
ABSTRACT
Purpose
To study crystal growth rates of amorphous griseofulvin (GSF) below its glass transition temperature (T g) and the effect of surface crystallization on the overall crystallization kinetics of amorphous GSF.
Methods
Amorphous GSF was generated by melt quenching. Surface and bulk crystal growth rates were determined using polarized light microscope. X-ray powder diffraction (XRPD) and Raman microscopy were used to identify the polymorph of the crystals. Crystallization kinetics of amorphous GSF powder stored at 40°C (T g−48°C) and room temperature (T g−66°C) was monitored using XRPD.
Results
Crystal growth at the surface of amorphous GSF is 10- to 100-fold faster than that in the bulk. The surface crystal growth can be suppressed by an ultrathin gold coating. Below T g, the crystallization of amorphous GSF powder was biphasic with a rapid initial crystallization stage dominated by the surface crystallization and a slow or suspended late stage controlled by the bulk crystallization.
Conclusions
GSF exhibits the fastest surface crystallization kinetics among the known amorphous pharmaceutical solids. Well below T g, surface crystallization dominated the overall crystallization kinetics of amorphous GSF powder. Thus, surface crystallization should be distinguished from bulk crystallization in studying, modeling and controlling the crystallization of amorphous solids.
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
INTRODUCTION
A large proportion of the new pharmaceutical small molecules under development today are found to have poor water solubility. This in turn may lead to low bioavailability, which can have a significant impact on the development of the compound. From the standpoint of maximizing exposure, the amorphous phase is of great interest for pharmaceutical scientists, since it is at higher energy and, as such, offers the promise of higher solubility and faster dissolution rate and thereby the potential to increase bioavailability (1,2). In spite of these advantages, amorphous materials are rarely used during drug development due to physical and chemical stability issues and processing difficulties (3). The amorphous form tends to be more chemically unstable than their crystalline counterparts (4). However, from a development standpoint, the physical instability is the most problematic. Physical instability leads to the transformation of the amorphous state to the thermodynamically favored crystalline state. One approach to predict the stability of amorphous phase against crystallization is to measure its molecular mobility that is believed to correlate with crystal growth in supercooled liquids (5). Although the quantitative correlation between molecular mobility and crystallization kinetics has not been well established, in practice, this correlation has been used to predict physical stability in lieu of complete understanding of crystallization mechanism and direct monitoring of crystallization kinetics below T g (6–9). It has been suggested that amorphous pharmaceutical solids are expected to be stable at temperatures about fifty degrees below their glass transition, where the molecular mobility is usually negligible according to the spectroscopic and calorimetric measurements. However, recent studies have found that crystal growth rates at the surface of amorphous materials can be much faster than that in the bulk at temperatures below their glass transition. For example, crystal growth at the surface of amorphous indomethacin (IMC) (10) and nifedipine (NIF) (11) are at least one order of magnitude faster than that of their bulk crystals. This faster surface crystallization can result in a crystalline shell surrounding the amorphous interior, thereby negating the advantages of amorphous pharmaceutical solids. Since most amorphous pharmaceutical solids will be subjected to intensive processing techniques that impact surface area, like milling and compression surface crystallization becomes a dominant problem in controlling the amorphous stability in the formulation. Thus, surface crystallization is an important factor that must be considered when studying physical stability of amorphous materials.
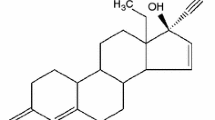
GSF, a poorly water soluble antifungal drug, is a model compound (Fig. 1) that has been used to study the crystallization kinetics of amorphous pharmaceutical solids (8,9,12). Yamamura et al. reported that the crystallization of amorphous GSF was biphasic with an initial fast crystallization stage followed by a slow crystallization stage. Zhou et al. showed that amorphous GSF was capable of rapid crystallization at 28°C below its glass transition. They reported that the extent of correlation between molecular mobility and the rate of crystallization was much weaker in the amorphous state when compared to the supercooled liquid state. This lack of correlation in the amorphous state was attributed to a change in the crystallization mechanism. In order to understand the rapid and biphasic crystallization kinetics of amorphous GSF below its T g, we fully examined its crystal growth at the surface and in the bulk. In addition, we have compared our results with those published for indomethacin and nifedipine; the possible mechanism is speculated.

