
Abstract
The goal of brain drug targeting technology is the delivery of therapeutics across the blood–brain barrier (BBB), including the human BBB. This is accomplished by re-engineering pharmaceuticals to cross the BBB via specific endogenous transporters localized within the brain capillary endothelium. Certain endogenous peptides, such as insulin or transferrin, undergo receptor-mediated transport (RMT) across the BBB in vivo. In addition, peptidomimetic monoclonal antibodies (MAb) may also cross the BBB via RMT on the endogenous transporters. The MAb may be used as a molecular Trojan horse to ferry across the BBB large molecule pharmaceuticals, including recombinant proteins, antibodies, RNA interference drugs, or non-viral gene medicines. Fusion proteins of the molecular Trojan horse and either neurotrophins or single chain Fv antibodies have been genetically engineered. The fusion proteins retain bi-functional properties, and both bind the BBB receptor, to trigger transport into brain, and bind the cognate receptor inside brain to induce the pharmacologic effect. Trojan horse liposome technology enables the brain targeting of non-viral plasmid DNA. Molecular Trojan horses may be formulated with fusion protein technology, avidin–biotin technology, or Trojan horse liposomes to target to brain virtually any large molecule pharmaceutical.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
INTRODUCTION
The limiting factor in the development of new drugs for the brain is the blood–brain barrier (BBB). The BBB phenomenon is illustrated in Fig. 1, which is a whole body autoradiogram of an adult mouse sacrificed 30 min after the intravenous injection of radiolabeled histamine, a small molecule just 111 Da (1). Despite the small size of this relatively lipid soluble molecule, there is no significant penetration of histamine into the brain or spinal cord, whereas histamine readily traverses the porous capillary beds perfusing all organs other than the central nervous system (CNS). The observation in Fig. 1 belies a widely held misconception, which is that small molecules readily cross the BBB. A small molecule does not cross the BBB in pharmacologically significant amounts, unless the molecule is both lipid soluble and has a molecular weight (MW) <400 Da. Most small molecules lack these dual molecular characteristics, and do not cross the BBB. Of the more than 7,000 molecules in the Comprehensive Medicinal Chemistry (CMC) database, <5% of all drugs are active in the CNS and the activity of these drugs is generally confined to just four conditions: affective disorders, chronic pain, insomnia, and epilepsy (2). In another study, if affective disorders are excluded, <1% of all drugs are pharmacologically active in the brain (3).

Whole body autoradiogram of mouse at 30 min following the intravenous administration of labeled histamine. The study shows this small molecule readily penetrates into all organs of the body, except for the brain or spinal cord. From (1).
The BBB problem is even more severe in the case of large molecule drugs. Essentially 100% of all large molecule drugs do not cross the BBB, including all of the products of biotechnology: recombinant proteins and enzymes, monoclonal antibodies (MAb), antisense drugs, short interfering RNA (siRNA), or gene therapy.
The delivery of drugs to the brain has been traditionally approached with either medicinal chemistry or neurosurgical-based invasive brain drug delivery. The trans-cranial approaches to brain drug delivery include intra-cerebroventricular (ICV) injection, intra-cerebral (IC) injection, and convection-enhanced diffusion (CED).
MEDICINAL CHEMISTRY AND BRAIN TARGETING OF SMALL MOLECULES
Lipid-mediated Transport
A long-standing goal of medicinal chemistry has been the lipidization of water soluble drugs, whereby medicinal chemical medications are used to block existing hydrogen bond-forming groups on the parent drug molecule. Despite the extensive application of medicinal chemistry, there presently is not a single FDA approved drug that exemplifies the conversion of a poorly brain-penetrating molecule into a high brain-penetrating molecule such that the medicinal chemical modification enabled pharmacologic activity in the brain and drug approval. There are several reasons why attempts to increase the lipid solubility of a parent drug molecule is difficult to implement. First, increasing lipid solubility increases permeation across all biological membranes in the body, including the BBB. Therefore, increasing the lipid solubility of a drug greatly increases the plasma clearance of the drug and in pharmacokinetic terms, the plasma area under the concentration curve (AUC) is reduced, which is illustrated with a lipidized form of chlorambucil (4). The pharmacologic action of the drug in brain is proportional to the brain uptake of the drug, as reflected in the percent of injected dose (ID)/g. The % ID/g is directly proportional to both the BBB permeation constant and to the plasma AUC (5). Increasing lipid solubility increases BBB permeation but decreases plasma AUC. These effects of increasing lipid solubility are offsetting, such that there can be little increase in brain uptake of the drug. Second, medicinal chemical modifications to the parent drug invariably lead to an increase in molecular weight of the drug. Any increase of the molecular weight above a 400 Da threshold can have deleterious effect on brain penetration. BBB permeability decreases 100-fold as the surface area of the drug is increased from 52 Å2 (molecular weight = 200 Da) to 105 Å2 (molecular weight = 450 Da) (6). The “threshold” effect between the molecular weight of a drug and membrane permeation has been known for many years, and is a general property of solute diffusion through biological membranes (7–9). Third, increasing the lipid solubility of a drug may enhance binding to plasma proteins, which could offset the enhanced membrane permeation caused by lipid solubility. However, many plasma protein-bound drugs are available for transport across the BBB in vivo via a mechanism of enhanced dissociation at the brain capillary endothelia surface (5).
Carrier-mediated Transport
An alternative use of medicinal chemistry to increase brain penetration is to modify the drug such that there is increased carrier-mediation of the drug via one of several endogenous carrier-mediated transporters (CMT), such as those listed in Fig. 2. For example, the α-carboxylation of the water-soluble catecholamine drug results in the formation of a neutral amino acid. Whereas the BBB penetration of the catecholamine is very low, the α amino acid may then penetrate the BBB at pharmacologically significant rates via CMT on the BBB large neutral amino acid transporter type 1 (LAT1). In addition to LAT1, there are numerous other BBB CMT systems that could be portals of entry for small molecule drugs to the brain, as discussed below. Medicinal chemical modifications which cause the drug to have a molecular structure that mimics the structure of an endogenous ligand for a BBB CMT system is an under-developed yet promising application of medicinal chemistry.

Twelve trans-membrane region model of rabbit LAT1 shows the position of the 12 cysteine (C) residues. Also highlighted are the Gly219 (G) and Trp234 (W) residues in the extracellular loop between domains 5 and 6, as mutation of these residues alters transporter activity. From (10).
