
ABSTRACT
Purpose
Herein, we designed a nanoparticulate combined delivery system decorated on the surface with RGD peptide, and encapsulating paclitaxel (PTX) and combretastatin A4 (CA4) as the respective anticancer and antiangiogenesis agent in the nanoparticle.
Methods
PTX and CA4 were co-encapsulated into the biocompatible PLGA, followed by solvent evaporation to form solid nanoparticle. The cRGDfK peptide was then conjugated onto the nanoparticle surface with EDC/NHS chemistry.
Results
The developed nanoparticles (NPs) were found uniform in size and well dispersed in buffers. The cellular uptake of such NPs could be efficiently detected as early as 20 min after incubation. In 24-h incubation, the encapsulated PTX could induce caspase 3/7-dependent apoptosis at 50 nM, whereas the CA4-loaded NPs could disrupt tubulin structure at 2.5 μM. The targeted dual drug-loaded nanoparticle achieved significant tumor growth suppression in vivo compared to the control from day 8 (P < 0.05). Histological results revealed that the targeted dual drug nanoparticle led to dramatic tumor vasculature disruption, significant cancer cell apoptosis and cell proliferation inhibition in the mouse model.
Conclusion
These findings indicate that the targeted dual drug nanoparticulate delivery system encompassing both antiangiogenesis and anticancer effects can be a potential candidate in cancer therapy.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
INTRODUCTION
Angiogenesis, the growth of new blood vessels from preexisting capillaries, is a fundamental process involved in the embryonic development and adult life. Physiological angiogenesis is vitally important in maintaining tissue functions and homeostasis (1,2), whereas the angiogenetic feature in the solid tumor contributes to the tumor proliferation, migration and metastasis (3). During tumor cell development, the vasculatures provide the necessary oxygen and nutrients supporting the highly proliferating cancerous cells' growth and invasion. Hence, angiogenesis-targeted therapy was proposed as an essential therapeutic modality for the suppression of tumor growth and metastasis (4). In addition, the specific feature of tumor vasculature and its less-likeliness of acquiring drug resistance owing to the genetic stability of endothelial cells makes the antiangiogenesis therapy a promising approach in cancer treatment (5). Unfortunately, it was evidenced that tumor angiogenesis is a comprehensive result of multiple pathways which are mutually associated and compensatory (6); this compensatory mechanism could limit the therapeutic effects from a single antiangiogenesis therapy. Thus, combining a conventional cytotoxic agent with an antiangiogenesis drug under optimal dosage could likely enhance the therapeutic effects in a synergistic manner and reduce the potential side effects. Some recent studies on the therapeutic effect of the combined antiangiogenesis and conventional chemotherapy have shown the promising effects of this strategy in fighting against cancer (7,8), especially in advanced metastatic cancer (9).
Amongst the conventional chemotherapeutic agents, paclitaxel (PTX) has emerged as a wide spectrum anticancer drug for treatment of various cancers, such as breast cancer, ovarian cancer, lung cancer, etc. PTX binds to the β-tubulin on cytoskeleton, and disables the microtubule disassembling during mitosis in cell cycle, and finally leads to apoptosis of the treated cancer cells. To disrupt the blood vessels supporting tumor growth and the spreading, vascular-disrupting agents such as combretastatin A4 (CA4) take full use of the differences in the pathophysiology of tumor versus normal tissue blood vessels and lead to selective and efficient shutdown of the blood vessels in tumor, resulting in the deprivation of oxygen and nutrients to tumor cells (10).
To date, although researchers have proved the encouraging therapeutic effect on tumors with the combination of paclitaxel and combretastatin analogue (11), there is no report on using tumor vasculature-targeting nanoparticulate delivery system for this combinatorial therapy. Nanotechnology has proven to be a valuable approach to improve drug delivery for the treatment of numerous diseases, including cancers (12). The therapeutic application of many anticancer drugs has been hampered by their poor water solubility and/or low bioavailability in clinical trials. Formulation of these drugs as nanomedicine could help to overcome some of these problems. In addition, nanoparticles with suitable surface properties could extend circulation time in the bloodstream, resulting in accumulation of these nanoparticles loaded with drug at the tumor site under the passive targeting effect. With addition of targeting moiety to the surface, the delivery of nanoparticles could be further improved by active targeting. In this study, we designed a nanoparticulate delivery system decorated on the surface with a small peptide containing Arg-Gly-Asp (RGD) sequence and encapsulating paclitaxel (PTX) and combretastatin A4 (CA4) as the respective anticancer and antiangiogenesis agent in the core of the nanoparticle. Biodegradable polyesters such as poly(lactic acid) (PLA) or poly(lactic-co-glycolic acid) (PLGA) are extensively used in biomedical application due to biocompatibility, ability of loading various therapeutic compounds, and sustained drug release manner. Here, we chose PLGA as the building-up matrix to encapsulate PTX and CA4 inside the nanoparticles. To enhance the tumor vasculature targeting selectivity and reduce the possible side effects arising from anticancer drugs, the NPs were surface modified with small peptides containing Arg-Gly-Asp (RGD) sequence. This RGD sequence was reported as a highly affinitive ligand to αvβ3 receptor overexpressed on many cancerous cells (13) and endothelial cells (14). Therefore, the objective of this study is to explore the delivery mechanism of this nanoparticulate formulation and investigate the combinatorial potency in treatment of malignant cancer.