Molecular structure of griseofulvin.
MATERIALS AND METHODS
Materials
Griseofulvin ((2S,6′R)- 7-chloro- 2′,4,6-trimethoxy- 6′-methyl- 3H,4′H-spiro [1-benzofuran-2,1′-cyclohexene]- 3,4′-dione, purity >98%) was obtained from Alfa Aesar (Ward Hill, MA) and used as received. The crystalline form of the as-received GSF was consistent with the polymorph reported in literature (12).
X-ray Powder Diffractometry (XRPD)
The diffractometer (PANanlytical X’pert, Philips) was equipped with a CuKα source (λ = 1.54056 Å) operating at a tube load of 45 kV and 40 mA. The divergence slit size was 1/4°, while the receiving slit and the detector slit, were 5.0 mm and 0.1 mm, respectively. A small amount of sample was loaded onto Si 510 zero-background sample holder and scanned between 3 and 40° (2θ) with a step size of 0.008 and a step time of 15.2 s/step in the continuous mode. Data was collected by a high-resolution sealed proportional detector. Si(111) with diffraction peak at 28.44 2θ was used as a standard to calibrate the instrument.
Differential Scanning Calorimetry (DSC)
DSC measurements were conducted in crimped aluminum pans using a Q1000 (TA Instruments, NewCastle, DE) unit under 50 mL/min N2 purge.
Hot Stage Optical Microscopy
Polarized Light Microscopy (PLM) was performed with a Nikon Eclipse E600 Pol microscope equipped with a Nikon Digital Camera DXM1200. The optical image was calibrated against a 1 mm stage micrometer (100 divisions). Temperature was controlled using a Linkam LTS350 hot stage.
Raman Microscopy
Raman microscope (RAMANRXN1™ Microprobe, RAMANRXN Systems) equipped with a 785 nm iodine laser was used to identify the form of crystal growing at the surface or in the bulk of melt-quenched amorphous film. The Raman system was calibrated using Cyclohexone, and a scanning range from 200 cm−1 to 4,000 cm−1 with the resolution 4 cm−1 was employed. The detector is a CCD camera cooled at −40°C.
Density
True density of amorphous GSF was measured using a AccuPyc helium pycnometer (Micormetrics, Norcross, GA). About 0.5 g sample was loaded into a cell volume of 11.9 cm3 for each measurement, and the results are reported as average from duplicate measurements.
Particle Size
Particle size distribution (PSD) analysis of hand-ground amorphous GSF was conducted using a Mastersizer 2000 particle size analyzer (Malvern Instruments, UK) equipped with a Hydro SM dispersing unit. A saturated aequous GSF solution containing 0.1% SDS was used as the dispersant.
Surface and Bulk Crystal Growth Rates of Amorphous GSF Below T g
The methods used are similar to those described by Wu et al. (10,13). To study the surface-enhanced crystallization of GSF, 3−6 mg of crystalline GSF was melted between cover glasses at 220°C for 1 min on a hot stage and quenched to room temperature on an aluminum block. One cover glass was gently detached from the amorphous GSF to expose the free surface. The amorphous GSF sample produced by melt quenching was about 15 μm thick and was confirmed to be free of crystals by polarized light microscopy. To study the bulk crystal growth, both cover glasses remain in contact with the amorphous GSF. Due to the low nucleation rate in the bulk at temperatures below T g, seeding was applied to initiate the bulk crystal growth. Seeding was achieved by contacting GSF crystals with the edge of the supercooled liquid GSF at 170°C. Subsequent to seeding, time was given for the bulk crystals to start growing. As soon as some obvious bulk crystal growth was observed, the whole sample was quenched to room temperature on an aluminum block for further study. The resulting amorphous GSF samples were then transferred to dessicators loaded with Drierite® to protect the samples from moisture. These desiccators were kept in ovens at different temperatures (40–75°C). For room temperature measurements, desiccators containing samples were kept at ambient lab conditions (22°C). The samples were then periodically withdrawn from the storage conditions and analyzed by optical microscopy to measure crystal growth rates. While surface growth rate was measured at a range of temperatures below T g, bulk crystal growth rate was measured only at 75°C.
