.
Meng F, Matteucci M, Har C. The antiproliferative activity of 3-Bromopyruvate is not due to selective inhibition of glycolysis. AACR Meeting Abstracts. 2008;2008:2714.
Ganapathy-Kanniappan S, Vali M, Kunjithapatham R, Buijs M, Syed LH, Rao PP, et al. 3-Bromopyruvate: a new targeted antiglycolytic agent and a promise for cancer therapy. Curr Pharm Biotechnol. 2010;11(5):510–7.
Flescher E. Jasmonates—a new family of anti-cancer agents. Anticancer Drugs. 2005;16(9):911–6.
Goldin N, Arzoine L, Heyfets A, Israelson A, Zaslavsky Z, Bravman T, et al. Methyl jasmonate binds to and detaches mitochondria-bound hexokinase. Oncogene. 2008;27(34):4636–43.
Cohen S, Flescher E. Methyl jasmonate: a plant stress hormone as an anti-cancer drug. Phytochemistry. 2009;70(13–14):1600–9.
Fingrut O, Flescher E. Plant stress hormones suppress the proliferation and induce apoptosis in human cancer cells. Leukemia. 2002;16(4):608–16.
Reischer D, Heyfets A, Shimony S, Nordenberg J, Kashman Y, Flescher E. Effects of natural and novel synthetic jasmonates in experimental metastatic melanoma. Br J Pharmacol. 2007;150(6):738–49.
Park C, Jin CY, Kim GY, Cheong J, Jung JH, Yoo YH, et al. A methyl jasmonate derivative, J-7, induces apoptosis in human hepatocarcinoma Hep3B cells in vitro. Toxicol Vitro. 2010;24(7):1920–6.
Fulda S. Betulinic acid for cancer treatment and prevention. Int J Mol Sci. 2008;9(6):1096–107.
Fulda S, Scaffidi G, Susin SA, Krammer PH, Kroemer G, Peter ME, et al. Activation of mitochondria and release of mitochondrial apoptogenic factors by betulinic acid. J Biol Chem. 1998;273(51):33942–8.
Fulda S, Susin SA, Kroemer G, Debatin KM. Molecular ordering of apoptosis induced by anticancer drugs in neuroblastoma cells. Cancer Res. 1998;58(19):4453–60.
Chiara F, Castellaro D, Marin O, Petronilli V, Brusilow WS, Juhaszova M, et al. Hexokinase II detachment from mitochondria triggers apoptosis through the permeability transition pore independent of voltage-dependent anion channels. PloS one. 2008;3(3).
Zuco V, Supino R, Righetti SC, Cleris L, Marchesi E, Gambacorti-Passerini C, et al. Selective cytotoxicity of betulinic acid on tumor cell lines, but not on normal cells. Cancer Lett. 2002;175(1):17–25.
Ren W, Qin L, Xu Y, Cheng N. Inhibition of betulinic acid to growth and angiogenesis of human colorectal cancer cell in nude mice. Chin Ger J Clin Oncol. 2010;9(3):153–7.
Shin YG, Cho KH, Chung SM, Graham J, Das Gupta TK, Pezzuto JM. Determination of betulinic acid in mouse blood, tumor and tissue homogenates by liquid chromatography-electrospray mass spectrometry. J Chrom B Biomed Sci Appl. 1999;732(2):331–6.
Smith PF, Ogundele A, Forrest A, Wilton J, Salzwedel K, Doto J, et al. Phase I and II study of the safety, virologic effect, and pharmacokinetics/pharmacodynamics of single-dose 3-O-(3′3′- dimethylsuccinyl)betulinic acid (bevirimat) against human immunodeficiency virus Infection. Antimicrob Agents Chemother. 2007;51(10):3574–81.
Ishitsuka K, Hideshima T, Hamasaki M, Raje N, Kumar S, Hideshima H, et al. Honokiol overcomes conventional drug resistance in human multiple myeloma by induction of caspase-dependent and -independent apoptosis. Blood. 2005;106(5):1794–800.
Fried LE, Arbiser JL. Honokiol, a multifunctional antiangiogenic and antitumor agent. Antioxid Redox Signal. 2009;11(5):1139–48.
Chen F, Wang T, Wu YF, Gu Y, Xu XL, Zheng S, et al. Honokiol: a potent chemotherapy candidate for human colorectal carcinoma. World J Gastroenterol. 2004;10(23):3459–63.