MATERIAL AND METHODS
Materials
PLGA (RG503H) was obtained from Boehringer Ingelheim (Germany). RGDfK cyclic peptide was purchased from AnaSpec (U.S.A.). CellTiter 96® AQueous Non-Radioactive Cell Proliferation Assay (MTS) was from Promega. Paclitaxel (microcrystalline white powder, purity >99%) was obtained from 21 CEC company (Oaklands, UK). Combretastatin A4 was from (Toronto Research Chemicals, Canada). N-hydroxysuccinimide (NHS), N-(3-Dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDC), polyvinyl alcohol (PVA, M.W. 30,000–70,000), Dulbecco’s Modified Eagle’s Medium (DMEM), and other necessary chemicals were of analytical grade and were purchased from Sigma Chemical Co. (St. Louis, MO, U.S.A.). Human umbilical vascular endothelia cells (HUVEC) was generously provided from Dr. Ruowen Ge from the Department of Biological Sciences, National University of Singapore. Anti-β tubulin primary monoclonal antibody was from Sigma Co. (MO, U.S.A.); other primary antibodies used in western blots were obtained from Cell Signaling Inc. All of the secondaries involved in this study were purchased from Pierce Biotechnology. Deionized water (Millipore, Bedford, MA, U.S.A.) was used throughout the entire study.
Preparation of Drug-Loaded Nanoparticles
Nanoparticles containing either single drug or dual drugs were prepared using emulsion solvent evaporation method. In brief, 100 mg PLGA and, if required, 10 mg PTX and/or 20 mg CA4, were dissolved in 5 ml of dichloromethane to form organic phase. This organic phase was emulsified in 40 ml of 1% (w/v) aqueous PVA solution by probe sonication (50 W out-put, 2 min, 2 s pulse) in an ice bath, followed by organic evaporation with magnetic stirring at 1,000 rpm for 6 h. The nanoparticles produced were collected and washed three times with deionized water by ultrafiltration (Millipore, Amicon®) at 4,000 rpm for 45 min. To make the nanoparticle fluorescent for uptake mechanism study, 6-coumarin, a widely hydrophobic fluorophore was added to the organic phase at 0.05% concentration.
To modify the nanoparticle surface with RGD peptide, the above prepared concentrated NPs were diluted with 0.1 M MES (pH 6.0), subjected to ultrafiltration, washed with 0.1 M MES twice, and finally dispersed in MES buffer. EDC and NHS were added into the nanoparticle solution at 0.1 M and 0.25 M, respectively, and this mixture was incubated for 2 h, followed by washing three times with PBS (pH 7.4) and separated by ultrafiltration. Then, 1 ml (2 mg/ml) RGD peptide solution was introduced into the NPs solution under mild vortex. The reaction was allowed to proceed overnight, and the unconjugated peptides were washed off by ultrafiltration with PBS.
Characterization of Nanoparticles
The mean diameter and polydispersity index were determined by dynamic light scattering (DLS) using Zeta Sizer (Nano 90s, Malvern Instruments, England). The zeta potential of NPs was also characterized by Zeta Sizer. To determine the drug loading efficiency (LE) and encapsulating efficiency (EE), the nanoparticle solution was first lyophilized by freeze-drier, and then the pre-weighted nanoparticles were dissolved in acetonitrile and incubated for 4 h to completely release the encapsulated drugs from nanoparticles. Then, the solution was diluted using acetonitrile and water mixture at 50:50% v/v ratio. The drug content was analyzed by HPLC. The mobile phase was 50:50 acetonitirle:water at flow rate of 1 mL/min, and detection was made at wavelength of 227 nm for PTX and 295 nm for CA4.
The conjugation efficiency of RGD peptide onto the nanoparticle surface was determined by a fluorescamine assay adopted in our previous study (13). Briefly, fixed concentration (0.2 mg/ml) of fluorescamine dissolved in acetone was allowed to react with the amine groups of the free RGD peptide in the filtrate of the original nanoparticle solution. The reaction was followed for 10 min at room temperature, and then the fluorescence signal was detected at 470 nm under the excitation wavelength of 390 nm. The amount of RGD peptides conjugated onto the nanoparticle surface was calculated by subtracting the amount of peptide detected in the filtrate from the initial amount of RGD peptide used for the conjugation. The morphology and microstructure of the NPs was visualized by scanning electron microscopy (SEM, JEOL, Japan). To prepare the sample for SEM imaging, the lyophilized NP powder was loosely scattered onto carbon conductor tape supported with copper holder, and then the NPs were coated with Pt/Au before observing under SEM at 5 kV voltage.