Coating the Free Surface of Amorphous GSF with Gold
A Pelco SC-7 Sputter Coater was used to coat the amorphous GSF with an ultrathin gold coating. The current was set at 30 mA, and deposition time was set at 40 s. To study the growth rate of the surface crystals under coating, the surface crystals were initiated at 75°C and then coated with gold. The samples were then kept in the oven at 75°C for growth rate measurement.
Isothermal Crystallization of Amorphous GSF Powder Below T g
The melt-quenched amorphous GSF was slightly ground in a mortar by hand to reduce its particle size. The resulting amorphous powders were sealed in desiccators that were stored in an oven at 40°C and at room temperature (22°C). Crystallization kinetics of the stored amorphous GSF particles were monitored using XRPD as described by Zhou et al. (9). Briefly, the average intensity of four peaks at 10.9, 13.4, 14.7, and 16.6° 2θ, was used to quantify the crystallinity to prevent the bias from preferred orientations. The percent crystallinity at each time interval t was calculated as
where I(t) is the average peak intensity for the sample at time t, and \( {I_\infty } \) is the average peak intensity for the fully crystallized sample. To determine \( {I_\infty } \), at the end of each study, the samples were heated to 135°C for 10 min to complete crystallization for XRPD measurement.
RESULTS AND DISCUSSION
Surface-Enhanced Crystallization of Amorphous GSF Below T g
The glass transition temperature of amorphous GSF prepared in this study was 88°C as determined by DSC at 10°C/min, which is in agreement with published data (9). Fig. 2a shows a representative crystal that has spontaneously nucleated and grown for 8 h on the amorphous griseofulvin without the top cover glass at 75°C (T g−13°C). The crystal had loose spherulite-like morphology and grew out radially from the center. A closer examination of the loose spherulite revealed that it is composed of fibrous crystals. As supercooled liquid GSF crystallizes easily, rapid cooling of the melt is essential to produce the pure amorphous phase. This rapid cooling resulted in stress build-up, which eventually produces cracks in the amorphous GSF (14). The faint birefringent lines seen across the sample in Fig. 2a are from the cracks in the sample formed during preparation of the amorphous GSF. Fig. 2b shows the same crystal as Fig. 2a, but under an ultrathin coating of gold and after incubation for another 8 h at 75°C. Only slight increase of the radii of the spherulite was observed after gold coating. In contrast, the sample without gold coating which was stored at 75°C for 16 h was almost completely covered by the surface crystals (Fig. 3a). The dark lines across the sample were due to the cracks and relatively large thickness of the sample compared to that shown in Fig. 2. These data confirmed that the crystal growth was significantly inhibited by gold coating. The result, in turn, indicates that the crystals are actually growing at the surface of amorphous GSF; otherwise, the thin layer of gold coating would not have produced a strong inhibitory effect on crystal growth (15). The surface crystals were found to conform to the original crystalline form of the as-received sample using XRPD (Fig. 3b). The X-ray powder pattern of the surface crystals has crystalline peaks overlaid on a prominent amorphous halo, indicating incomplete crystallization even after 16 h at 75°C. These data indicate that crystallization is much faster at the surface of amorphous GSF with the underneath bulk still being amorphous. For the sample coated with gold, some birefringent crystalline fibers were also found growing out of the major crystalline spherulite (Fig. 2b). This can be attributed to the continuous crystal growth along the cracks produced during the preparation of the amorphous GSF. It is speculated that these crack surfaces might not be coated by gold, thereby permitting surface crystal growth.

Photomicrographs of GSF crystals grown at the surface. a Crystals nucleated and grown at 75°C for 8 h. b The same crystal as in (a), under gold coating and grown for another 8 h at 75°C.

Photomicrograph and XRPD pattern of crystals grown at the surface of amorphous GSF. a The sample covered by surface crystals and b its XRPD pattern (red trace) compared with that of the as-received crystalline GSF (black trace).
