
ABSTRACT
Purpose
Using human lung cancer cells, we evaluated the involvement of plasminogen activator inhibitor-1 (PAI-1) in the anti-invasive action of cannabidiol, a non-psychoactive cannabinoid.
Methods
Invasion was quantified by a modified Boyden chamber assay. PAI-1 protein in cell culture media and PAI-1 mRNA were determined by immunoblotting and RT-PCR, respectively.
Results
Cannabidiol caused a profound inhibition of A549 cell invasion, accompanied by a decreased expression and secretion of PAI-1. Cannabidiol's effects on PAI-1 secretion and invasion were suppressed by antagonists to CB1 and CB2 receptors as well as to transient receptor potential vanilloid 1. Recombinant human PAI-1 and PAI-1 siRNA led to a concentration-dependent up- and down-regulation of invasiveness, respectively, suggesting a crucial role of PAI-1 in A549 invasiveness. Evidence for a causal link between cannabidiol's effects on PAI-1 and invasion was provided by experiments showing a reversal of its anti-invasive action by addition of recombinant PAI-1 at non-proinvasive concentrations. Key data were confirmed in two other human lung cancer cell lines (H460, H358). In vivo, a significant downregulation of PAI-1 protein by cannabidiol was demonstrated in A549 xenografts.
Conclusion
Our data provide evidence for a hitherto unknown mechanism underlying the anti-invasive action of cannabidiol on human lung cancer cells.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
INTRODUCTION
Although the anti-tumorigenic effects of cannabinoids were described in 1975 (1), comprehensive studies on cannabinoids' possible use as anticancer drugs during the last decade provided stronger evidence for anti-proliferative (2,3), proapoptotic (4,5), anti-metastatic (6), and anti-angiogenic effects (7,8). Furthermore, recent data support the view that the endocannabinoid system contributes to endogenous antitumorigenic defence mechanisms (9) and that cannabinoid receptor antagonists may promote carcinogenesis in people who are at high risk of developing human colorectal cancers (10). However, apart from the well-known antiproliferative and proapoptotic mechanisms of cannabinoids, their anti-invasive action on tumor cells is still poorly understood (for review, see 11). Recent evidence shows that inhibition of phosphorylation of focal adhesion kinase (12) and epidermal growth factor receptor (13), upregulation of tissue inhibitor of matrix metalloproteinases-1 (TIMP-1) (14,15) and downregulation of matrix metalloproteinase-2 (MMP-2) expression (16) contribute to cannabinoids' anti-invasive activity.
As the clinical use of cannabinoid-based drugs is limited by their psychoactive side effects, interest in non-psychoactive cannabinoid-mimetic compounds such as cannabidiol has substantially increased in recent years. Besides the beneficial effects of cannabidiol in the treatment of pain and spasticity associated with multiple sclerosis (17), several reports demonstrated anti-metastatic (18,19), proapoptotic (20–22), and anti-angiogenic (23) effects of this cannabinoid. A recent report demonstrated an inhibitor of basic helix-loop-helix transcription factors, Id-1, as a possible key signalling factor in the anti-invasive action of cannabidiol on breast cancer cells (19). Moreover, a recent study from our group revealed TIMP-1 as one potential target conferring the anti-invasive action of cannabidiol on human cancer cells (15). In the latter study, an involvement of cannabinoid receptors and the transient receptor potential vanilloid 1 (TRPV1) in the anti-invasive action of cannabidiol could be demonstrated.
Cancer cell invasion is a complex process of tumor-stroma interaction and cell migration through matrix components of tissues surrounding tumor cells, and represents a crucial event within the cascade leading to local growth and spreading of cancers. Besides the MMP/TIMP system, the plasminogen/plasmin system consisting of urokinase plasminogen activator (uPA), urokinase plasminogen activator receptor (uPAR) and the plasminogen activator inhibitors PAI-1 and PAI-2 has been demonstrated as an important factor in the regulation of cancer cell spreading (for review, see 24). Although high levels of PAI-1 should be intuitively beneficial in cancer by down-regulating uPA proteolytic activity, apparently contradictory results have been published regarding the impact of PAI-1 on tumor invasion, angiogenesis and metastasis with studies demonstrating either inhibitory (25–27) or stimulatory effects (25,28–30) on these events. In this context, differences in tumor models or PAI-1 concentrations as well as multiple uPA-dependent and -independent functions of PAI-1 have been suggested to contribute to the conflicting data (25). A rather pro-tumorigenic action of PAI-1 can be reasoned from numerous clinical studies implying high serum levels of PAI-1 as a predictor of poor prognosis for patients with breast, ovarian, gastric, colorectal, non-small-cell lung, renal cell, head and neck cancers as well as brain tumors (for review, see 31).
In light of these laboratory and clinical studies on PAI-1 and tumor progression, we investigated cannabidiol's action on the expression and release of PAI-1 and its possible impact on the invasiveness of human lung cancer cells.
MATERIALS AND METHODS
Materials
Cannabidiol was purchased from Tocris (Bad Soden, Germany) for in vitro experiments and from Biotrend Chemikalien (Cologne, Germany) for in vivo applications. AM-251, AM-630 and capsazepine were obtained from Alexis Deutschland GmbH, Grünberg, Germany. Dulbecco's Modified Eagle's medium (DMEM) with 4 mM L-glutamine and 4.5 g/L glucose and Roswell Park Memorial Institute medium (RPMI) was from Cambrex Bio Science Verviers S.p.r.l. (Verviers, Belgium). Fetal calf serum (FCS) and penicillin-streptomycin were obtained from PAN Biotech (Aidenbach, Germany) and Invitrogen (Karlsruhe, Germany), respectively. Recombinant PAI-1 was purchased from Calbiochem (Darmstadt, Germany).
Cell Culture
A549 cells were maintained in DMEM with 4 mM L-glutamine and 4.5 g/L glucose supplemented with 10% heat-inactivated FCS, 100 U/ml penicillin and 100 μg/ml streptomycin. H460 and H358 cells were maintained in RPMI supplemented with 10% heat-inactivated FCS, 100 U/ml penicillin and 100 μg/ml streptomycin.
The cells were grown in a humidified incubator at 37°C and 5% CO2. All incubations were performed in serum-free medium. Phosphate-buffered saline was used as a vehicle for the tested substances with a final concentration of 0.1% (v/v) ethanol (for cannabidiol) or 0.1% (v/v) DMSO (for AM-251, AM-630 and capsazepine). Recombinant PAI-1 was dissolved in phosphate-buffered saline only.