Cellular Uptake of NPs and Their Distribution in Cells
The cellular uptake of NPs by HUVEC was evaluated by confocal laser scanning microscope (CLSM). Frozen HUVEC cells (passage <4) were re-suspended in standard endothelial cell culture medium (Cell Systems. Inc. U.S.A.) with 1% penicillin and streptomycin, and cultured in the humidified atmosphere of 5% CO2 at 37°C. For every sub-culture, cells were harvested by trypsinization for 1 min at room temperature with an addition of 2 min incubation at 37°C before fresh medium was added. HUVECs were seeded in Lab-Tek® chambers (Nalgene, Nunc Inc., Denmark) at a density of 2 × 104 per well and allowed to adhere to the wells for 24 h. 6-coumarin-labeled NPs were diluted with serum-free cell culture medium to 100 μg/ml before adding to the cultured cells. At the designated time points (20 min, 1, 4, and 8 h after incubation), the cells were washed three times with PBS and fixed with 4% paraformaldehyde and Triton X-100 before staining with Texas Red® conjugated phalloidin; then the cells were washed with PBS, followed by adding DAPI to stain the cell nuclei. Optical images were obtained using Olympus Flowview FV1000 Confocal laser scanning microscope. A 60× objective aqueous lens was used equipped with 488 nm argon, 543 nm HeNe and 405 nm diode lasters. To investigate the cellular distribution of NPs in HUVEC, the cells were incubated with NPs (100 μg/ml) for 1 h before using endo/lysome tracker kit (Molecular Probes, CA) according to manufacturer’s instruction.
To further verify the internalization mechanism of NPs into cells through endocytosis pathway, we measured the cellular uptake of NPs in cold condition (4°C) versus normal cell culture condition (37°C). HUVECs were seeded on 24-well plate at density of 2 × 104 per well 24 h before experiment. 6-coumarin-labeled NPs were incubated with cells for different times (20, 40, and 60 min) at 4°C and 37°C, respectively. After incubation for the respective times, cells were washed three times with PBS, and 0.2 ml Trypan Blue aqueous solution (0.2 mg/ml) was added for 5 min to quench any non-internalized or non-specific absorbed NPs. After washing with PBS twice, the cells were lysed with 0.2 ml lysis buffer (0.1 N NaOH/5% SDS) for 30 min. Cell extracts were measured for fluorescent signal with Multiwell plate reader (Tecan Group, Ltd., Switzerland) at excitation/emission of 488 nm/530 nm.
Western Blot to Detect Cellular Apoptosis Induced by PTX NPs
B16F10 cells were cultured overnight in the 6-well plates at 100,000 per well with DMEM cell culture medium before they were treated with various concentrations of NPs containing PTX alone. The cells were then incubated with the NPs for 48 h before western blot assay. For western blot assay, protein was extracted from treated cancer cells (B16F10) with lysis buffer (1% Triton X-100 with Proteinase inhibitor cocktail) on ice. The extracted proteins were separated with SDS-PAGE and transferred to PVDF membrane. The membrane was blocked by TBS buffer containing 5% skim milk for 1 h. After washing with the washing buffer (TBS buffer with 0.1% Tween 20), the membrane was incubated with primary antibodies specific for cleaved caspases 3, caspase 7 and actin at room temperature for 2 h, respectively, and immunoblotted for 1 h with the corresponding secondary antibodies coupled with horseradish peroxidase. After extensive washing, bands were detected by SuperSignal West Pico Chemiluminescence Substrate (Pierce Biotechnology).
Immnunofluorescence Staining of Tubulin
To reveal the ability of NPs singly loaded with CA4 to disrupt endothelial microtubule cytoskeleton, cultured HUVECs were incubated with CA4 containing NPs (equivalent to 0.25 μM drug) for 24 h. After incubation, cells were fixed with acetone/methanol (1:1) mixture for 10 min at −20°C, followed by sequential incubation with anti-β-tubulin primary antibody and corresponding FITC coupled secondary antibody. Fluorescent images were taken using Nikon Eclipse TE200 inverted microscope equipped with cooled CCD camera.
In Vivo Antitumor Studies
Cell Culture
B16F10 mouse melanoma cells obtained from American Type Culture Collection (Rockville, MD) were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) supplemented with 10% fetal bovine serum and 1% streptomycin and penicillin. The cells were maintained in a humidified atmosphere of 5% CO2 at 37°C. Cells grown in monolayer were sub-cultured by 0.05% trypsin/EDTA. The viability of cells was routinely monitored using Trypan Blue dye exclusion method. The cells were harvested during the logarithmic growth phase and re-suspended in serum-free medium before inoculated in animals.
Subcutaneous Tumor Xenograft Experiment
Animal procedures were performed according to the protocols approved by the university institutional animal care and use committee. The experiments were conducted in the Animal Housing Unit, National University of Singapore using 6–8 weeks old C57BL6 male mice. For tumor development, B16F10 cells (0.5 × 106 in 0.1 ml serum-free medium) were implanted subcutaneously in 6–8-week-old C57BL6 male mice. The tumor was allowed to grow for 8 days when the tumor volume was about ~100 mm3 prior to the drug administration. Tumor-bearing mice were assigned randomly into six groups of five mice each as follows: 1) RGD surface modified with two drug-loaded NPs (T-Dual NP), 2) and 3) RGD surface modified with single drug-loaded NPs (T-PTX NP or T-CA4 NP), 4) RGD surface modified blank NPs (BLK NP), 5) two drug-loaded NPs without RGD surface modification (Dual NP), and 6) PBS solution as control. All formulations were diluted with sterile PBS solution. Each tumor-bearing mouse received PTX at 5 mg/kg and CA4 at 20 mg/kg by intravenous administration through the tail vein in the volume of 100 μl in PBS. The time interval in between the two drug administration was 6 days.