Bulk Crystallization of Amorphous GSF Below T g
Fig. 4 shows the photomicrographs of bulk crystals growing in the amorphous GSF sandwiched between two pieces of cover glass. A different interference mirror than the one used to observe surface crystals was used to improve the contrast for better visualization of bulk crystals. Since the spontaneous nucleation at 75°C was not observed after the sample was incubated for days, seeding at high temperature was introduced to initiate the crystallization as described in the “Materials and Methods” section. The newly grown crystals from the seeds displayed two kinds of morphologies: one appeared as the compact spherulite morphology similar to the seed with a smooth interface into the liquid (Fig. 4a), and the other appeared as loose fiber morphology significantly different from the seed (Fig. 4b). The growth of bulk crystal as spherulites could only be observed in the amorphous region where there were no cracks present. Since cracks are easily formed in amorphous GSF during cooling, only two out of ten samples examined in the study exhibited spherulitic morphology. The remainder of the samples exhibited fiber-like morphology growing in the amorphous region with cracks. Since the surface of the cracks can allow surface crystal growth, fibrous crystal growth should be considered as the combination of surface and bulk crystal growth. Both spehurilitc and fibrous bulk crystals were confirmed to be the same crystalline phase as the as-received material by Raman microscopy (Fig. 4). However, it was observed that these two kinds of crystal habits possess significantly different crystal growth rates (details provided later).

Photomicrographs and Raman spectra of crystals grown in the bulk amorphous sandwiched between two cover glasses. (a) Photomicrograph of bulk crystals grown into compact spherulites from the seeds at the edge of the cover glass. (b) Photomicrograph of bulk crystals grown into loose fibers from the seeds (S) prepared the same way as (a). The Raman spectra of the crystals corresponding to photomicrographs (a) and (b) are shown at the bottom and are compared with that of the as-received crystalline GSF. The baseline of the spectra was corrected for the convenience of visualization.
Growth Rates of Surface and Bulk Crystals Below T g
Fig. 5 compares the crystal growth rates at the surface, at the surface covered by an ultrathin gold coating and in the bulk of amorphous GSF. All the growth rates were measured at temperature below T g. Bulk crystal growth rate was only measured at 75°C. At lower temperatures, the growth rate of the spherulitic crystals was too slow to be measured, while the growth rate of the fibrous crystals could not be tracked because of interference from the cracks. At 75°C (T g−13°C), the crystal growth rate at the free surface is more than two orders of magnitude faster than that of the compact spherulitic bulk crystal and about one order of magnitude faster than that of the fibrous bulk crystal. In contrast, the crystal growth at the surface covered by an ultrathin gold coating decreased to a rate similar to that of the fibrous bulk crystal at 75°C due to the inhibition of crystal growth by gold coating (11). The fact that crystals under gold coating share a similar morphology with the bulk fibrous crystals may explain why they both have similar growth rates.

Crystal growth rates of GSF at the surface, in the bulk (sandwiched between two pieces of cover glass), and at the surface under an ultrathin gold coating (crystals first grown at the free surface at 75°C and then coated with an ultrathin gold coating to observe further growth). Error bars is one standard deviation (n = 3).
Contribution of Surface Crystallization to the Overall Crystallization Kinetics of Amorphous GSF Powder Below T g
The overall crystallization kinetics of amorphous GSF powder below T g were studied using the method outlined in the “Materials and Methods” section. The melt-quenched amorphous GSF was hand-ground to reduce particle size to d 50∼100 μm as determined by PSD measurement. Crystallinity of the ground amorphous GSF powder was monitored at 40°C and at 22°C (room temperature) for a period of 423 h and 163 days, respectively, using XRPD. Fig. 6 shows the representative XRPD patterns of the amorphous GSF powder as a function of time at 40°C (Fig. 6a) and at 22°C (Fig. 6b). The data shown in Fig. 6 were then used to estimate the degree of crystallinity using Eq. 1. Fig. 7 shows the degree of crystallinity as a function of storage time for samples stored at 40°C and at 22°C. At the end of the study, about 50% crystallization was observed for samples stored at 40°C and about 35% for the samples stored at 22°C. However, optical photomicrographs of the samples taken at the end of the study showed that they were almost completely birefringent (Fig. 6c and d) under polarized light microscope. This observation confirms that surface crystallization is responsible for the majority of the crystallinity observed for the two samples stored well below glass transition temperature. Surface crystallization leads to the formation of a crystalline shell around the cores of GSF particles that are still amorphous, thereby making them birefringent under PLM while retaining a significant amount of amorphous interior, which is detected as a halo background by XRPD. Fig. 7 also shows that biphasic kinetics are involved with the evolution of crystallinity in both samples. In the