Matrigel Invasion and Migration Assays
The invasiveness of cells was quantified using a modified Boyden chamber technique with Matrigel-coated membranes according to the manufacturer's instructions (BD Biosciences, Oxford, UK) as described recently (14,32). In this assay, cells must overcome a reconstituted basement membrane by proteolytic degradation of a Matrigel layer and active migration. In brief, the upper sides of the transwell inserts (8-μm pore size) were coated with 28.4 μg Matrigel per insert in a 24-well plate format. Cells were used at a final concentration of 5 × 105 cells per well in a volume of 500 μl serum-free medium in each insert and treated with test substances or vehicles for the indicated times. Medium containing 10% FCS was used as a chemo-attractant in the companion plate. Following incubation in a humidified incubator at 37°C and 5% CO2 for the indicated times, the non-invading cells on the upper surface of the inserts were removed with a cotton swab, and viability of invaded cells on the lower surface was measured by the colorimetric WST-1 test (Roche Diagnostics, Mannheim, Germany). For calculation of migration, the viability of cells on the lower side of uncoated invasion chambers was determined by WST-1 test. Invasion was expressed as the invasion index, which is calculated as the absorbance at 490 nm of cells that invaded through Matrigel-coated Boyden chambers divided by absorbance of cells that migrated through uncoated control inserts with equal treatment ([invasion/migration] ×100%).
Cellular Viability
To exclude the possibility that the effect of cannabidiol on invasion was an unspecific cytotoxicity-related phenomenon, cell viability was analyzed after cannabidiol exposure in quadruplicate. For this purpose, cells were seeded into 48-well plates at 5 × 105 cells per well to match conditions of invasion assays in a volume of 500 μl medium per well and treated with cannabidiol or ethanol vehicle for 72 h. Viability was measured subsequently using the WST-1 test.
MRNA Analysis
For RNA analysis, cells were seeded into 24-well plates at a density of 2 × 105 cells per well. Following an incubation for the indicated times, total RNA was isolated using the RNeasy total RNA Kit (Qiagen, Hilden, Germany). β-Actin- (internal standard), PAI-1, uPA and uPAR mRNA levels were determined by quantitative real-time RT-PCR using the TaqMan® RNA-to-CT™ 1-Step Kit from Applied Biosystems according to the manufacturer's instruction. Primers and probes for human β-Actin, PAI-1, uPA and uPAR were Gene Expression Assays™ (Applied Biosystems, Darmstadt, Germany). Percent control represents comparison with vehicle-treated cells (100%) in the absence of test substance.
Western Blot Analysis
Determination of PAI-1 in cell culture media was performed as described recently (14,32) using a specific antibody raised to PAI-1 (Oncogene Research Products, San Diego, CA) and a horseradish peroxidase-conjugated anti-mouse IgG as secondary antibody (New England BioLabs GmbH, Frankfurt, Germany). For determination of uPA in media, a specific antibody raised to uPA (American Diagnostica, Pfungstadt, Germany) and a horseradish peroxidase-conjugated anti-rabbit IgG as secondary antibody (New England BioLabs GmbH, Frankfurt, Germany) were used. PAI-1 and uPA detection revealed a band at 48 kDa and 52 kDa, respectively. To match identical experimental conditions of PAI-1/uPA protein analysis with mRNA quantification and invasion assays, cell culture media were obtained from 24-well plates (Fig. 2a) or upper Boyden chambers for Matrigel invasion assays (all other PAI-1 blots except that in Fig. 2c). A band stained with Ponceau Red or Coomassi Blue corresponding to a protein of about 65 kDa whose expression appeared unregulated was used as the loading control (LC) for all analyses of cell culture media.

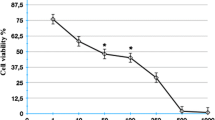
Influence of cannabidiol (CBD) on invasion of A549 cells. Concentration-dependent effect of cannabidiol on invasion, migration and cellular viability following a 72-hour incubation (A). Time-dependent effect of cannabidiol on invasion (B). Percent control represents comparison with vehicle-treated cells (100%) in the absence of test substance. Values are means ± SEM of n = 3–4 (A), n = 4–8 (B) experiments; *P < 0.05; **P < 0.01, vs. corresponding vehicle control (Student's t-test).

Impact of cannabidiol (CBD) on PAI-1, uPA and uPAR levels of A549 cells. Time-dependent effect of cannabidiol on PAI-1 release and PAI-1 mRNA expression (A). Time-dependent effect of cannabidiol on uPA release and mRNA expression and on uPAR protein and mRNA expression (B). The pictures on the right side show representative Western blots. Concentration-dependent effect of cannabidiol on PAI-1 and uPA protein levels in cell culture media and on uPAR in cell lysates following a 72-hour-incubation period (C). Percent control represents comparison with vehicle-treated cells (100%) in the absence of test substance. Values are means ± SEM of n = 4–12 (A, mRNA), n = 6 (A, protein), n = 5–7 (B, uPA), n = 4 (B, uPAR), n = 3–4 (C) experiments. *P < 0.05 vs. corresponding vehicle control (Student's t-test). Protein staining of Western blot membranes is shown as loading control (LC) when cell culture media were analysed.
For analysis of ß-actin and uPAR from whole cell lysates, cells were lysed in solubilization buffer (50 mM HEPES pH 7.4, 150 mM NaCl, 1 mM EDTA, 1% (v/v) Triton® X-100, 10% (v/v) glycerol, 1 mM phenylmethylsulfonyl fluoride, 1 μg/ml leupeptin, and 10 μg/ml aprotinin), homogenized by sonication, and centrifuged at 10,000 × g for 5 min. Specific antibodies raised to ß-actin and uPAR were purchased from Calbiochem (Bad Soden, Germany) and American Diagnostica (Pfungstadt, Germany), respectively. ß-Actin and uPAR detection revealed a band at 42 kDa and 60 kDa, respectively.
For Western blotting of PAI-1, uPA and uPAR in tumor samples, tissues were homogenized in liquid nitrogen, resuspended in solubilization buffer, and centrifuged at 10,000 × g for 5 min.