Tumor diameters were recorded every 2 days with a digital caliper in two dimensions. Tumor volumes were calculated using the following formula: \( {\hbox{Tumor Volume}} = \left[ {{\hbox{Length}} \times {{\left( {\hbox{Width}} \right)}^{{2}}}} \right]/{2} \), where length is the longest diameter, and width is the shortest diameter perpendicular to the length (15).
Immunohistochemical Staining
Xenograft tissues were fixed in 4% paraformaldehyde for 2 days and stored in 70% ethanol solution before experiment. Serial sections (5 μm thick) were deparaffinized in xylene and rehydrated through descending percentages of ethanol to water. Antigen retrieval was done with 10 mM citrate buffer (pH 6.1) for CD31 and Ki-67 antibodies. Endogenous peroxidase was blocked by 1% H2O2 methanol solution. For cell proliferation analysis, slides were incubated with Ki-67 antibody (Abcam, 1:100), and CD31 antibody (Abcam, 1:100) was used for micro-vessel staining. Detection was done with diaminobenzidine chromogen, which results in brown color staining. The nuclei were counterstained with haematoxylin. Cellular apoptosis was carried out by reacting sliced tissues with terminal transferase dUTP nick end labeling (TUNEL) assay (Roche) according to the protocol from the manufacturer.
RESULTS
Preparation and Characterization of Targeted Nanoparticles
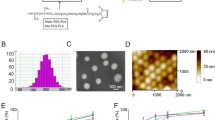
Figure 1A presents the scheme of formulation process. Since both CA4 and PTX are hydrophobic compounds, both drugs can be encapsulated simultaneously into PLGA matrix with a solvent-evaporation method. The encapsulation efficiency of PTX and CA4 was 60%–65% and 53%–57%, respectively. The drug-loading ratio was fixed at 1–4 for PTX and CA4 so as to maintain a relatively low dose of PTX (5 mg/kg). In a single drug-loaded nanoparticle, PTX could be loaded up to 68–72 μg/mg NP. Surface modification of the nanoparticle was accomplished with ECD/NHS chemistry to conjugate RGD peptide onto the particle surface. The conjugation efficiency was found to be about 15%–20% in this study. The resultant nanoparticle was uniform in size as shown in Fig. 1B, and the nanoparticle size was about 200 nm as determined by dynamic light scattering method (Fig. 1C).

Poly(lactic-co-glycolic acid) (PLGA) nanoparticles decorated on the surface with RGDfK peptide and encapsulating paclitaxel and combretastatin A4 in the core. Schematic illustration of the targeted nanoparticulate dual-drug delivery system (A); the scanning electron micrograph image of the resultant nanoparticles with smooth surface, clear boundary and uniform size distribution (B) and dynamic light scattering analysis of the nanoparticles (C).
Table I summarizes the physical properties of NPs prepared in different formulations used in this study. It shows no significant particle size changes between blank NPs and drug-loaded NPs, nor did the surface modification step significantly increase the NP size of the T-Dual NP in comparison to the Dual-NP. Although the drug loading and/or RGD modification slightly altered the particle size, the resultant NPs remained uniform in the size distribution as indicated by the polydispersity values (Table I). All of as-prepared NPs presented negative surface charge due to the carboxylic end groups on PLGA polyesters. The surface charge tended to become lower when drugs were loaded and RGD was conjugated to the particle surface. This is attributed to the inevitable drug absorption on the particle surface and/or the positive charge groups of RGD partially neutralizing the negative charge of NP. Fortunately, all of these minor changes did not adversely influence the physical properties of NPs, which makes this novel targeted dual drug delivery system robustly applicable for cancer therapy.
Cellular Uptake of Nanoparticles
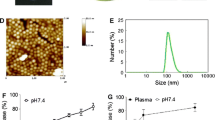
We investigated the cellular uptake manner of NPs by HUVEC through confocal laser scanning microscope (CLSM). Figure 2(a–d) presents the time-dependent uptake of NPs at 100 μg/ml concentration. The uptake of nanoparticles by HUVEC was prominent as early as 20 min (Fig. 2a and e) and is the most significant after a 4-h incubation (Fig. 2c). A longer incubation time (8 h) could not further increase the nanoparticle uptake significantly (Fig. 2d and f). Thus, the resultant nanoparticles could be effectively taken up by HUVEC, and this provided a solid foundation to the high efficacy of the dual drug encapsulating nanoparticle for antivasculature and antitumor activities.

Cellular uptake of targeted nanoparticle. Confocal images showing uptake of nanoparticles by HUVEC at different time points: (a) 20 min; (b) 1 h; (c) 4 h; and (d) 8 h. Red: actin filaments; Blue: cell nuclei; Green: targeted nanoparticles. (e) (with both bright and fluorescent illumination) and (f) (with fluorescent illumination only) show the distribution of nanoparticles in cells upon 1 h incubation. Red: endosome/lysosome (black arrow); Green: nanoparticles (white arrow); Yellow/orange: overlay. The concentration of nanoparticle was set at 0.1 mg/ml for all uptake experiments.
To confirm the targeting ability of the RGD-modified NPs was from the affinity of RGD to HUVEC, a higher concentration of free RGD peptides (50 μM) was added into the cell culture medium before initiating the uptake experiment. The cellular uptake of RGD-modified NPs was inhibited in the presence of free RGD peptide in the medium (Fig. 3).