early stage (labeled as E in the plots), the degree of crystallinity increases rapidly, but in the late stage (labeled as L in the plots), the degree of crystallinity ascends slowly for the samples at 40°C while it becomes suspended for the samples at 22°C. These biphasic crystallization kinetics are similar to those observed for amorphous indomethacin below T g (10). The rapid increase of crystallinity in the early stage (E) is attributed to the dominant, fast surface crystallization at the beginning. After the surface is covered with crystals, a second late stage takes over, whose kinetics are dictated by slow bulk crystallization. Thus, the crystallization rates of the surface and bulk can be estimated by linearly fitting the data points in the early and late stages, respectively (solid lines superimposed on the data points in the plots). The data from the fitting is summarized in Table I. Surface-controlled crystallization rate is about 20 times faster than bulk-controlled crystallization rate at 40°C, whereas this difference further increased to about 300-fold when the temperature dropped to 22°C. The surface crystallization rate only decreased about 10-fold going from 40 to 22°C, while the bulk crystallization rate decreased about 160-fold for the same temperature range. This obvious difference indicates that the surface crystallization rate has much weaker temperature dependence than bulk crystallization rate below T g. This result is similar as that observed for surface and bulk crystallization of nifedipine below T g (11,16). By linearly extrapolating the late-stage crystallization fitting line to time zero (the dashed lines in the plots of Fig. 7), we can estimate the degree of crystallinity due to surface crystallization, which is 40% at 40°C and 34% at 22°C. Therefore, the contribution of surface crystallization to the overall crystallization is similar for samples stored at 40°C and 22°C. This result is not surprising considering both samples share the same particle size distribution with a d 50∼100 μm.

Representative XRPD traces of amorphous GSF powder stored at 40°C (a) and room temperature, 22°C (b). Y axis is shifted for comparison. The peaks denoted as “*” were used for quantification. Typical particles of amorphous GSF after 423 h at 40°C (c) and 163 days at 22°C (d).

Degree of crystallinity vs. time for ground amorphous GSF powder stored at 40°C (left) and 22°C (right). Solid lines are the linear fitting of the crystallization at early (E) and late stages (L), and dashed lines are linear extrapolation of the late stage crystallization to time zero. Error bar covers the variation between samples (n = 2).
Furthermore, we can theoretically estimate the contribution of surface crystallization to the overall crystallization kinetics assuming spherical particles using the following equation:
and
where C surface is the degree of crystallinty due to surface crystallization, V surface is the volume fraction of particles that undergo surface crystallization, and V total is the total volume of particles. V surface is estimated as the difference between the total volume of particles with a radius R T and the volume available for bulk crystallization (V bulk). V bulk is calculated from the internal bulk radius R B, which is the difference between R T and penetration depth of surface crystallization (D surface). For hand-ground amorphous particles used in this study, a d 50∼100 μm was estimated from PSD measurement. For theoretical estimation of C surface, we assume the spherical particles with the 50 μm radius (half of the d 50) and a 5-μm penetration depth for surface crystallization. The penetration depth was assumed to be the same as that estimated from surface crystallization of indomethacin (10). The calculated value of C surface was found to be 27%, which is in reasonable agreement with our experimental results of 40 and 34% given the assumption of spherical particles. This result is comparable to that reported by Yamamura et al. (12). They reported that amorphous GSF powder with similar particle size (63 μm sieve pass) rapidly crystallized to about 30% within 10 days for samples stored at room temperature. This was followed by a later slow crystallization stage. In their study, the moisture was not controlled, and this could have resulted in faster crystallization kinetics than observed in our study.
It is anticipated that if the particle size of amorphous GSF can be further reduced (leading to increased surface area), a higher degree of crystallinity could be reached in shorter times due to the surface crystallization as demonstrated in the study of indomethacin surface crystallization (10). Although we do not have experimental data within our study to prove this hyphothesis, it is interesting to compare our data with the previous crystallization study of amorphous GSF by Zhou et al. (9). They reported that melt-quenched GSF took about 1 month at 60°C to reach 50% degree of crystallinity, while we show that it took only 15 days at 40°C to reach the same level of crystallinity. This apparent contradiction in data is resolved when the method of preparation of the amorphous samples is taken into account. While Zhou et al. used melt-quenched amorphous GSF confined to XRPD sample holder with a very limited surface area in their study, we used hand-ground amorphous GSF with increased surface area. This difference in surface area accounts for the faster crystallization kinetics observed in our study.