For all blots shown, antibody binding was visualized by enhanced chemiluminescence Western blotting detection reagents (Amersham Biosciences, Freiburg, Germany). Densitometric analysis of PAI-1, uPA and uPAR band intensities was achieved by using the ChemiDoc XRS imaging system and quantified using the Quantity One 1-D Analysis Software (Biorad, Muenchen, Germany).
SiRNA Transfections
A549 cells were transfected with siRNA targeting PAI-1 using RNAiFect® as the transfection reagent (Qiagen GmbH, Hilden, Germany) or negative control siRNA (Eurogentec, Seraing, Belgium; Cat. No. OR-0030-neg). The target sequence of PAI-1 siRNA (Qiagen GmbH, Hilden, Germany) was as follows: 5′–ttg gag gac ctt tag gtc aaa–3′. A BLAST search revealed that the sequence selected did not show any homology to other known human genes. Transfections were performed according to the manufacturer's instructions. For invasion assays, cells grown to confluence were transfected with 1 or 2.5 μg/mL siRNA or non-silencing siRNA as negative control with an equal ratio (w/v) of RNA to transfection reagent for 24 h in DMEM supplemented with 10% FCS. Subsequently, cells were trypsinized, centrifuged at 200 ×g, resuspended to a final density of 5 × 105 cells in 500 μl of serum-free DMEM containing the same amounts of siRNA or non-silencing siRNA to provide constant transfection conditions, and seeded for invasion analysis as described above. Cells were then incubated for an additional 72 h before determination of invasiveness and collection of cell culture media from the upper Boyden chambers for analysis of PAI-1 levels.
Induction of A549 Xenografts in Nude Mice
Tumors were induced in NMRI (nu/nu) mice (Charles River, Margate, Kent, UK) by subcutaneous flank inoculation of 1 × 107 A549 lung tumor cells. Animals were assigned randomly to a vehicle and a cannabidiol group and were injected intraperitoneally 3 times per week with vehicle or cannabidiol (5 mg/kg). The treatment started 24 h after subcutaneous induction of tumors into the dorsal right side. On day 41, tumors were measured with an external caliper, and the volume was calculated as (4π/3) × (width/2)2 × (length/2). After 42 days, animals were sacrificed, and tumors were explanted for protein analysis. Therefore, tissue parts were quick-frozen in liquid nitrogen and proceeded as described under “Western Blot Analysis.”
Statistics
Statistical analyses were undertaken using GraphPad Prism 4.00 (GraphPad Software, San Diego, CA). Student's t-test was used to evaluate statistical differences between the groups. Results were considered to be statistically significant at P < 0.05.
RESULTS
Effect of Cannabidiol on Invasion, Migration and Viability of A549 Cells
In A549 cells (human lung carcinoma) cannabidiol caused a concentration-dependent decrease of invasion that was even significant at concentrations as low as 0.1 μM (Fig. 1a). Time-course experiments further showed that the diminished invasion occurred within the first 48 h of incubation with 0.1 and 1 μM cannabidiol and continued to decrease over the investigated 72-hour incubation period (Fig. 1b).
Experiments addressing a possible impact of cannabidiol on cellular viability demonstrated the anti-invasive effect of cannabidiol as independent of drug toxicity (Fig. 1a). Furthermore, cellular motility through uncoated Boyden chambers was virtually unaltered in the presence of the test substance (Fig. 1a).
Effect of Cannabidiol on PAI-1 Expression and Secretion by A549 Cells
To investigate a possible impact of altered PAI-1 expression to the anti-invasive action of cannabidiol, cell culture media and cell lysates of A549 cells were analyzed for changes in PAI-1, uPA and uPAR protein and mRNA levels, respectively. According to Fig. 2a, treatment of cells with cannabidiol at 1 μM was associated with a decreased release of PAI-1 protein into the cell culture media at 24 h and 48 h post-administration. Real-time RT-PCR analyses of PAI-1, uPA and uPAR mRNA expression over a 48-hour incubation period revealed a transient short-lived down-regulation of PAI-1 mRNA expression after a 2-hour incubation with 1 μM cannabidiol (Fig 2a, left), whereas uPA and uPAR mRNA levels remained virtually unaltered (Fig 2b, left). PAI-1 protein secretion was reduced by 33% and 41% subsequent to a 72-hour treatment with cannabidiol at 0.1 and 1 μM, respectively (Fig. 2c). Western blot analyses of cell culture media did not show a significant modulation of uPA secretion by cannabidiol-treated cells at the indicated times (Fig. 2b, right) and concentrations (Fig. 2c). Similar results were obtained from protein analysis of uPAR levels in cell lysates (Fig. 2b, right, and c).
Involvement of Cannabinoid Receptors and TRPV1 in the Anti-invasive and PAI-1-Lowering Action of Cannabidiol
To investigate the role of cannabinoid receptors and transient receptor potential vanilloid 1 (TRPV1) in cannabidiol-mediated reduction of A549 cell invasiveness, the impact of antagonists to CB1 receptor (AM-251), CB2 receptor (AM-630) and TRPV1 (capsazepine) was tested. Inhibitors were used at a concentration of 1 μM, which has been reported to be within the range of concentrations inhibiting CB1-, CB2- and TRPV1-dependent events (2,33). As shown in Fig. 3a, cannabidiol-induced inhibition of cell invasion was significantly suppressed by a 1-hour preincubation with AM-251, AM-630 or the combination of both antagonists. Likewise, the TRPV1 antagonist capsazepine diminished the anti-invasive effect of cannabidiol (Fig. 3a). Noteworthy, incubation of cells with the receptor antagonists alone did not result in significant changes of cell invasion (data not shown). Experiments using cannabinoid receptor and TRPV1 antagonists were also performed to determine the receptor targets of cannabidiol involved in impaired PAI-1 release. According to Western blot analyses from cell culture media obtained from the upper Boyden chambers of the respective invasion experiments, cannabidiol-mediated decrease of PAI-1 secretion versus vehicle (72.9% ± 21.4%, mean difference ± SEM) was prevented by antagonists to either CB1 or CB2 receptors as well as to TRPV1, whereas the secretion of uPA into cell culture media as well as the expression of uPAR measured in cellular lysates remained virtually unaltered (Fig. 3b).