The application of medicinal chemistry to brain targeting of small molecules via the CMT systems requires knowledge about (a) the specific gene encoding a given BBB CMT system, (b) the structure-transport relationships (STR) for the transporter, and (c) an expression system for the BBB CMT system. If the goal is to target drugs that have a neutral amino acid structure via the BBB large neutral amino acid transporter, then it is necessary to know that the principal large neutral amino acid transporter at the BBB is LAT1. Next, detailed information about the STRs for this transporter are required, much like it is necessary to define structure-activity relationships (SAR) for drug receptors. Ultimately, 3-dimensional models of the BBB CMT system will aid in defining the STRs for the transporter. At present, 2-dimensional models of BBB transporters can be produced, such as that generated by hydropathy plots, which are based on the amino acid sequence of the transporter deduced from the nucleotide sequence of the cloned cDNA. The predicted 2-dimensional structure of LAT1 is illustrated in Fig. 2 (10). The transporter is comprised of 12 trans-membrane regions (TMR) (11). The cysteine (C) residue in the extracellular loop between the third and fourth TMRs forms a disulfide bond with the 4F2hc heavy chain to comprise the functional hetero-dimeric transporter (12). Site-directed mutagenesis (SDM) studies implicate the role of glycine (G) and tryptophan (W) residues in the extracellular loop between fifth and sixth TMRs (13). The 12 cysteine residues are phylogenetically conserved across species (13). LAT1 is inhibited by Hg+2, and SDM studies implicate Cys-439 as the mercuric-sensitive cysteine residue of LAT1 (10).
TRANS-CRANIAL AND TRANS-NASAL BRAIN DRUG DELIVERY
Intra-cerebroventricular Injection
The ICV approach injects drug into the cerebrospinal fluid (CSF) compartment, which is 140 ml in humans. The entire CSF pool in the human brain is turned over every 4–5 h and four to five times per day (5). CSF exits the brain via bulk flow and is absorbed into the peripheral blood stream at the superior sagittal sinus. Whereas CSF exits brain via bulk flow, drug penetrates into brain parenchyma from the CSF compartment via diffusion. The kinetics of bulk flow is log orders greater than the kinetics of diffusion. For example, 4 days are required for albumin to diffuse 5 mm in water (5). Consequently, drug exits the CSF compartment faster than drug can diffuse into the brain. Following ICV drug administration, the concentration of the drug in brain parenchyma decreases exponentially with each mm of distance removed from the ependymal surface of brain. Drug concentration in brain is only 1–2% of the CSF concentration at just 1–2 mm from the surface (14). The paradox of ICV drug administration is that the drug distributes preferentially to the blood, rather than the brain. Christy and Fishman (15) noted more than 40 years ago that “a CSF injection is like a slow intravenous infusion.” The dose of barbiturate that induces pharmacologic activity in brain is the same whether the drug is given by intravenous or ICV injection (16). Following ICV injection, the drug readily distributes to the general circulation, where the drug then enters brain parenchyma following transport across the BBB. The pathway from CSF to brain parenchyma is via the bloodstream. Certain drugs may exert a pharmacologic effect in the brain following ICV administration without first passing through the blood, if the target receptor of the drug is located near the ependymal surface of the brain. For example, opioid peptides induce central analgesia following ICV administration as the opioid receptors are located in the peri-aqueductal gray, just beneath the path of CSF flow.
Intra-cerebral Injection
Drug, or gene vector, distributes into brain via diffusion following IC injection. Diffusion decreases exponentially with the diffusion distance. Consequently, the brain concentration of a small molecule decreases 90%, relative to the injection concentration, at just 500 μm removed from the injection site (17). It is not possible to achieve significant distribution of a drug or gene in the rat brain (18), must less in the 1,200 g human brain, following an IC injection.
Convection-enhanced Diffusion
The ICV and IC approaches rely on diffusion for drug penetration into the brain, whereas drug distributes via bulk flow in the case of CED. A catheter implanted in the brain is connected to a pump, which drives fluid flow in the brain at a prescribed infusion rate. The brain is the only organ of the body that lacks a lymphatic system, which normally serves to return extracellular fluid to the general circulation. The brain does have a “microcirculation,” whereby fluid flows along paravascular pathways in proportional to arterial pressure (19, 20). However, this fluid flow is low, and about 0.1 μl/min in the rat brain (21). In contrast, the rate of CSF flow in the rat brain is about 3 μl/min (22), or 30-fold faster than the paravascular pathway. A concern about CED is the intense astrogliotic reaction in the region of the fluid flow, which is demonstrated by glial fibrillary acidic protein (GFAP) immunocytochemistry in the primate brain following CED (23).
Trans-nasal Delivery
The trans-nasal administration of a lipid soluble small molecule, progesterone, was shown over 20 years ago to achieve a higher distribution in the CSF than in the blood (24). Once the molecule crosses the nasal epithelial barrier and enters the submucous space of the nose, the drug may diffuse across the arachnoid membrane and enter the CSF compartment of the olfactory region, and then distribute along the usual CSF flow tracks to be absorbed into the general circulation. Drugs that are lipid soluble and have a MW < 400 Da, will not only cross the BBB via lipid-mediation, but also cross the nasal epithelial barrier and the arachnoid membrane. Lipid soluble small molecules enter olfactory CSF from the nose in proportion to lipid solubility (25). If the drug is water soluble, or has a MW > 400 Da, then the drug crosses biological membranes poorly via free diffusion. In this case, it would be necessary to disrupt the epithelial barrier to achieve transport (26). The instillation of a volume of >100 μl/nares in the human is sufficient to cause local injury (27). Most studies in rodents that demonstrate transport of drug from the nose to the olfactory lobe involve the instillation of volumes that cause local injury. The movement of drug from the nose to the olfactory lobe begins with transport across the olfactory region of the nasal epithelium. Whereas the olfactory region of the nasal epithelium is 50% of the total nasal surface area in the rat, the olfactory region is only 3–8% in the human nose (28). If drug is infused into the nose of humans without local injury, then no flux of drug from the nose to the CSF is observed for drugs such as melatonin, a lipid soluble small molecule, or for vitamin B12, a water soluble molecule with a MW of 1,355 Da (27, 29).
The direct pathway to the brain is trans-vascular-through the BBB. Drugs that do not cross the BBB via lipid-mediation, which is >98% of all drugs (28), can cross the BBB via transport on one of many endogenous transporters. Both small molecule and large molecule drugs can be re-formulated to enable transport on the BBB endogenous transporters. The use of the endogenous brain transporters is called brain drug targeting, since the structure of the drug is specifically altered to target a BBB endogenous transporter. The endogenous BBB transporters fall into three categories: carrier-mediated transport (CMT), active efflux transport (AET), and receptor-mediated transport (RMT).