Comparison of Surface Crystallization Rate of GSF with Other Systems that Exhibit Surface-Enhanced Crystallization
Griseofulvin, a neutral compound without any ionic group, is the third pharmaceutical system to exhibit the surface-enhanced crystallization in line with the other two pharmaceutical systems indomethacin, a free acid, and nifedipine, a free base. The comparison of surface crystallization rates of GSF, IMC, and NIF are shown in Fig. 8. At T g−13°C, the surface-enhanced growth rate of GSF is about 10-fold faster than NIF and more than 100-fold faster than IMC. The lowest temperature at which the surface crystal growth rate could be measured in a conventional experimental time window were T g−66°C for GSF, T g−38°C for NIF and T g−20°C for IMC. GSF obviously displays the fastest surface crystallization kinetics among the three systems.

In order to rationalize the differences in surface crystallization kinetics between GSF, IMC, and NIF, we compared the molecular and thermal properties of these systems. They are summarized in Table II. All three molecules have relatively low molecular weight (MW) of about 350 and have similar surface tension (γ) of around 50 dyn/cm. GSF and IMC have fewer free rotatable bonds (FRB) than NIF, but NIF has higher molecular symmetry. Thus, all three molecules can be considered rigid structures with the low conformational flexibility (17). In comparison with NIF and IMC, GSF has a relatively higher T g, but all three molecules have low configurational entropy. These observations from molecular and thermal properties suggest that they can readily undergo crystallization. However, compared to NIF and IMC, GSF has a much larger free volume in the amorphous phase as indicated from the true density difference between amorphous and crystalline forms and also lacks hydrogen bonding capability as it has no hydrogen bonding donors (HD). These properties of GSF suggest that it would crystallize more easily than NIF or IMC in the bulk or at the surface, even though it has the highest glass transition temperature among three systems. However, the relatively large difference between surface and bulk crystallization kinetics in these molecules cannot be rationalized using the available molecular and thermal properties. Some studies have indicated that the free surface of amorphous solids have a much faster mobility than the bulk (18,19); thereby, the detailed studies of molecular motions at the surface of these three systems can shed light on the fast surface crystallization mechanism. The result of the fast surface crystal growth suppressed by an ultrathin layer of gold coating suggests that a thin layer of molecules at the surface of amorphous GSF might have higher mobility than the bulk molecules. However, it is noteworthy that the activation energy of surface crystallization below T g for the three systems are similar, so the crystallization mechanism could be the same for all three system at the surface.
Significance of Surface Crystallization for the Stability of Amorphous Drugs
The stability of amorphous drug has been commonly correlated with molecular mobility, which is thought to be a key factor that reflects the molecular diffusion-controlled crystallization. However, a quantitative correlation between molecular mobility and crystallization kinetics is rarely established among known amorphous systems, especially at the temperature near or below T g (5,7,9). The experimentally measured bulk crystallization kinetics is usually several orders faster than that predicted by molecular mobility (13), and the correlation between molecular mobility and surface crystallization is even weaker if surface and bulk mobility are assumed to be the same. This lack of correlation is illustrated in the current study, where significant crystallization (about 30%) occurred within a month at temperature well below glass transition (T g−66°C), where the molecular mobility as estimated by the α, relaxation dynamics are negligible (8,9). Therefore, the molecular mobility, mainly a bulk property, cannot be responsible for the observed fast surface crystallization kinetics below T g. Thus, as previously postulated, negligible bulk molecular motion at temperatures well below T g cannot guarantee the stability of amorphous solids. Moreover, in addition to surface crystallization, diffusionless glassy crystal growth has also been documented in several organic systems near and below T g (20–22). These modes of crystallization cause a sudden increase in crystallization rate near or below T g compared to the crystallization rate from the supercooled liquid. Thus, in order to predict the physical stability of amorphous solids, a complete understanding of the crystallization mechanism is important.