Effect of antagonists to CB1, CB2 and TRPV1 on cannabidiol (CBD)-mediated inhibition of invasion, modulation of PAI-1 and uPA in cell culture media and expression of uPAR in cell lysates by A549 cells. Effect of AM-251 (CB1 antagonist; 1 μM), AM-630 (CB2 antagonist; 1 μM) and capsazepine (Capsa; TRPV1 antagonist; 1 μM) on the anti-invasive (A) and PAI-1-lowering (B) action of cannabidiol. Cells were pretreated with the indicated antagonists for 1 h and incubated with cannabidiol (1 μM) or vehicle for a further 72 h. Analyses of PAI-1 and uPA protein levels were performed in cell culture media collected from the upper Boyden chambers. Western blots of uPAR were performed using cell lysates. Values are means ± SEM of n = 8 (A) or n = 3–4 (B) experiments. Panel B shows representative blots with densitometric data, respectively. ***P < 0.01, vs. corresponding vehicle control (Student's t-test). ##P < 0.01; ###P < 0.001, vs. cannabidiol (Student's t-test). Protein staining of Western blot membranes is shown as loading control (LC) when cell culture media were analysed.
Effect of PAI-1 Knockdown on the Invasiveness of A549 Cells
To prove an essential role of PAI-1 in mediating a proinvasive action on A549 cells, experiments using PAI-1 siRNA were performed. As shown in Fig. 4a, knockdown of PAI-1 by siRNA led to a significant decrease of A549 cell invasion, whereas cultures treated with a non-silencing sequence exhibited no significant alteration in invasion as compared to controls treated with transfection agent only. Monitoring of PAI-1 levels in culture media of the upper Boyden chambers confirmed a substantial inhibition of PAI-1 release from A549 cells transfected with PAI-1 siRNA, whereas the non-silencing control was inactive in this respect (Fig. 4b). The specificity of PAI-1 silencing was further substantiated by hybridisation of the membranes used for PAI-1 analysis with an uPA antibody and analyses of uPAR in lysates of cells treated with PAI-1 siRNA. As a result of these approaches, a modulation of both parameters by PAI-1 silencing was excluded (Fig. 4b).

Impact of PAI-1 knockdown on invasiveness of A549 cells. Effect of PAI-1 siRNA and non-silencing siRNA (both at 1 and 2.5 μg/ml) on the invasiveness of (A) or PAI-1, uPA and uPAR production (B) by A549 cells following a 72-hour incubation with the substances at the indicated concentrations. Analyses of PAI-1 and uPA protein levels were performed in cell culture media collected from the upper Boyden chambers. Western blots of uPAR were performed using cell lysates. Invasion data are means ± SEM of n = 4–8 experiments (A), **P < 0.01, vs. corresponding vehicle control (Student's t-test); values above the blots (B) are means ± SEM obtained from densitometric analysis of n = 4 blots and represent percent control in comparison with vehicle-treated cells (100%) in the absence of test substance. Protein staining of Western blot membranes is shown as loading control (LC) when cell culture media were analysed.
Effect of Recombinant PAI-1 on the Invasiveness of A549 Cells and on the Anti-invasive Action of Cannabidiol
To further confirm a pivotal role of PAI-1 in modulating A549 cell invasion, we next investigated the impact of recombinant human PAI-1 on A549 cell invasiveness. According to Fig. 5a, Matrigel invasion was up-regulated by recombinant PAI-1 in a concentration-dependent manner.

Impact of exogenous PAI-1 on invasiveness of A549 cells. Effect of recombinant human PAI-1 on the invasiveness of A549 cells (A) and on cannabidiol's (CBD) anti-invasive action (B). Data are means ± SEM of n = 7–8 (A) or n = 3–4 (B) experiments. *P < 0.05, **P < 0.01, vs. corresponding vehicle control (Student's t-test); #P < 0.05, vs. cannabidiol (Student's t-test).
In further experiments addressing a possible causal link between cannabidiol's effects on PAI-1 secretion and invasion, the influence of recombinant PAI-1 on the anti-invasive action of cannabidiol was assessed using PAI-1 at concentrations that did not significantly alter the baseline invasiveness of A549 cells. Fig. 5b illustrates that treatment of A549 cells with PAI-1, at concentrations that did not significantly change basal invasiveness, reversed the anti-invasive effect of cannabidiol.
Role of PAI-1 in the Anti-invasive Action of Cannabidiol in Other Human Lung Cancer Cell Lines
To exclude that the observed effects are restricted to A549 cells, we corroborated key findings using two other human lung cancer cell lines: H460 (a large-cell lung cancer cell line derived from carcinoma) and H358 (a non-small cell lung cancer cell line derived from a bronchioalveolar carcinoma). Again, cannabidiol was shown to elicit both inhibition of invasion (Fig. 6a and 7a) and decrease of PAI-1 secretion versus vehicle (60.2% ± 37.5%, mean difference ± SEM for H460, and 67.5% ± 21.7%, mean difference ± SEM for H358, [Fig. 6b and 7b]) with both events being significantly suppressed by antagonists to cannabinoid receptors and TRPV1. As shown for A549 cells, uPA and uPAR were not regulated by cannabidiol or receptor antagonists (Fig. 6b and 7b). Likewise, a 72-hour treatment with cannabidiol at 1 μM elicited no significant change in cellular motility (H460: 82% ± 14%, n = 4, vs. vehicle [100% ± 9%]; H358: 88% ± 7%, n = 4, vs. vehicle [100% ± 13%]) and viability (H460: 112% ± 6%, n = 4, vs. vehicle [100% ± 16%]; H358: 119% ± 11%, n = 4, vs. vehicle [100% ± 3%]) as compared to its pronounced anti-invasive action. Furthermore, treatment of both cell lines with recombinant PAI-1 was associated with increased cellular invasiveness (Fig. 6c and 7c). The threshold concentrations causing significant increases of invasion were 10 ng/ml (H460) and 1 ng/ml (H358), respectively. Finally, the decrease of invasion by cannabidiol was inhibited by cotreatment with recombinant PAI-1 at non-proinvasive concentrations (Fig. 6d and 7d), suggesting that also in these cells restoration of cannabidiol-attenuated PAI-1 levels in cell culture media may reverse the anti-invasive properties of this cannabinoid.