Competition assay of cellular uptake of the targeted nanoparticles. Targeted or non-targeted NPs (0.1 mg/ml) were incubated with HUVECs for 1 h. Then, the cells were washed 2 times, followed by adding trypan blue to bleach any non-specific absorbed NPs on cell membrane. Cells were lysed in lysis buffer before measurement of fluorescence at 480/530 ex/em. For the competition, 50 μM free RGDfK peptide was added into the medium before incubation with targeted nanoparticles. Data are presented as mean ± s.e.m (n = 3) (*P < 0.05).
In the live-cell analysis, the co-localization of RGD-modified NPs with endo/lysosome tracker staining suggested that the receptor-mediated endocytosis underlined the cellular uptake mechanism (Fig. 2g and h). This mechanism was further confirmed by incubation of NPs with HUVECs in cold (4°C) and normal (37°C) conditions, respectively. The uptake of NPs was reduced by nearly 50% at 4°C in the first 20 min incubation. Whereas the NPs uptake became higher after longer incubation (60 min), the uptake of NPs in cold condition still remained much lower (31.5% less) than that in the normal condition (Fig. 4).

Mechanism study of cellular uptake of nanoparticle. The cellular uptake of nanoparticles is a time- and energy-dependent process. At cold condition (4 degree), the uptake of nanoparticles was decreased by nearly 50% of that in normal condition (37 degree) at 20 min, and even after 60 min incubation, the uptake in cold condition was still 68.5% of that in normal condition. Data are shown as mean ± s.e.m (n = 3). (*, P < 0.05; #, P < 0.01).
In Vitro Evaluation of Efficacy of Nanoparticles
We investigated the potency of the formulated nanoparticles to induce apoptosis in cancer cells by PTX and the ability of CA4-loaded nanoparticle to disrupt the HUVEC β-tubulin structure, respectively. To avoid the potential interference of two drugs in one formulation, we individually formulated each drug into nanoparticles. Figure 5 demonstrates the western blot results of PTX-loaded nanoparticle to malignant melanoma cancer cells (B16F10). It can be seen that 50 nM equivalent PTX is potent to induce the caspase 3 and 7 activation, indicating the undergoing apoptosis pathway upon the exposure of 50 nM PTX formulation, whilst a lower drug concentration (10 nM) could not function so efficiently. At 50 nM drug concentration, the PTX-encapsulated nanoparticles dispersed in aqueous cell culture medium are as effective and efficient as the PTX directly dissolved in DMSO leading to cancer cell death within 24 h.

Western blot results show the potency of T-PTX NPs in inducing caspase 3/7 apoptosis pathway in B16F10 cancer cells at different drug concentration upon 24 h incubation. Control: culture medium; Blank NP: blank nanoparticle with RGDfK modification but no drug loading; PTX (DMSO): pure paclitaxel directly dissolved in DMSO; PTX NP: targeted paclitaxel loaded nanoparticle.
Figure 6 shows the immufluorescent staining results of β-tubulin structure in HUVEC after incubation with the respective nanoparticulated CA4 at an equivalent concentration of 2.5 μM (Fig. 6a) and the blank nanoparticles (Fig. 6b). Upon a 24-h incubation, the blank nanoparticle-treated HUVEC cells did not show any obvious β-tubulin disruption, whilst the HUVECs treated with 2.5 μM CA4 containing nanoparticle presented significant β-tubulin disruption.

Immunofluorescent staining of HUVEC illustrates β-tubulin disruption by combretastatin A4 (CA4) formulation. A presents the unchanged tubulin filament (white arrows) upon incubation with blank nanoparticle for 24 h; B shows the disrupted tubulin structure (white arrows) and changed cell morphology (white arrow head) after treatment by equivalent 2.5 μM CA4 encapsulated NPs for 24 h.
Antitumor Evaluation of Different Formulations
We evaluated the biological activities of different formulated NPs for antiangiogenesis and anticancer activities in tumor-bearing mice. Figure 7 presents the results of the tumor volume changes as a function of treatment time with PTX and CA4 therapeutics either as single drug or dual drug-loaded nanoparticles. It can be seen that the tumor growth was significantly inhibited by the targeted dual drug-loaded NPs from day 8 onward (p < 0.05), and from day 10 onward by the non-targeted dual drug-loaded nanoparticle (p < 0.05). In contrast, the two targeted single drug-loaded nanoparticles at the given dose (5 mg/kg PTX, 20 mg/kg CA4) showed discernable but statistically insignificant tumor growth inhibition throughout the whole study period. No mice died from the treatment in the entire experiment. However, it may be noted that the control groups and the respective treatment groups on the single drug-loaded nanoparticle were terminated 2 days earlier than the treatment groups on the dual drug-loaded nanoparticle, as their tumor sizes started to grow beyond the limit stipulated by the institutional animal care and use committee.

Changes in tumor growth as a function of time in C57BL6 mice after intravenous treatment with PTX and CA4. The injection dose of PTX and CA4 was set at 5 mg/kg and 20 mg/kg. The drugs were administrated on day 0 and 6, as indicated by arrows (* P < 0.05, # P < 0.05 vs. control).