BLOOD–BRAIN BARRIER ENDOGENOUS TRANSPORTERS
Carrier-mediated Transporters
The carrier-mediated transport of nutrients, vitamins, or hormones across the BBB was initially investigated with in vivo physiologic techniques such as the brain uptake index (BUI) method developed by Oldendorf (30). This work allowed for determination of the Michaelis–Menten kinetic parameters of BBB nutrient transport (31), and the K m and V max of representative substrates of several BBB CMT systems are shown in Table I. Generally, as the affinity of the transporter increases, the capacity (V max) decreases. The (V max / K m) + K D is equal to the BBB permeability-surface area (PS) product of BBB transport for the given substrate, where K D is the constant of non-saturable transport.
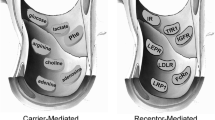
An important goal in BBB biology is the molecular cloning of the specific transporters that function as CMT systems at the BBB. For example, there are over a half-dozen members of the GLUT glucose transporter gene family within the Solute Carrier (SLC) sub-group, SLC2, and almost all of these glucose transporter genes have, in various reports, been localized to the BBB. However, a comparison of the total glucose transporter sites, as determined with a cytochalasin B binding assay, and the total GLUT1 sites at the BBB, as determined with a quantitative Western blot using purified human erythrocyte glucose transporter as an assay standard, shows that GLUT1 constitutes >90% of BBB glucose transporters (32). The principal large neutral and cationic amino acid transporters at the BBB are LAT1 and CAT1, respectively (Fig. 3). The major adenosine transporter at the rat BBB is CNT2 (33); this is a concentrative, sodium-dependent transporter, which correlates with in vivo observations on the sodium dependency of adenosine influx from blood to brain (34). The BBB choline transporter (CHT), or the BBB adenine nucleobase transporter (NBT), has not yet been cloned, as indicated by the CHT-X and NBT-X designation in Fig. 3.

The endogenous BBB transporters are grouped into three categories: carrier-mediated transport (CMT), active efflux transport (AET), and receptor-mediated transport (RMT). GLUT1 = glucose transporter, member 1 (SLC2); LAT1 = large neutral amino acid transporter, member 1 (SLC7); CAT1 = cationic amino acid transporter, member 1(SLC7); MCT1 = monocarboxylic acid transporter, member 1 (SLC16); CNT2 = concentrative nucleoside transporter, member 2 (SLC28); CHT = choline transporter (SLC5); NBT = nucleobase transporter; ABCB1 = adenosine triphosphate binding cassette (ABC) transporter, sub-family B, member 1, also called P-glycoprotein; ABCC = ABC transporter, sub-family C; ABCG = ABC transporter, sub-family G, member 2; OAT = organic anion transporter (SLC22); OATP = organic anion-transporting polypeptide (SLC21); EAAT = glutamic acid amino acid transporter (SLC1); TAUT = taurine transporter (SLC6); INSR = insulin receptor; TFR = transferrin receptor; IGFR = insulin-like growth factor receptor; LEPR = leptin receptor; FCGRT = Fc fragment of IgG receptor transporter, also called neonatal Fc receptor (FCRN); SCARB1 = scavenger receptor, class B, member 1. The X designation for the CHT and NBT transporter indicates these transporters have not yet been cloned, or identified at the molecular level for the BBB. From (28).
Active Efflux Transporters
Many drugs and metabolites are actively effluxed from brain to blood (35–37). The kinetics of this efflux can be quantitated with the Brain Efflux Index (BEI) method developed by Terasaki (38). P-glycoprotein is the classic BBB active efflux transport system. Although there has been intense investigation of BBB p-glycoprotein function, the field of BBB active efflux transporters needs to expand in two directions. First, there are many other members of the ATP-binding cassette (ABC) transporter gene family other than p-glycoprotein that are energy dependent BBB active efflux transporters, and a partial list is shown in Fig. 3. Second, the active efflux transport of a drug from blood to brain is mediated by the coordinate interaction of at least two transporters functioning at opposite poles of the brain capillary endothelial cell. For example, an ABC-type energy dependent transporter may function at the luminal membrane in concert with an energy independent transporter at the abluminal membrane. The energy independent transporters are typically members of the SLC gene family and a partial list of SLC-type transporters at the BBB is shown in Fig. 3. It is also possible that the energy-dependent transporters are expressed at the luminal membrane of the brain capillary endothelium, while the energy-independent transporters are expressed at the abluminal membrane. It is generally not possible to differentiate the luminal and abluminal membranes of the brain capillary endothelial cell with confocal microscopy because these membranes are separated by an endothelial cytoplasm of only 200 nm, which is a distance too small to resolve with confocal microscopy. What is needed in the field of BBB active efflux transport is the ultrastructural localization of ABC or SLC transporters at the luminal and abluminal membranes of the brain capillary endothelium using techniques such as immunogold electron microscopy, similar to prior ultrastructural localization of BBB GLUT1 (39).
Receptor-mediated Transport
A third class of endogenous BBB transporter is the receptor-mediated transport (RMT) system (5). A partial list of BBB RMT systems is shown in Fig. 3. The BBB insulin receptor (INSR) is responsible for the brain uptake of circulating insulin (40). Similarly, the brain uptake of circulating transferrin, insulin-like growth factors (IGF), or leptin is mediated by the BBB TFR, IGF1R, IGF2R, or LPR, respectively. The complicating factor in the BBB transport of the IGFs is that these molecules are >99.9% bound by specific plasma proteins (41). Although IgG molecules do not undergo significant transport in the blood to brain direction (42), there is rapid reverse transcytosis of IgG molecules in the brain to blood direction (43). Following intra-cerebral injection, albumin effluxes from brain with a half time of 10–12 h (44). Conversely, IgG molecules efflux from brain with a half time of 40 min following intra-cerebral injection, and this selective efflux, or reverse transcytosis, of IgG molecules from brain back to blood is mediated by a BBB Fc receptor (FcR) (43). Confocal microscopy shows high expression of the neonatal FcR (FcRn) (45), and it is assumed the BBB FcR responsible for IgG efflux is the FcRn. The brain capillary endothelial cell also expresses an acetylated low density lipoprotein (LDL) receptor, which is also called a scavenger receptor or SCARB1 (Fig. 3). However, this receptor only mediates the endocytosis of acetylated LDL, and not the transcytosis of acetyl-LDL, through the endothelial compartment from blood to brain (46).