Effect of AM-251 (CB1 antagonist; 1 μM), AM-630 (CB2 antagonist; 1 μM) and capsazepine (Capsa; TRPV1 antagonist; 1 μM) on the anti-invasive (A) and PAI-1-lowering action (B) of cannabidiol (CBD) in H460 cells. Effect of recombinant human PAI-1 on the invasiveness of H460 cells (C) and on cannabidiol's anti-invasive action (D) following a 72-hour incubation with the substances at the indicated concentrations. (A,B) Cells were pretreated with the indicated antagonists for 1 h and incubated with cannabidiol (1 μM) or vehicle for a further 72 h. Analyses of PAI-1 and uPA protein levels were performed in cell culture media collected from the upper Boyden chambers. Analysis of uPAR was performed using cellular lysates. Invasion data are means ± SEM of n = 4 experiments. **P < 0.01, vs. corresponding vehicle control (Student's t-test); #P < 0.05; ##P <0.01, vs. cannabidiol (Student's t-test). Values above the blot (B) are means ± SEM obtained from densitometric analysis of n = 3 (uPA, uPAR) and n = 4 blots (PAI-1) and represent percent control in comparison with vehicle-treated cells (100%) in the absence of test substance. (C,D) Cells were incubated with cannabidiol (1 μM) and/or recombinant PAI-1 for 72 h. Invasion data are means ± SEM of n = 4 experiments. *P < 0.05, **P < 0.01, vs. corresponding vehicle control (Student's t-test); ###P < 0.001, vs. cannabidiol (Student's t-test). Protein staining of Western blot membranes is shown as loading control (LC) when cell culture media were analysed.

Effect of AM-251 (CB1 antagonist; 1 μM), AM-630 (CB2 antagonist; 1 μM) and capsazepine (Capsa; TRPV1 antagonist; 1 μM) on the anti-invasive (A) and PAI-1-lowering action (B) of cannabidiol (CBD) in H358 cells. Effect of recombinant human PAI-1 on the invasiveness of H358 cells (C) and on cannabidiol's anti-invasive action (D) following a 72-hour incubation with the substances at the indicated concentrations. (A,B) Cells were pretreated with the indicated antagonists for 1 h and incubated with cannabidiol (1 μM) or vehicle for a further 72 h. Analyses of PAI-1 and uPA protein levels were performed in cell culture media collected from the upper Boyden chambers. Analysis of uPAR was performed using cellular lysates. Invasion data are means ± SEM of n = 4 experiments. ***P < 0.001, vs. corresponding vehicle control (Student's t-test); ###P < 0.001, vs. cannabidiol (Student's t-test). Values above the blot (B) are means ± SEM obtained from densitometric analysis of n = 3 (uPA, uPAR) and n = 4 blots (PAI-1) and represent percent control in comparison with vehicle-treated cells (100%) in the absence of test substance. (C,D) Cells were incubated with cannabidiol (1 μM) and/or recombinant PAI-1 for 72 h. Invasion data are means ± SEM of n = 4 experiments. **P < 0.01, ***P < 0.001, vs. corresponding vehicle control (Student's t-test); #P < 0.05; ##P < 0.01, vs. cannabidiol (Student's t-test). Protein staining of Western blot membranes is shown as loading control (LC) when cell culture media were analysed.
Impact of Cannabidiol on PAI-1 Levels in A549 Xenografts of Nude Mice
To verifiy the cannabidiol-mediated reduction of PAI-1 levels in vivo, nude mice were xenografted with A549 cells, randomised into two groups and treated intraperitoneally with vehicle or 5 mg/kg cannabidiol 3 times per week. Mice treated with cannabidiol exhibited a significant reduction of tumor size as compared to vehicle-treated animals at day 41 post-induction of xenografts (Fig. 8a). Monitoring of tissue protein on day 42 revealed significantly reduced PAI-1 levels in tumors obtained from mice treated with cannabidiol as compared to vehicle-treated animals, whereas uPA and uPAR were left virtually unaltered (Fig. 8c).

In vivo action of cannabidiol (CBD) on PAI-1 levels in tumors from athymic nude mice xenografted with A549 cells. Tumors were generated by flank inoculation of A549 cells in nude mice. Animals were treated with either vehicle or cannabidiol 3 times per week (5 mg/kg) for up to 42 days. (A,B). Tumor size at day 41 was measured with an external caliper and calculated as described in the Materials and Methods section. Tumor volumes are means ± SEM of n = 4 animals per group. **P < 0.01, vs. corresponding vehicle control (Student's t-test). Photographs were taken from representative tumors. (C) Protein samples for Western blot analysis were obtained from animals after a 42-day treatment with vehicle or cannabidiol. Quantification of PAI-1, uPA and uPAR protein levels (C, left panel) was achieved by videodensitometric analysis. Each Western blot (C) shows one representative of 3–4 experiments. Data represent means ± SEM of n = 4 (PAI-1, uPAR) or n = 3 (uPA) different tumors per group. **P < 0.01, vs. corresponding vehicle control (Student's t-test).
DISCUSSION
Cannabinoids are currently used for the treatment of pain and to palliate wasting in AIDS and cancer patients. Only recently, cannabinoids have received attention with respect to their anti-tumorigenic effects. In this context, the present study demonstrates a pivotal role of PAI-1 in the anti-invasive action of the non-psychoactive cannabinoid compound cannabidiol.
Using Matrigel-coated Boyden chambers, cannabidiol was found to decrease invasiveness of human lung cancer cells (A549) in a concentration- and time-dependent manner, which is in line with recent observations on human cervical cancer cells (15). In the present study, cancer cell invasion was inhibited by 29% and 63% following a 72-hour incubation with 0.01 μM or 0.1 μM cannabidiol, respectively. In comparison, plasma peak levels of approximately 0.01 μM and up to 0.05 μM cannabidiol were measured in humans after administration of 10 mg buccal sativex® (1:1 ratio of THC and cannabidiol approved for the pharmacotherapy of multiple sclerosis-related spasticity and pain in Canada) and self-titrated doses during chronic therapy, respectively (34). In another study, mean cannabidiol plasma levels of 0.036 μM were analyzed following a 6-week oral treatment with cannabidiol at doses of 10 mg/kg/day (35). Thus, effects of cannabidiol on cell invasion occurred in a range of therapeutically relevant concentrations. Experiments addressing a possible impact of cannabidiol on cellular viability demonstrated the anti-invasive effect of cannabidiol as independent of drug toxicity. Furthermore, cellular motility through uncoated Boyden chambers was virtually unaltered in the presence of the test substance, suggesting that a cannabidiol-induced upstream degradation of matrix scaffold confers transmigration through matrigel.