Li Z, Liu Y, Zhao X, Pan X, Yin R, Huang C, et al. Honokiol, a natural therapeutic candidate, induces apoptosis and inhibits angiogenesis of ovarian tumor cells. Eur J Obstet Gynecol Reprod Biol. 2008;140(1):95–102.
Liu J, Bartesaghi A, Borgnia MJ, Sapiro G, Subramaniam S. Molecular architecture of native HIV-1 gp120 trimers. Nature. 2008;455(7209):109–13.
Zheng X, Kan B, Gou M, Fu S, Zhang J, Men K, et al. Preparation of MPEG-PLA nanoparticle for honokiol delivery in vitro. Int J Pharm. 2010;386(1–2):262–7.
Floridi A, Paggi MG, Marcante ML. Lonidamine, a selective inhibitor of aerobic glycolysis of murine tumor cells. J Natl Cancer Inst. 1981;66(3):497–9.
Belzacq AS, El Hamel C, Vieira HLA, Cohen I, Haouzi D, Métivier D, et al. Adenine nucleotide translocator mediates the mitochondrial membrane permeabilization induced by lonidamine, arsenite and CD437. Oncogene. 2001;20(52):7579–87.
Ravagnan L, Marzo I, Costantini P, Susin SA, Zamzami N, Petit PX, et al. Lonidamine triggers apoptosis via a direct, Bcl-2-inhibited effect on the mitochondrial permeability transition pore. Oncogene. 1999;18(16):2537–46.
Oudard S, Carpentier A, Banu E, Fauchon F, Celerier D, Poupon MF, et al. Phase II study of lonidamine and diazepam in the treatment of recurrent glioblastoma multiforme. J Neurooncol. 2003;63(1):81–6.
Notario B, Zamora M, Viñas O, Mampel T. All-trans-retinoic acid binds to and inhibits adenine nucleotide translocase and induces mitochondrial permeability transition. Mol Pharmacol. 2003;63(1):224–31.
Robert C, Delva L, Balitrand N, Nahajevszky S, Masszi T, Chomienne C, et al. Apoptosis induction by retinoids in eosinophilic leukemia cells: Implication of retinoic acid receptor-α signaling in all-trans-retinoic acid hypersensitivity. Cancer Res. 2006;66(12):6336–44.
Barna G, Sebestyén A, Weischede S, Peták I, Mihalik R, Formelli F, et al. Different ways to induce apoptosis by fenretinide and all-trans-retinoic acid in human B lymphoma cells. Anticancer Res. 2005;25(6 B):4179–85.
Marchetti P, Zamzami N, Joseph B, Schraen-Maschke S, Méreau-Richard C, Costantini P, et al. The novel retinoid 6-[3-(1-adamantyl)-4-hydroxyphenyl]-2-naphtalene carboxylic acid can trigger apoptosis through a mitochondrial pathway independent of the nucleus. Cancer Res. 1999;59(24):6257–66.
Lotan R. Retinoids in cancer chemoprevention. FASEB J. 1996;10(9):1031–9.
Tallman MS, Nabhan C, Feusner JH, Rowe JM. Acute promyelocytic leukemia: evolving therapeutic strategies. Blood. 2002;99(3):759–67.
Milligan DW, Wheatley K, Littlewood T, Craig JI, Burnett AK, Group NHOCS. Fludarabine and cytosine are less effective than standard ADE chemotherapy in high-risk acute myeloid leukemia, and addition of G-CSF and ATRA are not beneficial: results of the MRC AML-HR randomized trial. Blood. 2006;107(12):4614–22.
Burnett AK, Hills RK, Green C, Jenkinson S, Koo K, Patel Y, et al. The impact on outcome of the addition of all-trans retinoic acid to intensive chemotherapy in younger patients with nonacute promyelocytic acute myeloid leukemia: overall results and results in genotypic subgroups defined by mutations in NPM1, FLT3, and CEBPA. Blood. 2010;115(5):948–56.
Dilda PJ, Hogg PJ. Arsenical-based cancer drugs. Cancer Treat Rev. 2007;33(6):542–64.
Dang CV. Glutaminolysis: supplying carbon or nitrogen or both for cancer cells? Cell Cycle. 2010;9(19):3884–6.