MOLECULAR TROJAN HORSES FOR BRAIN DRUG TARGETING
Peptidomimetic Monoclonal Antibodies as Molecular Trojan Horses
In addition to the endogenous ligands, certain peptidomimetic monoclonal antibodies (MAb) also undergo RMT across the BBB via the endogenous peptide receptor/transporters (Fig. 3). The MAb binds an exofacial epitope on the receptor that is spatially removed from the binding site of the endogenous ligand and this receptor binding allows the MAb to “piggy back” across the BBB via the endogenous RMT system within the BBB. The receptor-specific MAb may act as a molecular Trojan horse (MTH) and ferry across the BBB any attached drug or non-viral plasmid DNA. A panel of MAb-based MTHs has been developed for brain drug delivery in different species. For brain drug transport in mice, the rat 8D3 MAb to the mouse TfR is used (47). For brain drug delivery in rats, the murine OX26 MAb to the rat TfR is used (42); the OX26 MAb is not active in mice or in other species. Brain drug delivery in Old World primates such as the Rhesus monkey is accomplished with the murine 83-14 MAb to the human insulin receptor (HIR) (48, 49).
In Vivo CNS Pharmacologic Effects with Molecular Trojan Horses
The definitive test of whether a given BBB delivery system is effective is the demonstration of in vivo CNS pharmacologic effects following the intravenous administration of low doses of the drug attached to the BBB delivery system. It is assumed that the BBB delivery system mediates the transport of the drug through the BBB without disrupting the BBB. BBB disruption is toxic to the brain, and is associated with chronic neuropathologic changes, cerebral vasculopathy, and seizures (50–52).
Examples of in vivo CNS pharmacologic effects with BBB molecular Trojan horses are given in Table II. Vasoactive intestinal peptide (VIP) is a potent cerebral vasodilator when applied topically to pial vessels (53). However, the infusion of VIP into the carotid artery causes no increase in cerebral blood flow (CBF) (54), because the neuropeptide does not cross the BBB. Following conjugation of VIP to the TfRMAb, the intravenous administration of low doses, 10–20 μg/kg, of VIP in conscious rats resulted in a 65% increase in hemispheric CBF (55). The intravenous administration of brain-derived neurotrophic factor (BDNF) resulted in complete neuroprotection of pyramidal neurons in the CA1 sector of the hippocampus in rats subjected to 10 min of transient forebrain ischemia and isoelectric electroencephalograms, providing the BDNF was conjugated to a BBB molecular Trojan horse (56). The intravenous administration of BDNF alone resulted in no neuroprotection of brain, because the neuropeptide does not cross the BBB (57). The intravenous administration of BDNF or fibroblast growth factor (FGF)-2 to rats subjected to regional brain ischemia with a middle cerebral artery occlusion (MCAO) model resulted in a 65–80% reduction in stroke volume, providing the neurotrophin was conjugated to a BBB molecular Trojan horse (58–60). The intravenous administration of BDNF alone or FGF-2 alone resulted in no reduction in stroke volume because the neurotrophins cannot cross the BBB, and because the BBB is not disrupted in the early phases of stroke when neuroprotection is still possible (61). The intravenous administration of epidermal growth factor (EGF) conjugated with a radionuclide allowed for early detection of intra-cranial brain cancer, providing EGF was conjugated to a BBB molecular Trojan horse (62). The intravenous administration of the EGF peptide radiopharmaceutical alone did not result in any imaging of brain cancer, because the EGF does not cross the BBB (63). The intravenous administration of an Aβ1–40 peptide radiopharmaceutical allowed for imaging of brain amyloid in transgenic mice providing the Aβ1–40 peptide radiopharmaceutical was conjugated to a BBB molecular Trojan horse (64). The intravenous administration of the Aβ1–40 peptide radiopharmaceutical alone did not result in any imaging of brain amyloid, because the neuropeptide does not cross the BBB (65). In vivo expression of the caveolin (CAV)-1α gene or the glial fibrillary acidic protein (GFAP) gene in vivo in brain cancer and brain ischemia was possible following the intravenous administration of sequence specific antisense radiopharmaceuticals using peptide nucleic acids (PNA), providing the antisense PNA was conjugated to a BBB molecular Trojan horse (66, 67). Intravenous administration of the PNA alone did not result in imaging gene expression in vivo, because the PNA does not cross the BBB (68). The intravenous administration of a 116 kDa enzyme, β-galactosidase, in mice did not increase brain enzyme activity, because the enzyme does not cross the BBB (69). However, conjugation of the β-galactosidase to the TfRMAb did increase β-galactosidase enzyme activity in brain (69).
In all of the cases described in Table II, the pharmaceutical was attached to the MTH with avidin–biotin technology. In this approach, the drug is mono-biotinylated in parallel with the production of a 1:1 conjugate of the MTH and streptavidin (SA). Owing to the very high binding of biotin to SA, or avidin (K D = 10−15 M, dissociation half-time = 89 days), there is instantaneous capture of the biotinylated drug by the MTH/SA conjugate. In order to advance the use of molecular Trojan horses to neurotherapeutic development in humans, it was necessary to (a) genetically engineer the human-specific MTH, which is the MAb to the human insulin receptor (HIR), and (b) genetically engineer fusion proteins of the HIRMAb and the recombinant protein or therapeutic antibody.
Genetic Engineering of Molecular Trojan Horses for Delivery Across the Human BBB
The murine HIRMAb cannot be used in humans owing to immunologic reactions. However, both chimeric and humanized forms of the HIRMAb have been genetically engineered and shown to undergo transport across the primate BBB in vivo (70, 71). The affinity of the chimeric HIRMAb for the human insulin receptor was compared to the affinity of the original murine HIRMAb using a binding assay with purified HIR extracellular domain (ECD). Chinese hamster ovarian (CHO) cells that were permanently transfected with the gene encoding the HIR ECD were immunoreactive using the murine HIRMAb (Fig. 4a, left panel). The HIR ECD produced by these CHO cells was purified by lectin affinity chromatography and this allowed for establishment of the HIR binding assay (70). The binding isotherms for the murine HIRMAb and the genetically engineered chimeric HIRMAb overlap and there was no significant change in affinity following the genetic engineering (Fig. 4a, right panel). The chimeric HIRMAb bound to the human BBB insulin receptor (70). Human brain capillaries can be isolated free of the adjoining brain tissue, as shown in Fig. 4b (left panel). The chimeric HIRMAb was radioiodinated and the binding of the [125I] chimeric HIRMAb to human BBB was linear with incubation time and reached high values of binding (Fig. 4b, right panel). This binding was nearly completely suppressed by 10 μg/ml concentrations of unlabeled murine HIRMAb (70). The chimeric HIRMAb was conjugated with diethylenetriaminepentaacetic acid (DTPA) and radiolabeled with 111-Indium and injected intravenously into the adult Rhesus monkey (70). The brain scan taken 2 h after the intravenous injection of the labeled chimeric HIRMAb is shown in Fig. 4c. This study shows global penetration of the chimeric HIRMAb into the Rhesus monkey brain, with increased uptake in gray matter relative to white matter. Gray matter has greater vascular density and thus greater microvascular density of insulin receptor.