Histological Evaluation of Different Formulation Treatment
Tumor tissues with small and large vessels are delineated by CD 31 immunostaining (Fig. 8). Untreated tumor (PBS) showed apparent lumen structure with large blood vessels, T-PTX NPs-treated tumor resulted in discontinued vascular capillaries, and T-CA4 NPs presented a significant disruption of vasculatures in the tumor. Moreover, the most prominent disruption of tumor vasculature occurred in the T-Dual NPs-treated group, accompanied by necrosis within tumor tissues (as indicated by black arrows). Tumor in the group treated with T-Dual NPs showed the least proliferative activity, and other drug-treated groups presented different level of proliferation reduction, as compared with the PBS-treated group (Fig. 8). TUNEL examination of DNA fragmentation in tumor cell nuclei presented in Fig. 8 revealed the potency of each formulation in inducing apoptosis to cancer cells. Tumor of the T-Dual NPs-treated group showed the most significant TUNEL staining, with notable deformed cellular morphology. Interestingly, tumor of the T-PTX NPs-treated group showed less apoptotic effect than that of the T-CA4 NPs-treated group. This might be attributed to the relative low PTX dosage as a single agent used in this study (5 mg/kg).

Histology of tumor tissues with treatment of different formulations. CD 31 staining reveals the tumor vasculature; Ki 67 staining shows the extent of cell proliferation inhibition of different formulations; and TUNEL staining demonstrates the degree of apoptosis induced by various formulations. The paclitaxel and combretastatin A4 were given at dose of 5 mg/kg and 20 mg/kg, respectively. All images are 10× of magnification.
The change of body weight was recorded (data not shown). There was no significant body weight change in all of the treated and non-treated groups. The slight increase of body weight may be due to the natural body growth.
DISCUSSION
Numerous studies have evidenced that the combination of antiangiogenic agents with conventional chemotherapeutic agents (16,17) or radiotherapy (18) could have additive or synergistic effects against malignant cancers. In clinic, antiangiogenetic agents as monotherapeutics have limited, if any, discernable efficacy, especially in the treatment of advanced malignant cancers. The conventional chemotherapy can result in the well-known drug resistance effect after long-term administration. This drug resistance sets a barrier for better treatment of cancer. In addition, due to plasticity, tumors are likely to evade a single-targeted therapeutic method designated to control the proliferation and survival of tumor cells (19). Hence, formulation of drugs with the respective antivasculature and anticancer activities could hold great potential for cancer therapy.
In this study, PLGA was chosen as the matrix building-up material to encapsulate drugs into nanoparticles, followed by decorating the nanoparticle surface with targeting moiety. The PLGA or PLA have been extensively used in biomedical applications from drug delivery system to tissue engineering. Thus, this biocompatible and degradable material is more feasible to be translated to clinical practice than any other newly discovered biomaterials (12). Because of the hydrophobic feature of PLGA, it is of ease to encapsulate hydrophobic drugs, like PTX or CA4, into nanoparticulate form. In addition, the availability of carboxyl end group of PLGA facilitates the further conjugation of targeting moieties for disease-specific drug/gene delivery (20).
As shown in Table I, the resulted NPs presented negative surface charges that could enhance nanoparticle stability by decreasing the nonspecific absorption of protein onto the NPs during circulation (15). The RGDfK peptide conjugated onto the nanoparticle surface is supposed not only to target some specific cancer cells which overexpress integrin receptors on cell membrane, but also orient the NPs to the proliferating tumor vascular cells where integrin receptors were specifically overexpressed (14). In this sense, nanoparticulated drugs could be more selectively accumulated at the tumor site.
Efficient cellular uptake of NPs plays a vital role in the delivery of drugs to cancer cells so as to initiate the apoptotic pathways. Figure 2 shows that the RGD-modified NPs were taken up by HUVECs as early as 20 min. This uptake trend could be, to some extent, inhibited by prior addition of free RGDfK peptides (Fig. 3). This highly efficient cellular uptake may explain the efficient onset of cytotoxicity of T-PTX NPs to cancer cells (Fig. 5). Since PLGA-based drug delivery system usually presents a sustained-release manner for encapsulated drugs (21), it takes some time (usually 48 h) for the sequestered anticancer drugs to show significant cytotoxic effect. However, in our system, the encapsulated PTX presented effective cytotoxic effect in 24 h at equivalent drug concentration of 50 nM, which is comparable to the PTX directly dissolved in DMSO (Fig. 5).
The CA4-loaded targeted NPs also presented good potency in disruption of β-tubulin of HUVECs in 24 h (Fig. 6). Upon release from the NPs, CA4 can bind to tubulin at or close to the colchicines-binding site (22) and result in rapid re-organization of the actin cytoskeleton, mediated by disruption of the tubulin cytoskeleton. This process was reported to be associated with the Rho/Rho-kinase, as well as stress-activated protein kinase 2 (SAPK2)-dependent pathways (23). Unlike other antiangiogenetic agents, such as bevacizumab (Avastin), a recombinant monoclonal antibody targeted to VEGF that leads to transient “normalization” of the tumor vasculature (24), CA4 is a vascular disrupting agent that induces tumor vasculature permeability, leading to further shutdown of tumor blood flow, and finally resulting in increased tumor hemorrhage and necrosis (25).