From a practical point of view, understanding the crystallization kinetics above and below T g, provides guidance about the physical stability of amorphous drugs during normal shelf life. If surface crystallization exists for an amorphous solid, it is usually faster than other crystallization modes, such as diffusionless glassy crystallization and bulk crystallization. Thus, surface crystallization kinetics should always be considered as a part of modeling and prediction of the long-term stability regardless of how slow the other crystallization modes may be. Although surface crystallization may only be limited to surface of the particles leaving the internal core or majority of the sample still amorphous, it can nevertheless negate the solubility/dissolution advantage provided by amorphous solids. For example, a recent study has shown that a thin layer of crystalline shell at the surface of IMC reduces the dissolution rate significantly (23). In addition, the different crystallization kinetics between surface and bulk of amorphous materials indicate that at least two domains exist in the amorphous solids. These two types of domains might exhibit significantly different reaction kinetics (24) and moisture absorption kinetics (25).
Recent advances in understanding surface crystallization have provided alternative ways to stabilize amorphous drugs. As an example, nanocoating the surface of amorphous drugs with hydrophilic polymers could inhibit surface crystallization while maintaining its high dissolution rate (15). Moreover, combination of nanocoated amorphous drugs with a small amount of polymer can mitigate effects of both surface and bulk crystallization while making it possible to have a high drug load formulation. In summary, understanding the kinetics of all modes of crystallization is critical for predicting the stability of amorphous solids and stabilizing them in the long-term.
CONCLUSION
Surface-enhanced crystallization was observed in amorphous GSF below its glass transition temperature. Crystal growth rate at the surface of amorphous GSF was found to be 10- to 100-fold faster than in the bulk at temperatures below T g. Surface crystallization kinetics were shown to have much weaker temperature dependence than bulk crystallization below T g. The surface-enhanced crystallization could be suppressed by an ultrathin coating of gold. This supports the view that the molecules at the free surface of amorphous solids might have higher mobility than bulk molecules. The overall crystallization kinetics of amorphous GSF powder at T g−48°C and T g−66°C showed a biphasic process. There is a rapid early stage dominated by surface crystallization and a slow or suspended late stage controlled by the bulk crystallization.
Amorphous GSF shows the fastest surface crystallization kinetics compared to IMC and NIF. Earlier studies of amorphous IMC and NIF suggested that surface-enhanced crystallization can be a problem for stabilizing amorphous pharmaceutical solids that have T g close to room temperature. Herein, we show that even for the materials, like GSF, that have a T g much higher than room temperature, surface crystallization can still be a significant problem. Therefore, surface crystallization should be distinguished from bulk crystallization and used for modeling and controlling the crystallization of amorphous solids.
REFERENCES
Yu L. Amorphous pharmaceutical solids: preparation, characterization and stabilization. Adv Drug Deliv Rev. 2001;48:27–42.
Kennedy M, Hu J, Gao P, Li L, Ali-Reynolds A, Chal B, et al. Enhanced bioavailability of a poorly soluble VR1 antagonist using an amorphous solid dispersion approach: a case study. Mol Pharm. 2008;5:981–93.
Abu TMS. Solid dispersion of poorly water-soluble drugs: Early promises, subsequent problems, and recent breakthroughs. J Pharm Sci. 1999;88:1058–66.
Guo Y, Byrn SR, Zografi G. Physical characteristics and chemical degradation of amorphous quinapril hydrochloride. J Pharm Sci. 2000;89:128–43.
Ediger MD, Harrowell P, Yu L. Crystal growth kinetics exhibit a fragility-dependent decoupling from viscosity. J Chem Phys. 2008;128:034709/1–6.
Hancock BC, Shamblin SL, Zografi G. Molecular mobility of amorphous pharmaceutical solids below their glass transition temperatures. Pharm Res. 1995;12:799–806.
Hancock BC, Zograti G. Characteristics and significance of the amorphous state in pharmaceutical systems. J Pharm Sci. 1997;86:1–12.
Tombari E, Presto S, Johari G, Shanker R. Molecular mobility, thermodynamics and stability of Griseofulvin’s ultraviscous and glassy states from dynamic heat capacity. Pharm Res. 2008;25:902–12.
Zhou D, Geoff G, Law D, Grant DJW, Schmitt EA. Thermodynamics, molecular mobility and crystallization kinetics of amorphous griseofulvin. Mol Pharm. 2008;5:927–36.
Wu T, Yu L. Surface crystallization of indomethacin below Tg. Pharm Res. 2006;23:2350–5. Epub 2006 Aug 2323.