Development of a genetically engineered chimeric MAb to the human insulin receptor (HIR) for BBB drug delivery in humans. a CHO cells, permanently transfected with a gene encoding the extracellular domain (ECD) of the HIR is used to produce soluble HIR ECD to establish an ELISA showing retention of high affinity of the chimeric MAb for the HIR. b The chimeric HIRMAb was labeled with 125I and added to isolated human brain capillaries. The chimeric HIRMAb binds with high affinity to the human BBB HIR, and the binding is suppressed by the murine HIRMAb. c The chimeric HIRMAb was labeled with 111In and injected intravenously in the adult Rhesus monkey, and the brain was scanned 2 h later. The antibody rapidly crosses the primate BBB in vivo. From (70).
Neurotrophin-antibody Fusion Protein
Recombinant protein neurotherapeutics can be delivered across the human BBB following the genetic engineering, expression, and purification of recombinant fusion proteins. In this approach, the non-transportable protein therapeutic, e.g. a neurotrophin, is fused to the carboxyl or amino terminus of either the heavy chain or light chain of the genetically engineered HIRMAb. A fusion protein of the chimeric HIRMAb and a neuroprotective neurotrophin was genetically engineered and expressed and shown to retain the bifunctional properties of the fusion protein (72). The fusion protein both bound the HIR with high affinity, to enable transport across the human BBB in vivo, and bound the cognate neurotrophin receptor on brain cells, to induce neuroprotection (72).
Antibody-antibody Fusion Protein
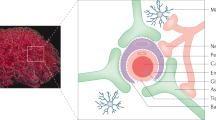
MAb-based therapeutics may also be delivered across the human BBB with fusion protein technology. This is exemplified with the recent genetic engineering, expression, and validation of a fusion protein of the chimeric HIRMAb and a single chain Fv (ScFv) antibody to the Aβ amyloid peptide of Alzheimer’s disease (AD) (73). Anti-Aβ antibodies are potential therapeutics of AD, as these agents disaggregate the amyloid plaque of AD following the intra-cerebral injection of the anti-Aβ antibody. The aim of either active or passive immunization therapy of AD is to use MAb-based therapeutics to deplete the brain of Aβ amyloid plaque. However, in the case of either active or passive immune therapy of AD, it is necessary for the anti-Aβ MAb to cross the BBB in both the blood to brain and brain to blood direction (Fig. 5). There is no IgG transporter at the BBB to mediate the blood to brain transport of these large molecules. Therefore, IgG therapeutics do not cross the BBB in the blood to brain direction, since these molecules lack affinity for any BBB receptor/transporter. However, MAb-therapeutics can be made to cross the BBB via RMT following the re-engineering of these molecules as fusion proteins with a BBB MTH (73). The structure of a genetically engineered fusion antibody of the HIRMAb and the anti-Aβ ScFv is shown in Fig. 6. This fusion antibody is a tri-functional molecule that is comprised of three domains. The first domain is the HIRMAb at the “head” of the molecule, which mediates the RMT of the fusion antibody from blood to brain across the BBB via the endogenous BBB HIR (Fig. 5). The second domain is the anti-Aβ ScFv at the “tail” of the fusion antibody, which allows for binding to, and disaggregation of, Aβ amyloid plaque within the brain behind the BBB (Fig. 5). The third domain is the CH2–CH3 interface in the midsection of the molecule, which is the binding site for the BBB FcRn. The BBB FcR mediates the efflux of the fusion antibody in the brain to blood direction via reverse transcytosis across the BBB in vivo (Fig. 5). All three functionalities of the fusion antibody shown in Fig. 6 are retained following genetic engineering and expression of this molecule (73). The intra-cerebral injection of the fusion antibody into double transgenic AD mouse brain results in a 40% clearance of Aβ amyloid plaque within 48 hours of the single intra-cerebral injection (73). The fusion antibody is rapidly transported across the primate BBB in vivo (73). Other therapeutic MAb-based molecules could be delivered across the human BBB with the MTH fusion protein technology.

The fusion antibody clears amyloid from brain in Alzheimer’s disease (AD) via three sequential steps, and each of these three sequential steps uses separate parts of the fusion antibody molecule shown in Fig. 6. Step 1 is the influx of the fusion antibody from blood to brain across the BBB, which is mediated by binding of the fusion antibody to the BBB human insulin receptor (HIR). Step 2 is binding of the fusion antibody to the amyloid plaque in AD, which promotes disaggregation of the amyloid plaque, and this binding to the plaque is mediated by the anti-Aβ ScFv part of the fusion antibody. Step 3 is the efflux of the fusion antibody from brain to blood across the BBB, which is mediated by binding of the fusion antibody to the BBB FcR at the CH2–CH3 interface of the Fc region of the fusion antibody. From (73).

The fusion antibody is comprised of three domains. The first domain, the HIRMAb, binds the BBB HIR to trigger influx across the BBB. The second domain, the CH2/CH3 interface of the Fc region, binds to the BBB FcR to trigger efflux from brain back to blood. The third domain, the anti-Aβ ScFv fused to the CH3 region, binds to the Aβ amyloid peptide of AD to cause disaggregation of amyloid in brain. From (73).