Even though PTX was used at a relatively small dose (5 mg/kg), the combination of such low dose of PTX with CA4 (20 mg/kg) in targeted NPs achieved substantial therapeutic effect in the B16F10 tumor-bearing mice (Fig. 7) than the single-agent-loaded NPs. Besides, the tumor in the group treated with the targeted dual-drug-loaded NPs demonstrated substantial antivasculature and anticancer therapy as evidenced by the immunohisochemical (IHC) staining results (Fig. 8). Figure 8 shows that the T-Dual NPs were able to induce widespread tumor vasculature disruption (CD 31 staining), extensive cytotoxicity (TUNEL), and strong proliferation inhibition of tumor cells (Ki 67 stainings).
In cancer therapy, the commonly employed strategy is to use high dose of drugs for patients so as to achieve maximum treatment efficacy. Nevertheless, the side effects associated with such high dose treatment are concerning for the benefit of patients. Thus, individualized low dose anticancer drug administration should be a plausible method for cancer treatment, but this low dose strategy may bear the risk of not achieving effective therapeutic effects, especially in those malignant cancers. It has been recently reported that low dose of PTX could be delivered to tumor site to achieve encouraging therapeutic effect by nanotubes, because of the long circulation time and high tumor cell uptake of that nanostructured material (26). With our developed delivery system, although there existed the therapeutic effect by T-PTX NPs as single agent administrated at 5 mg/kg, the average tumor growth was still 11.5-fold of the initial tumor size, whilst that in the T-Dual NPs group was much lower, just 5.4-fold. Meanwhile, the equivalent concentration of CA4 in T-CA4 NPs group presented even higher tumor growth (13.3 fold). This significant therapeutic effect of T-Dual NPs may be due to the fact that CA4 was released to the tumor microenvironment upon T-Dual NPs anchoring to either tumor cell or tumor vasculatures, leading to the disruption of tumor vasculature, and the simultaneously released PTX efficiently induced apoptosis of cancer cells. Since CA4, a vascular disrupting agent, resulted in the higher vascular permeation, the resultant leaky tumor vasculature might, in turn, facilitate the penetration of NPs into the tumor microenvironment to accumulate drug at high concentration within the tumor. This enabled enhanced cytotoxic effect of PTX to cancer cells. Furthermore, with more cancer cells dead, the growth factors secreted by cancer cells could also be dramatically reduced. The available low concentration of growth factors would not be able to support the subsequent neovasculature growth. Then, our targeted nanoparticulated combination therapy could exert both antivasculature and anticancer activities.
To date, various antiangiogenesis therapies have been proposed for cancer treatment, ranging from regulation of transcription factor expression (27) to combining antiangiogenetic agent with anticancer drugs (28) or treatment with combination of drugs in addition to radiotherapy (29). Each strategy possesses its own distinctive feature for effective treatment of cancer. Unfortunately, most of the strategies involved the complex drug administration routes and schedules, which may make the combination therapy difficult to follow in practice. From a practical point of view, it will be more convenient if the combination therapy method could be realized in a simple way. Our targeted nanoparticulate combination therapy aimed at neovasculature and malignant cancer cells could simultaneously, to some extent, simplify the drug administration route, as well as the schedule. More importantly, the appropriate combination of drugs could significantly enhance the therapeutic efficacy as an organic whole.
CONCLUSION
In this study, we designed and prepared a combined drug delivery system with surface RGD peptide modification for active targeting purpose. The cellular uptake was efficient, and the mechanism was evidenced to be a receptor-mediated endocytosis pathway. The single-agent-encapsulated nanoparticle could effectively induce cellular apoptosis by PTX and cytoskeleton disruption by CA4, respectively. The in vivo tumor model showed the tumor suppression by targeted dual-drug-loaded nanoparticle at day 8 and onwards, via vascular disruption, cellular proliferation inhibition, and apoptosis induction in tumor microenvironment. Thus, we envision that this combined drug delivery system could potentially be useful for treatment of cancer.
REFERENCES
Coultas L, Chawengsaksophak K, Rossant J. Endothelial cells and VEGF in vascular development. Nature. 2005;438:937–45.
Shojaei F, Ferrara N. Antiangiogenesis to treat cancer and intraocular neovascular disorders. Lab Invest. 2007;87:227–30.
Carmeliet P. Angiogenesis in life, disease and medicine. Nature. 2005;438:932–6.
Holmgren L, Oreilly MS, Folkman J. Dormancy of micrometastases—balanced proliferation and apoptosis in the presence of angiogenesis suppression. Nat Med. 1995;1:149–53.
Kerbel RS, Kamen BA. The anti-angiogenic basis of metronomic chemotherapy. Nat Rev Cancer. 2004;4:423–36.
Dorrell MI, Aguilar E, Scheppke L, Barnett FH, Friedlander M. Combination angiostatic therapy completely inhibits ocular and tumor angiogenesis. Proc Natl Acad Sci USA. 2007;104:967–72.
Uno T, Takeda K, Kojima Y, Yoshizawa H, Akiba H, Mittler RS, et al. Eradication of established tumors in mice by a combination antibody-based therapy. Nat Med. 2006;12:693–8.
Klement G, Baruchel S, Rak J, Man S, Clark K, Hicklin DJ, et al. Continuous low-dose therapy with vinblastine and VEGF receptor-2 antibody induces sustained tumor regression without overt toxicity. J Clin Invest. 2000;105:R15–24.
Hurwitz H, Fehrenbacher L, Novotny W, Cartwright T, Hainsworth J, Heim W, et al. Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. New Engl J Med. 2004;350:2335–42.