The involvement of cannabinoid receptors in the anti-invasive effect of cannabidiol was demonstrated by use of the CB1 antagonist AM-251 and the CB2 antagonist AM-630 confirming the results of a recent study from our laboratory that used higher concentrations of cannabidiol to demonstrate a receptor-dependent anti-invasive action of cannabidiol in diverse human cancer cells (15). Due to the low affinity of cannabidiol to either CB1 and CB2 receptors (36), this finding was rather surprising, and the pathways involved here still have to be elucidated. However, in line with our data, several effects of cannabidiol, such as modulation of cytokine release and macrophage chemotaxis (37) as well as antiproliferative (20) and proapoptotic properties (21), have been shown to be mediated via cannabinoid receptors. A possible explanation for the involvement of cannabinoid receptors in cannabidiol's action may lie in an inhibitory effect of this compound on the enzyme fatty acid amidohydrolase (18,38,39). As inhibition of this enzyme reduces both cellular uptake and degradation of anandamide, the observed effects may be a result of a prolonged effect of this endocannabinoid on cannabinoid receptors.
Additionally, the non-selective cation channel TRPV1 seems to be involved in cannabidiol's anti-invasive effect, which is in line with recent findings demonstrating antihyperalgesic (40) and antiproliferative effects (18) of cannabidiol to be mediated via TRPV1.
In the present study, several findings suggest a crucial role of PAI-1 in the anti-invasive action of cannabidiol. First, inhibition of invasion by cannabidiol was accompanied by a decrease of PAI-1 expression and release. With respect to other investigated components of the plasminogen/plasmin system, this effect was specific in that the expression of uPA and uPAR was not regulated by cannabidiol. Second, besides cannabidiol's anti-invasive effect, the associated attenuation of PAI-1 formation was reversed by antagonists to CB1 and CB2 receptors, as well as to TRPV1. Third, PAI-1 was proven to be functionally involved in invasiveness by findings demonstrating a proinvasive action of exogenously added recombinant PAI-1 and, conversely, an anti-invasive impact of PAI-1 knockdown with the respective siRNA. Most convincingly, however, the observed decrease of invasion by cannabidiol was inhibited by cotreatment with recombinant PAI-1 at non-proinvasive concentrations, suggesting that restoration of cannabidiol-attenuated PAI-1 levels in cell culture media may reverse the anti-invasive properties of cannabidiol. Finally, a PAI-1-dependent anti-invasive action was confirmed in two other human lung cancer cell lines, implying the observed signalling as a more general anti-invasive mechanism of cannabinoids.
There are some issues that merit special reference. First, the observed inhibition of PAI-1 mRNA expression was transient with a short-lived suppression of mRNA occurring 2 h post-administration of cannabidiol but not at later time points when a sustained decrease of PAI-1 protein levels was observed. Therefore, we cannot definitely exclude additional post-transcriptional events such as a decrease of PAI-1 protein by activation of proteasomal degradation contributing to the diminished PAI-1 secretion by cannabidiol. In fact, a posttranscriptional regulation of PAI-1 has been shown for several substances. Accordingly, glucocorticoids (41), testosterone (42), thrombin (43), and rosiglitazone (44) were demonstrated to confer alterations in PAI-1 secretion without causing concomitant changes in PAI-1 mRNA expression. Second, the finding that transfection of cells with PAI-1 siRNA at a concentration of 1.0 μg/ml elicited a profound inhibition of invasion but only a modest reduction of PAI-1 deserves further investigation. However, use of a 2.5-fold higher concentration of PAI-1 siRNA led to a further decrease of both PAI-1 levels and cellular invasiveness. This data together with the fact that a non-silencing control tested under the same experimental conditions did not confer a comparable suppression of both PAI-1 and invasion strongly supports a functional role of PAI-1 in basal A549 invasiveness.
To confirm down-regulation of PAI-1 by cannabidiol under in vivo conditions, additional experiments using thymic-aplastic nude mice were performed. In a recent study from our laboratory, a significant inhibition of A549 lung metastasis by cannabidiol was demonstrated in these animals (15). As this metastasation model is, however, not suitable for analysis of protein release from tumor cells before growing of metastasis into lung tissue, we chose a solid tumor xenograft model for our purposes. Here we found that the PAI-1 protein content in xenografts was significantly suppressed in cannabidiol-treated mice as compared to vehicle-treated animals. In agreement with the in vitro data, cannabidiol did not exert any effect on the levels of uPA and uPAR. The profound suppression of tumor volume by cannabidiol in this model is in line with a previous report using nude mice xenografted with human breast carcinoma and rat v-K-ras-transformed thyroid cells (18).
Collectively, the findings presented in this study are in line with studies reporting a positive influence of PAI-1 on tumor invasion, angiogenesis and metastasis (25,28–30), suggesting a pro- rather than an anti-tumorigenic action of PAI-1. A possible explanation for the tumor-promoting effect of PAI-1 may lie in its ability to orchestrate cell adhesion, migration and tumor neovascularisation by interference with extracellular matrix components in an uPA-independent manner (28,45). Accordingly, PAI-1 dissociates the vitronectin-integrin αVß3 and vitronectin-uPAR binding, thereby promoting tumor attachment and invasive properties of tumor cells (41,46,47). Clearly, more research is needed to understand the complete mode of action underlying the anti-invasive effect of cannabidiol. Thus, besides PAI-1, modulation of other proteolytic components, such as members of the MMP family, contribute to the anti-invasive action as well. A recent study from our laboratory demonstrated an involvement of TIMP-1 induction in the anti-invasive properties of cannabidiol accordingly (15). Future studies have to address a possible connection between both parameters.
In conclusion, our results suggest that cannabidiol is capable of inhibiting PAI-1 release, which is responsible, at least in part, for the anti-invasive action of this cannabinoid. To our knowledge, this is the first report of a PAI-1-dependent anti-invasive cannabinoid effect. Inhibition of PAI-1 may play an important role in the anti-metastatic action of cannabidiol, whose potential therapeutic benefit in the treatment of highly invasive cancers should be addressed in clinical trials.