MOLECULAR TROJAN HORSES AND BRAIN TARGETING OF NON-VIRAL PLASMID DNA
Trojan Horse Liposomes
In attempting to design a formulation for targeting of non-viral plasmid DNA across the BBB with molecular Trojan horses, an important design consideration was the need to eliminate the pro-inflammatory effects of exposed DNA. The intravenous injection of cationic liposome/DNA complexes results in a cytokine release, owing to the DNA component (74). The DNA part of cationic polyplex mixtures is still susceptible to nuclease digestion (75). If the DNA in a formulation is exposed to nuclease in vitro, then it is also exposed to the immune system in vivo. However, if the plasmid DNA is encapsulated in the interior of a 100 nm liposome, and if all external DNA is removed from the preparation by endo/exonuclease digestion, then the proinflammatory character of exogenous DNA is eliminated. Therefore, single plasmid DNA molecules were encapsulated in the interior of 100 nm liposomes (76). The surface of the liposome was conjugated with several thousand strands of 2,000 Da polyethylene glycol (PEG). The tips of 1–2% of the PEG strands were conjugated with a receptor (R)-specific MAb used as a molecular Trojan horse to ferry the Trojan horse liposome (THL), also called a pegylated immunoliposome, across the BBB via RMT. Owing to the expression of the TfR or insulin receptor on the brain cell membrane, the MTH also triggers the receptor-mediated endocytosis of the THL into brain cells, subsequent to RMT across the BBB (77).
Brain Targeting of Reporter Genes with THLs
Luciferase or β-galactosidase cDNAs have been subcloned into eukaryotic expression plasmids which were encapsulated in THLs targeted to mouse or rat brain with TfRMAbs or to Rhesus monkey brain with the HIRMAb (77–79). The expression plasmids may be engineered with either a widely read promoter, such as the SV-40 promoter, or a tissue-specific promoter, such as the 5′-flanking sequence (FS) of either the human glial fibrillary acidic protein (GFAP) gene or the bovine opsin gene. The TfR or insulin receptor is also expressed in peripheral tissues, as well as the BBB. Therefore, transgenes delivered with THLs are expressed in certain peripheral tissues, if the transgene is under the influence of a widely read promoter, such as the SV40 promoter (78, 79). However, if a brain, or eye specific promoter is used, then expression of the transgene is restricted to brain or eye (78, 80). This was demonstrated in the Rhesus monkey (77, 80). THLs were encapsulated with the β-galactosidase expression plasmid under the influence of either the SV40 promoter or the opsin promoter, and targeted with the HIRMAb. Organs were removed at 48 hr after the intravenous injection of the THL, and β-galactosidase histochemistry was performed. As shown in Fig. 7a–e, the β-galactosidase gene, under the influence of the SV40 promoter, is widely expressed in primate eye (Fig. 7a), cerebrum (Fig. 7b), cerebellum (Fig. 7c), liver (Fig. 7d), or spleen (Fig. 7e). However, if the identical formulation was injected intravenously in the Rhesus monkey, except the SV40 promoter was replaced by the opsin promoter, then transgene expression was eliminated in all organs, except the primate eye (Fig. 7f–j). Within the eye, the transgene was expressed in both the retinal pigmented epithelium, and the photoreceptor cells. The trangene was also expressed in other ocular structures, including the lens, cornea, ciliary body, and iris (77). These results in adult animals, parallel findings in mice transgenic for the opsin promoter, which enabled transgene expression in multiple structures of the eye (81, 82). Confocal microscopy with antibodies to both β-galactosidase and the neuronal neuN antigen demonstrated neuronal expression of the transgene in brain following intravenous administration of THLs (77).

Beta galactosidase histochemistry of adult Rhesus monkey organs removed 48 h after an intravenous injection of a β-galactosidase expression plasmid DNA encapsulated in Trojan horse liposomes (THLs) targeted with the monoclonal antibody to the human insulin receptor. The β-galactosidase cDNA was under the influence of either the widely read SV40 promoter (a–e) or the eye-specific promoter taken from the 5′-flanking sequence of the bovine opsin gene (f–j). Eye (a, f); cerebrum (b, g); cerebellum (c, h); liver (d, i); and spleen (e, j). The combined use of the THL gene transfer technology, and tissue-specific promoters, restricts expression of the exogenous gene to a specific region of the body. From (77, 80).
The insulin receptor is also expressed in peripheral tissues and the transgene was expressed in some but not all insulin receptor positive peripheral tissues in the Rhesus monkey (Fig. 7d and e). The liposomes must cross two barriers in vivo. The first barrier is the microvascular endothelial barrier, which in brain is the BBB. The second barrier is the parenchymal cell membrane barrier. In brain or retina, the insulin receptor is expressed on both barriers. In liver and spleen, the insulin receptor is expressed on only the second barrier. However, no targeting system is needed for the first barrier in liver and spleen since these organs are perfused by sinusoidal vessels that are freely permeable to 100 nm liposomes (79). There is abundant insulin receptor on the second barrier in heart, skeletal muscle, or fat. However these peripheral tissues are perfused by vessels with continuous endothelium, which are impermeable to liposomes. Moreover, the endothelial in these peripheral tissues do not express insulin receptor. Therefore, the THLs cannot traverse the first barrier in heart, skeletal muscle, or fat, and transgene expression was not detected in these organs (77–79).
THL Targeting of a Therapeutic Gene in Parkinson’s Disease
Parkinson’s disease (PD) is characterized by a deficiency in striatal tyrosine hydroxylase (TH). Experimental PD is produced by the intra-cerebral injection of a neurotoxin, 6-hydroxydopamine, into the median forebrain bundle. This results in atrophy of the TH fibers originating in the substantia nigra and terminating in the striatum. There is a >95% reduction in striatal TH in this model in the side ipsilateral to the neurotoxin injection, as demonstrated by either immunocytochemistry, confocal microscopy, or measurements of striatal TH enzyme activity (83). However, when the rats are administered an intravenous administration of TfRMAb-targeted THLs carrying a TH expression plasmid, there is a 100% normalization of striatal TH immunoreactivity (83). There is also 100% normalization in striatal TH enzyme activity and an 80% reduction in apomorphine-induced rotation behavior in experimental PD rats treated with TfRMAb-targeted THLs (83). If the rats are treated with an identical THL formulation, except the TfRMAb on the THL is replaced a mouse IgG2a isotype control antibody, then there is no restoration of striatal TH in experimental PD (83). These results confirm that the targeting of the TH transgene to the brain is strictly a function of the receptor specificity of the MTH on the THL.