Thorpe PE. Vascular targeting agents as cancer therapeutics. Clin Cancer Res. 2004;10:415–27.
Yeung SCJ, She MR, Yang HL, Pan JX, Sun LL, Chaplin D. Combination chemotherapy including combretastatin A4 phosphate and paclitaxel is effective against anaplastic thyroid cancer in a nude mouse xenograft model. J Clin Endocrinol Metab. 2007;92:2902–9.
Farokhzad OC, Cheng JJ, Teply BA, Sherifi I, Jon S, Kantoff PW, et al. Targeted nanoparticle-aptamer bioconjugates for cancer chemotherapy in vivo. Proc Natl Acad Sci USA. 2006;103:6315–20.
Wang Z, Chui WK, Ho PC. Design of a multifunctional PLGA nanoparticulate drug delivery system: evaluation of its physicochemical properties and anticancer activity to malignant cancer cells. Pharm Res. 2009;26:1162–71.
Green JJ, Chiu E, Leshchiner ES, Shi J, Langer R, Anderson DG. Electrostatic ligand coatings of nanoparticles enable ligand-specific gene delivery to human primary cells. Nano Lett. 2007;7:874–9.
Devalapally H, Duan ZF, Seiden MV, Amiji MM. Paclitaxel and ceramide co-administration in biodegradable polymeric nanoparticulate delivery system to overcome drug resistance in ovarian cancer. Int J Cancer. 2007;121:1830–8.
Kumar P, Benedict R, Urzua F, Fischbach C, Mooney D, Polverini P. Combination treatment significantly enhances the efficacy of antitumor therapy by preferentially targeting angiogenesis. Lab Invest. 2005;85:756–67.
Minischetti M, Vacca A, Ribatti D, Iurlaro M, Ria R, Pellegrino A, et al. TNP-470 and recombinant human interferon-alpha 2a inhibit angiogenesis synergistically. Br J Haematol. 2000;109:829–37.
Abdollahi A, Griggs DW, Zieher H, Roth A, Lipson KE, Saffrich R, et al. Inhibition of alpha(v)beta(3) integrin survival signaling enhances antiangiogenic and antitumor effects of radiotherapy. Clin Cancer Res. 2005;11:6270–9.
Poeck H, Besch R, Maihoefer C, Renn M, Tormo D, Morskaya SS, et al. 5′-triphosphate-siRNA: turning gene silencing and Rig-I activation against melanoma. Nat Med. 2008;14:1256–63.
McCarron PA, Marouf WM, Quinn DJ, Fay F, Burden RE, Olwill SA, et al. Antibody targeting of camptothecin-loaded PLGA nanoparticles to tumor cells. Bioconjug Chem. 2008;19:1561–9.
Dhar S, Gu FX, Langer R, Farokhzad OC, Lippard SJ. Targeted delivery of cisplatin to prostate cancer cells by aptamer functionalized Pt(IV) prodrug-PLGA-PEG nanoparticles. Proc Natl Acad Sci USA. 2008;105:17356–61.
McGown AT, Fox BW. Structural and biochemical-comparison of the anti-mitotic agents colchicine, Combretastatin-A4 AND amphethinile. Anticancer Drug Des. 1989;3:249–54.
Kanthou C, Tozer GM. The tumor vascular targeting agent combretastatin A-4-phosphate induces reorganization of the actin cytoskeleton and early membrane blebbing in human endothelial cells. Blood. 2002;99:2060–9.
Winkler F, Kozin SV, Tong RT, Chae SS, Booth MF, Garkavtsev I, et al. Kinetics of vascular normalization by VEGFR2 blockade governs brain tumor response to radiation: role of oxygenation, angiopoietin-1, and matrix metal loproteinases. Cancer Cell. 2004;6:553–63.
Tozer GM, Kanthou C, Baguley BC. Disrupting tumour blood vessels. Nat Rev Cancer. 2005;5:423–35.
Liu Z, Chen K, Davis C, Sherlock S, Cao QZ, Chen XY, et al. Drug delivery with carbon nanotubes for in vivo cancer treatment. Cancer Res. 2008;68:6652–60.
Henke E, Perk J, Vider J, de Candia P, Chin Y, Solit DB, et al. Peptide-conjugated antisense oligonucleotides for targeted inhibition of a transcriptional regulator in vivo. Nat Biotechnol. 2008;26:91–100.
Stoeltzing O, Liu WB, Reinmuth N, Fan F, Parry GC, Parikh AA, et al. Inhibition of integrin alpha(5)beta(1) function with a small peptide (ATN-161) plus continuous 5-FU infusion reduces colorectal liver metastases and improves survival in mice. Int J Cancer. 2003;104:496–503.
Bozec A, Formento P, Lassalle S, Lippens C, Hofman P, Milano G. Dual inhibition of EGFR and VEGFR pathways in combination with irradiation: antitumour supra-additive effects on human head and neck cancer xenografts. B J Cancer. 2007;97:65–72.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Wang, Z., Chui, WK. & Ho, P.C. Nanoparticulate Delivery System Targeted to Tumor Neovasculature for Combined Anticancer and Antiangiogenesis Therapy. Pharm Res 28, 585–596 (2011). https://doi.org/10.1007/s11095-010-0308-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11095-010-0308-2