DeBerardinis RJ, Mancuso A, Daikhin E, Nissim I, Yudkoff M, Wehrli S, et al. Beyond aerobic glycolysis: transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc Natl Acad Sci USA. 2007;104(49):19345–50.
Wise DR, Thompson CB. Glutamine addiction: a new therapeutic target in cancer. Trends Biochem Sci. 2010;35(8):427–33.
Cheng T, Sudderth J, Yang C, Mullen AR, Jin ES, Mates JM, et al. Pyruvate carboxylase is required for glutamine-independent growth of tumor cells. Proc Natl Acad Sci USA. 2011 May 24;108(21):8674–9.
Yang C, Sudderth J, Dang T, Bachoo RM, McDonald JG, DeBerardinis RJ. Glioblastoma cells require glutamate dehydrogenase to survive impairments of glucose metabolism or Akt signaling. Cancer Res. 2009 Oct 15;69(20):7986–93.
Meng M, Chen S, Lao T, Liang D, Sang N. Nitrogen anabolism underlies the importance of glutaminolysis in proliferating cells. Cell Cycle. 2010;9(19):3921–32.
Qin JZ, Xin H, Nickoloff BJ. Targeting glutamine metabolism sensitizes melanoma cells to TRAIL-induced death. Biochem Biophys Res Commun. 2010;398(1):146–52.
Thornburg J, Nelson K, Clem B, Lane A, Arumugam S, Simmons A, et al. Targeting aspartate aminotransferase in breast cancer. Breast Cancer Res. 2008;10(5):R84.
Mohanti BK, Rath GK, Anantha N, Kannan V, Das BS, Chandramouli BAR, et al. Improving cancer radiotherapy with 2-deoxy-D-glucose: Phase I/II clinical trials on human cerebral gliomas. Int J Radiat Oncol Biol Phys. 1996;35(1):103–11.
Tallman MS, Andersen JW, Schiffer CA, Appelbaum FR, Feusner JH, Woods WG, et al. All-trans retinoic acid in acute promyelocytic leukemia: long-term outcome and prognostic factor analysis from the North American Intergroup protocol. Blood. 2002;100(13):4298–302.
Arrieta O, González-De la Rosa CH, Aréchaga-Ocampo E, Villanueva-Rodríguez G, Cerón-Lizárraga TL, Martínez-Barrera L, et al. Randomized phase II trial of All-trans-retinoic acid with chemotherapy based on paclitaxel and cisplatin as first-line treatment in patients with advanced non-small-cell lung cancer. J Clin Oncol. 2010;28(21):3463–71.
Kumaran G, Middleton MR, Zee YK, McGuigan L, Clamp AR, Hogg PJ, et al. Selective inhibition of proliferating endothelial cells: a phase I study of the novel organoarsenical compound GSAO in patients with advanced solid tumors. ASCO Meeting Abstracts. 2010;28(15 Suppl):TPS167.
Maroun JA, Maksymiuk A, Eisenhauer E. Phase II study of acivicin in non-small cell lung cancer: a National Cancer Institute of Canada study. Cancer Treat Rep. 1986;70(11):1327–8.
Maroun JA, Fields AL, Pater JL. Phase II study of acivicin in colorectal carcinoma: a National Cancer Institute of Canada study. Cancer Treat Rep. 1984;68(9):1121–3.
Bonomi P, Finkelstein D, Chang A. Phase II trial of acivicin versus etoposide-cisplatin in non-small cell lung cancer: an Eastern Cooperative Oncology Group study. Am J Clin Oncol. 1994;17(3):215–7.
Maroun JA, Stewart DJ, Verma S, Evans WK, Eisenhauer E. Phase I study of acivicin and cisplatin in non-small-cell lung cancer: a National Cancer Institute of Canada study. Am J Clin Oncol. 1990;13(5):401–4.
Adolphson CC, Ajani JA, Stroehlein JR. Phase II trial of acivicin in patients with advanced colorectal carcinoma. Am J Clin Oncol. 1986;9(3):189–91.
Author information
Authors and Affiliations
Corresponding author
Additional information
Pierre J. Dilda and Philip J. Hogg are co‐senior authors.
Rights and permissions
About this article
Cite this article
Ramsay, E.E., Hogg, P.J. & Dilda, P.J. Mitochondrial Metabolism Inhibitors for Cancer Therapy. Pharm Res 28, 2731–2744 (2011). https://doi.org/10.1007/s11095-011-0584-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11095-011-0584-5