Abbreviations
- AM-251:
-
N-(Piperidin-1-yl)-5-(4-iodophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole-3-carboxamide
- AM-630:
-
(6-Iodo-2-methyl-1-[2-(4-morpholinyl)ethyl]-1H-indol-3-yl) (4-methoxyphenyl)methanone
- CB1 :
-
cannabinoid receptor 1
- CB2 :
-
cannabinoid receptor 2
- RT-PCR:
-
reverse transcriptase-polymerase chain reaction
- siRNA:
-
small-interfering RNA
- TRPV1:
-
transient receptor potential vanilloid 1
- WST-1:
-
4-[3-(4-Iodophenyl)-2-(4-nitrophenyl)-2H-5-tetrazolio]-1.6-benzene disulfonate
REFERENCES
Munson AE, Harris LS, Friedman MA, Dewey WL, Carchman RA. Antineoplastic activity of cannabinoids. J Natl Cancer Inst. 1975;55:597–602.
Jacobsson SO, Wallin T, Fowler CJ. Inhibition of rat C6 glioma cell proliferation by endogenous and synthetic cannabinoids. Relative involvement of cannabinoid and vanilloid receptors. J Pharmacol Exp Ther. 2001;299:951–9.
Blázquez C, Carracedo A, Barrado L, Real PJ, Fernández-Luna JL, Velasco G, et al. Cannabinoid receptors as novel targets for the treatment of melanoma. FASEB J. 2006;20:2633–5.
Galve-Roperh I, Sánchez C, Cortés ML, Gómez del Pulgar T, Izquierdo M, Guzmán M. Anti-tumoral action of cannabinoids: involvement of sustained ceramide accumulation and extracellular signal-regulated kinase activation. Nat Med. 2000;6:313–9.
Hinz B, Ramer R, Eichele K, Weinzierl U, Brune K. Up-regulation of cyclooxygenase-2 expression is involved in R(+)-methanandamide-induced apoptotic death of human neuroglioma cells. Mol Pharmacol. 2004;66:1643–51.
Portella G, Laezza C, Laccetti P, De Petrocellis L, Di Marzo V, Bifulco M. Inhibitory effects of cannabinoid CB1 receptor stimulation on tumor growth and metastatic spreading: actions on signals involved in angiogenesis and metastasis. FASEB J. 2003;17:1771–3.
Blázquez C, Casanova ML, Planas A, Gómez Del Pulgar T, Villanueva C, Fernández-Aceñero MJ, et al. Inhibition of tumor angiogenesis by cannabinoids. FASEB J. 2003;17:529–31.
Blázquez C, González-Feria L, Alvarez L, Haro A, Casanova ML, Guzmán M. Cannabinoids inhibit the vascular endothelial growth factor pathway in gliomas. Cancer Res. 2004;64:5617–23.
Kishimoto S, Muramatsu M, Gokoh M, Oka S, Waku K, Sugiura T. Endogenous cannabinoid receptor ligand induces the migration of human natural killer cells. J Biochem. 2005;137:217–23.
Wang D, Wang H, Ning W, Backlund MG, Dey SK, DuBois RN. Loss of cannabinoid receptor 1 accelerates intestinal tumor growth. Cancer Res. 2008;68:6468–76.
Freimuth N, Ramer R, Hinz B. Antitumorigenic effects of cannabinoids beyond apoptosis. J Pharmacol Exp Ther. 2010;332:336–44.
Grimaldi C, Pisanti S, Laezza C, Malfitano AM, Santoro A, Vitale M, et al. Anandamide inhibits adhesion and migration of breast cancer cells. Exp Cell Res. 2006;312:363–73.
Preet A, Ganju RK, Groopman JE. Delta9-Tetrahydrocannabinol inhibits epithelial growth factor-induced lung cancer cell migration in vitro as well as its growth and metastasis in vivo. Oncogene. 2008;27:339–46.
Ramer R, Hinz B. Inhibition of cancer cell invasion by cannabinoids via increased expression of tissue inhibitor of matrix metalloproteinases-1. J Natl Cancer Inst. 2008;100:59–69.
Ramer R, Merkord J, Rohde H, Hinz B. Cannabidiol inhibits cancer cell invasion via upregulation of tissue inhibitor of matrix metalloproteinases-1. Biochem Pharmacol. 2010;79:955–66.
Blázquez C, Salazar M, Carracedo A, Lorente M, Egia A, González-Feria L, et al. Cannabinoids inhibit glioma cell invasion by down-regulating matrix metalloproteinase-2 expression. Cancer Res. 2008;68:1945–52.
Rog DJ, Nurmikko TJ, Young CA. Oromucosal Δ9- tetrahydrocannabinol/cannabidiol for neuropathic pain associated with multiple sclerosis: an uncontrolled, open-label, 2-year extension trial. Clin Ther. 2007;29:2068–79.
Ligresti A, Moriello AS, Starowicz K, Matias I, Pisanti S, De Petrocellis L, et al. Antitumor activity of plant cannabinoids with emphasis on the effect of cannabidiol on human breast carcinoma. J Pharmacol Exp Ther. 2006;318:1375–87.
McAllister SD, Christian RT, Horowitz MP, Garcia A, Desprez PY. Cannabidiol as a novel inhibitor of Id-1 gene expression in aggressive breast cancer cells. Mol Cancer Ther. 2007;6:2921–7.
Vaccani A, Massi P, Colombo A, Rubino T, Parolaro D. Cannabidiol inhibits human glioma cell migration through a cannabinoid receptor-independent mechanism. Br J Pharmacol. 2005;144:1032–6.
McKallip RJ, Jia W, Schlomer J, Warren JW, Nagarkatti PS, Nagarkatti M. Cannabidiol-induced apoptosis in human leukemia cells: a novel role of cannabidiol in the regulation of p22phox and Nox4 expression. Mol Pharmacol. 2006;70:897–908.
Massi P, Valenti M, Vaccani A, Gasperi V, Perletti G, Marras E, et al. 5-Lipoxygenase and anandamide hydrolase (FAAH) mediate the antitumor activity of cannabidiol, a non-psychoactive cannabinoid. J Neurochem. 2008;104:1091–100.
Kogan NM, Blázquez C, Alvarez L, Gallily R, Schlesinger M, Guzmán M, et al. Cannabinoid quinone inhibits angiogenesis by targeting vascular endothelial cells. Mol Pharmacol. 2006;70:51–9.