Zhu L, Wong L, Yu L. Surface-enhanced crystallization of amorphous nifedipine. Mol Pharm. 2008;5:921–6.
Yamamura S, Takahira R, Momose Y. Crystallization kinetics of amorphous griseofulvin by pattern fitting procedure using X-ray diffraction data. Pharm Res. 2007;24:880–7.
Wu T, Yu L. Origin of enhanced crystal growth kinetics near Tg probed with indomethacin polymorphs. J Phys Chem B Condens Matter Mater Surf Interfaces Biophys. 2006;110:15694–9.
Matsumo T, Bogue DC. Stress birefringence in amorphous polymers under nonisothermal conditions. J Polym Sci Polym Phys Ed. 1977;15:1663–74.
Wu T, de Villiers M, Yu L. Inhibiting surface crystallization of amorphous indomethacin by nanocoating. Langmuir. 2007;23:5148–53.
Ishida H, Wu T, Yu L. Sudden rise of crystal growth rate of nifedipine near T g without and with polyvinylpyrrolidone. J Pharm Sci. 2007;96:1131–8.
Yu L, Reutzel-Edens SM, Mitchell CA. Crystallization and polymorphism of conformationally flexible molecules: problems, patterns, and strategies. Org Process Res Dev. 2000;4:396–402.
Swallen S, Kearns K, Mapes M, Kim Y, McMahon R, Ediger M, et al. Organic glasses with exceptional thermodynamic and kinetic stability. Science. 2007;315:353–6.
Fakhraai Z, Forrest JA. Measuring the surface dynamics of glassy polymers. Science. 2008;319:600–4.
Sun Y, Xi H, Chen S, Ediger MD, Yu L. Crystallization near glass transition: transition from diffusion-controlled to diffusionless crystal growth studied with seven polymorphs. J Phys Chem B. 2008;112:5594–601.
Sun Y, Xi H, Ediger MD, Richert R, Yu L. Diffusion-controlled and “diffusionless” crystal growth near the glass transition temperature: relation between liquid dynamics and growth kinetics of seven ROY polymorphs. J Chem Phys. 2009;131:074506–9.
Xi H, Sun Y, Yu L. Diffusion-controlled and diffusionless crystal growth in liquid o-terphenyl near its glass transition temperature. J Chem Phys. 2009;130:094508–9.
Savolainen M, Heinz A, Strachan C, Gordon KC, Yliruusi J, Rades T, Sandler N. Screening for differences in the amorphous state of indomethacin using multivariate visualization. Eur J Pharm Sci. 2007;30:113–23.
Luo D, Anderson BD. Application of a two-state kinetic model to the heterogeneous kinetics of reaction between cysteine and hydrogen peroxide in amorphous lyophiles. J Pharm Sci. 2008;97:3907–26.
Dawson KJ, Kearns KL, Ediger MD, Sacchetti MJ, Zografi GD. Highly stable indomethacin glasses resist uptake of water vapor. J Phys Chem B. 2009;113:2422–7.
Forster A, Hempenstall J, Tucker I, Rades T. The potential of small-scale fusion experiments and the Gordon–Taylor equation to predict the suitability of drug/polymer blends for melt extrusion. Drug Dev Ind Pharm. 2001;27:549–60.
Yoshioka M, Hancock BC, Zografi G. Crystallization of indomethacin from the amorphous state below and above its glass transition temperature. J Pharm Sci. 1994;83:1700–5.
Bhugra C, Shmeis R, Krill S, Pikal M. Predictions of onset of crystallization from experimental relaxation times I-correlation of molecular mobility from temperatures above the glass transition to temperatures below the glass transition. Pharm Res. 2006;23:2277–90.
ACKNOWLEDGEMENTS
The work is supported by Amgen summer internship program. We thank Dr.Yuan-hon Kiang, Dr. Darren Reid in Amgen and Professor Lian Yu in University of Wisconsin—Madison for helpful discussions and valuable comments.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article
Cite this article
Zhu, L., Jona, J., Nagapudi, K. et al. Fast Surface Crystallization of Amorphous Griseofulvin Below T g . Pharm Res 27, 1558–1567 (2010). https://doi.org/10.1007/s11095-010-0140-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11095-010-0140-8