THL Targeting of an Antisense RNA Gene in Brain Cancer
Experimental brain cancer is produced following the intra-cerebral injection of human U87 glioma cells in the brain of severe combined immunodeficient (scid) mice (84). These tumors over-express the human epidermal growth factor receptor (EGFR), which has a pro-oncogenic function in tumor cell growth. Therefore, knockdown of brain cancer EGFR could have a therapeutic effect in brain cancer. This was initially confirmed by targeting an expression plasmid to brain cancer in vivo with THLs. The plasmid DNA encoded a 700 nucleotide (nt) RNA antisense to a specific region of the human EGFR mRNA (84). Since the human brain cancer was perfused by vessels of mouse brain origin, it was necessary to conjugate two different MTHs to the THLs. One MTH was the rat MAb to the mouse TfR; this MTH targeted the THL across the mouse vascular endothelium perfusing the human brain cancer. However, the rat MAb to the mouse TfR does not recognize the human TfR on the human U87 glial cell. Therefore, the THL was also formulated with the mouse MAb to the human insulin receptor (HIR). Once the THL crosses the mouse BBB, the HIRMAb binds the HIR on the human brain cancer cell to initiate receptor-mediated endocytosis into the cancer cell. Weekly intravenous administration of 5 μg/mouse of the plasmid DNA encoding the EGFR antisense RNA resulted in a 100% increase in survival time of the mice with the intra-cranial brain cancer (84).
THL Targeting of an RNAi Gene in Brain Cancer
A new form of antisense therapy is RNA interference (RNAi). A plasmid DNA was engineered to encode a short hairpin RNA (shRNA) against a defined sequence of the human EGFR mRNA (85). The shRNA gene was under the influence of the U6 RNA polymerase. The plasmid DNA was encapsulated in THLs, which were doubly targeted with the anti-mouse TfRMAb and the anti-human HIRMAb. Weekly intravenous injections of 5 μg/mouse of the THL targeted plasmid DNA resulted in a 90% increase in survival time of scid mice implanted with intra-cerebral human U87 brain cancer (85). This was the first demonstration of an increase in survival in experimental cancer with intravenous RNAi of the brain, which was enabled by the development of the THL gene transfer technology.
Lack of Toxicity on Repeat Injection of THLs
The TH expression plasmid DNA was encapsulated in THLs targeted with either the mouse TfRMAb or a mouse IgG isotype control antibody, and injected intravenously weekly for six consecutive weeks (87). Chronic administration of THLs caused no change in body weight, organ histology, or serum chemistries. Chronic administration showed no evidence of inflammatory reactions based on immunocytochemistry with a panel of antibodies directed against antigens representing various arms of the immune system (87).
SUMMARY
The molecular Trojan horses that cross the BBB via RMT can be formulated in one of three technology platforms to target virtually any large molecule pharmaceutical across the BBB in vivo (Fig. 8). Recombinant proteins and monoclonal antibody-based therapeutics can be targeted with fusion protein technology (Fig. 6). Oligopeptides, antisense agents, or siRNA may be delivered with avidin-biotin technology (Fig. 8). In this approach, the therapeutic is monobiotinylated in parallel with the genetic engineering and expression of a fusion protein of avidin and the MTH. Non-viral plasmid DNA and genes encoding shRNA may be targeted to brain with THLs (Fig. 8). Molecular Trojan horses have been genetically engineered for human use. The genetically engineered HIRMAb crosses the primate BBB in vivo at rates comparable to many neuroactive small molecules. The goal of BBB drug targeting technology is the development of human neurotherapeutics, which are formulated specifically to cross the human BBB in vivo.

Molecular Trojan horses are incorporated into one of three technology platforms, depending on the type of pharmaceutical to be delivered to the brain. Recombinant proteins or monoclonal antibodies are delivered with the fusion protein technology. Peptides, antisense, or short interfering RNA (siRNA) are delivered with the avidin–biotin technology, wherein the drug is mono-biotinylated in parallel with the production of a fusion protein of avidin and the Trojan horse. Plasmid DNA, which can encode short interfering RNA (shRNA), is delivered across the BBB with Trojan horse liposomes.
References
W. M. Pardridge. Biochemistry of the human blood–brain barrier. Blood–brain barrier: interface between internal medicine and the brain. Ann. Intern. Med. 105:82–95 (1986).
A. K. Ghose, V. N. Viswanadhan, and J. J. Wendoloski. A knowledge-based approach in designing combinatorial or medicinal chemistry libraries for drug discovery. 1. A qualitative and quantitative characterization of known drug databases. J. Com. Chem. 1:55–68 (1999).
C. A. Lipinski. Drug-like properties and the causes of poor solubility and poor permeability. J. Pharmacol. Toxicol. Methods. 44:235–249 (2000).
N. H. Greig, E. M. Daly, D. J. Sweeney, and S. I. Rapoport. Pharmacokinetics of chlorambucil–tertiary butyl ester, a lipophilic chlorambucil derivative that achieves and maintains high concentrations in brain. Cancer Chemother. Pharmacol. 25:320–325 (1990).
W. M. Pardridge. Brain Drug Targeting: The Future of Brain Drug Development. Cambridge University Press, Cambridge, UK, 2001.
H. Fischer, R. Gottschlich, and A. Seelig. Blood–brain barrier permeation: molecular parameters governing passive diffusion. J. Membr. Biol. 165: 201–211 (1998).
D. J. Hingson and J. M. Diamond. Comparison of nonelectrolyte permeability patterns in several epithelia. J. Membr. Biol. 10:93–135 (1972).
B. E. Cohen and A. D. Bangham. Diffusion of small non-electrolytes across liposome membranes. Nature. 236:173–174 (1972).
W. R. Lieb and W. D. Stein. Non-Stokesian nature of transverse diffusion within human red cell membranes. J. Membr. Biol. 92:111–119 (1986).
R. J. Boado, J. Y. Li, C. Chu, F. Ogoshi, P. Wise, and W. M. Pardridge. Site-directed mutagenesis of cysteine residues of large neutral amino acid transporter LAT1. Biochim. Biophys. Acta 1715:104–110 (2005).
Y. Kanai, H. Segawa, K. Miyamoto, H. Uchino, E. Takeda, and H. Endou. Expression cloning and characterization of a transporter for large neutral amino acids activated by the heavy chain of 4F2 antigen (CD98). J. Biol. Chem. 273:23629–23632 (1998).
R. Pfeiffer, B. Spindler, J. Loffing, P. J. Skelly, C. B. Shoemaker, and F. Verrey. Functional heterodimeric amino acid transporters lacking cysteine residues involved in disulfide bond. FEBS Lett. 439:157–162 (1998).
R. J. Boado, J. Y. Li, and W. M. Pardridge. Site-directed mutagenesis of rabbit LAT1 at amino acids 219 and 234. J. Neurochem. 84:1322–1331 (2003).