Andreasen PA, Egelund R, Petersen HH. The plasminogen activation system in tumor growth, invasion, and metastasis. Cell Mol Life Sci. 2000;57:25–40.
Soff GA, Sanderowitz J, Gately S, Verrusio E, Weiss I, Brem S, et al. Expression of plasminogen activator inhibitor type 1 by human prostate carcinoma cells inhibits primary tumor growth, tumor-associated angiogenesis, and metastasis to lung and liver in an athymic mouse model. J Clin Invest. 1995;96:2593–600.
Roca C, Primo L, Valdembri D, Cividalli A, Declerck P, Carmeliet P, et al. Hyperthermia inhibits angiogenesis by a plasminogen activator inhibitor 1-dependent mechanism. Cancer Res. 2003;63:1500–7.
Chen SC, Henry DO, Hicks DG, Reczek PR, Wong MK. Intravesical administration of plasminogen activator inhibitor type-1 inhibits in vivo bladder tumor invasion and progression. J Urol. 2009;181:336–42.
Bajou K, Noël A, Gerard RD, Masson V, Brunner N, Holst-Hansen C, et al. Absence of host plasminogen activator inhibitor 1 prevents cancer invasion and vascularization. Nat Med. 1998;4:923–8.
Bajou K, Maillard C, Jost M, Lijnen RH, Gils A, Declerck P, et al. Host-derived plasminogen activator inhibitor-1 (PAI-1) concentration is critical for in vivo tumoral angiogenesis and growth. Oncogene. 2004;23:6986–90.
Gutierrez LS, Schulman A, Brito-Robinson T, Noria F, Ploplis VA, Castellino FJ. Tumor development is retarded in mice lacking the gene for urokinase type plasminogen activator or its inhibitor, plasminogen activator inhibitor-1. Cancer Res. 2000;60:5839–47.
Binder BR, Mihaly J. The plasminogen activator inhibitor “paradox” in cancer. Immunol Lett. 2008;118:116–24.
Ramer R, Eichele K, Hinz B. Upregulation of tissue inhibitor of matrix metalloproteinases-1 confers the anti-invasive action of cisplatin on human cancer cells. Oncogene. 2007;26:5822–7.
Mukherjee S, Adams M, Whiteaker K, Daza A, Kage K, Cassar S, et al. Species comparison and pharmacological characterization of rat and human CB2 cannabinoid receptors. Eur J Pharmacol. 2004;505:1–9.
Sativex Product Monograph; GW Pharma Ltd. Salisbury, Wiltshire U.K. SP4 0JQ Submission Control No: 091289.
Consroe P, Kennedy K, Schram K. Assay of plasma cannabidiol by capillary gas chromatography/ion trap mass spectroscopy following high-dose repeated daily oral administration in humans. Pharmacol Biochem Behav. 1991;40:517–22.
Thomas BF, Gilliam AF, Burch DF, Roche MJ, Seltzman HH. Comparative receptor binding analyses of cannabinoid agonists and antagonists. J Pharmacol Exp Ther. 1998;285:285–92.
Sacerdote P, Martucci C, Vaccani A, Bariselli F, Panerai AE, Colombo A, et al. The nonpsychoactive component of marijuana cannabidiol modulates chemotaxis and IL-10 and IL-12 production of murine macrophages both in vivo and in vitro. J Neuroimmunol. 2005;159:97–105.
Watanabe K, Kayano Y, Matsunaga T, Yamamoto I, Yoshimura H. Inhibition of anandamide amidase activity in mouse brain microsomes by cannabinoids. Biol Pharm Bull. 1996;19:1109–11.
de Filippis D, Iuvone T, d'Amico A, Esposito G, Steardo L, Herman AG, et al. Effect of cannabidiol on sepsis-induced motility disturbances in mice: involvement of CB receptors and fatty acid amide hydrolase. Neurogastroenterol Motil. 2008;20:919–27.
Costa B, Giagnoni G, Franke C, Trovato AE, Colleoni M. Vanilloid TRPV1 receptor mediates the antihyperalgesic effect of the nonpsychoactive cannabinoid, cannabidiol, in a rat model of acute inflammation. Br J Pharmacol. 2004;143:247–50.
Uddén J, Eriksson P, Hoffstedt J. Glucocorticoid-regulated adipose tissue secretion of PAI-1, but not IL-6, TNFalpha or leptin in vivo. Horm Metab Res. 2002;34:698–702.
Jin H, Lin J, Fu L, Mei YF, Peng G, Tan X, et al. Physiological testosterone stimulates tissue plasminogen activator and tissue factor pathway inhibitor and inhibits plasminogen activator inhibitor type 1 release in endothelial cells. Biochem Cell Biol. 2007;85:246–51.
Yamamoto C, Sugato M, Fujiwara Y, Kaji T. Selective promotion of plasminogen activator inhibitor-1 secretion by activation of proteinase-activated receptor-1 in cultured human brain microvascular pericytes: comparison with endothelial cells. Biol Pharm Bull. 2005;28:208–11.
Yki-Järvinen H, Sutinen J, Silveira A, Korsheninnikova E, Fisher RM, Kannisto K, et al. Regulation of plasma PAI-1 concentrations in HAART-associated lipodystrophy during rosiglitazone therapy. Arterioscler Thromb Vasc Biol. 2003;23:688–94.
Stefansson S, Lawrence DA. The serpin PAI-1 inhibits cell migration by blocking integrin αvβ3 binding to vitronectin. Nature. 1996;383:441–3.
Kanse SM, Kost C, Wilhelm OG, Andreasen PA, Preissner KT. The urokinase receptor is a major vitronectin-binding protein on endothelial cells. Exp Cell Res. 1996;224:344–53.
Dong Z, Saliganan AD, Meng H, Nabha SM, Sabbota AL, Sheng S, et al. Prostate cancer cell-derived urokinase-type plasminogen activator contributes to intraosseous tumor growth and bone turnover. Neoplasia. 2008;10:439–49.
ACKNOWLEDGEMENTS
This study was supported by grants from the Deutsche Krebshilfe e.V. (Bonn, Germany).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Ramer, R., Rohde, A., Merkord, J. et al. Decrease of Plasminogen Activator Inhibitor-1 May Contribute to the Anti-Invasive Action of Cannabidiol on Human Lung Cancer Cells. Pharm Res 27, 2162–2174 (2010). https://doi.org/10.1007/s11095-010-0219-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11095-010-0219-2