R. G. Blasberg, C. Patlak, and J. D. Fenstermacher. Intrathecal chemotherapy: brain tissue profiles after ventriculocisternal perfusion. J. Pharmacol. Exp. Ther. 195:73–83 (1975).
N. P. Christy and R. A. Fishman. Studies of the blood–cerebrospinal fluid barrier to cortisol in the dog. J. Clin. Invest. 40:1997–2006 (1961).
R. B. Aird. A study of intrathecal, cerebrospinal fluid-to-brain exchange. Exp. Neurol. 86:342–358 (1984).
L. K. Fung, M. Shin, B. Tyler, H. Brem, and W. M. Saltzman. Chemotherapeutic drugs released from polymers: distribution of 1,3-bis(2-chloroethyl)-1-nitrosourea in the rat brain. Pharm. Res. 13:671–682 (1996).
C. E. Krewson, M. L. Klarman, and W. M. Saltzman. Distribution of nerve growth factor following direct delivery to brain interstitium. Brain. Res. 680:196–206 (1995).
M. L. Rennels, T. F. Gregory, O. R. Blaumanis, K. Fujimoto, and P. A. Grady. Evidence for a ‘paravascular’ fluid circulation in the mammalian central nervous system, provided by the rapid distribution of tracer protein throughout the brain from the subarachnoid space. Brain Res. 326:47–63 (1985).
M. T. Krauze, R. Saito, C. Noble, J. Bringas, J. Forsayeth, T. R. McKnight, J. Park, and K. S. Bankiewicz. Effects of the perivascular space on convection-enhanced delivery of liposomes in primate putamen. Exp. Neurol. 196:104–111 (2005).
I. Szentistvanyi, C. S. Patlak, R. A. Ellis, and H. F. Cserr. Drainage of interstitial fluid from different regions of rat brain. Am. J. Physiol. 246:F835–844 (1984).
H. Davson, K. Welch, and M.B. Segal. Secretion of cerebrospinal fluid. In The Physiology and Pathophysiology of the Cerebrospinal Fluid. Churchill Livingstone, London, 1987, p. 201.
Y. Ai, W. Markesbery, Z. Zhang, R. Grondin, D. Elseberry, G. A. Gerhardt, and D. M. Gash. Intraputamenal infusion of GDNF in aged rhesus monkeys: distribution and dopaminergic effects. J. Comp. Neurol. 461:250–261 (2003).
T. C. Anand Kumar, G. F. David, A. Sankaranarayanan, V. Puri, and K. R. Sundram. Pharmacokinetics of progesterone after its administration to ovariectomized rhesus monkeys by injection, infusion, or nasal spraying. Proc. Natl. Acad. Sci. U. S. A. 79:4185–4189 (1982).
T. Sakane, M. Akizuki, S. Yamashita, T. Nadai, M. Hashida, and H. Sezaki. The transport of a drug to the cerebrospinal fluid directly from the nasal cavity: the relation to the lipophilicity of the drug. Chem. Pharm. Bull. (Tokyo). 39:2456–2458 (1991).
H. Yamazumi. Infiltration of India ink from subarachnoid space to nasal mucosa along olfactory nerves in rabbits. Nippon Jibi Inkoka Gakkai Kaiho. 92:608–616 (1989).
P. Merkus, H. J. Guchelaar, D. A. Bosch, and F. W. Merkus. Direct access of drugs to the human brain after intranasal drug administration? Neurology. 60:1669–1671 (2003).
W. M. Pardridge. Blood–brain barrier delivery. Drug Discov Today. 12:54–61 (2007).
M. P. van den Berg, P. Merkus, S. G. Romeijn, J. C. Verhoef, and F. W. Merkus. Uptake of melatonin into the cerebrospinal fluid after nasal and intravenous delivery: studies in rats and comparison with a human study. Pharm. Res. 21:799–802 (2004).
W. H. Oldendorf. Brain uptake of radiolabeled amino acids, amines, and hexoses after arterial injection. Am. J. Physiol. 221:1629–1639 (1971).
W. M. Pardridge. Brain metabolism: a perspective from the blood–brain barrier. Physiol. Rev. 63:1481–1535 (1983).
W. M. Pardridge, R. J. Boado, and C. R. Farrell. Brain-type glucose transporter (GLUT-1) is selectively localized to the blood–brain barrier. Studies with quantitative western blotting and in situ hybridization. J. Biol. Chem. 265:18035–18040 (1990).
J. Y. Li, R. J. Boado, and W. M. Pardridge. Cloned blood–brain barrier adenosine transporter is identical to the rat concentrative Na+ nucleoside cotransporter CNT2. J. Cereb. Blood Flow Metab. 21:929–936 (2001).
W. M. Pardridge, T. Yoshikawa, Y. S. Kang, and L. P. Miller. Blood–brain barrier transport and brain metabolism of adenosine and adenosine analogs. J. Pharmacol. Exp. Ther. 268:14–18 (1994).
A. Tsuji and I. I. Tamai. Carrier-mediated or specialized transport of drugs across the blood–brain barrier. Adv. Drug. Deliv. Rev. 36:277–290 (1999).
T. Terasaki and K. Hosoya. The blood–brain barrier efflux transporters as a detoxifying system for the brain. Adv. Drug Deliv. Rev. 36:195–209 (1999).
H. Kusuhara and Y. Sugiyama. Efflux transport systems for drugs at the blood–brain barrier and blood–cerebrospinal fluid barrier (Part 1). Drug Discov. Today. 6:150–156 (2001).
T. Terasaki. Development of Brain Efflux Index (BEI) method and its application to the blood–brain barrier efflux transport study. In Introduction to the Blood–Brain Barrier; Methodology, Biology and Pathology, Cambridge University Press, Cambridge, 1998, pp. 24–31.
E. M. Cornford, S. Hyman, M. E. Cornford, and M. J. Caron. Glut1 glucose transporter activity in human brain injury. J. Neurotrauma. 13:523–536 (1996).
K. R. Duffy and W. M. Pardridge. Blood–brain barrier transcytosis of insulin in developing rabbits. Brain Res. 420:32–38 (1987).
J. Holly and C. Perks. The role of insulin-like growth factor binding proteins. Neuroendocrinology. 83:154–160 (2006).
W. M. Pardridge, J. L. Buciak, and P. M. Friden. Selective transport of an anti-transferrin receptor antibody through the blood–brain barrier in vivo. J. Pharmacol. Exp. Ther. 259:66–70 (1991).
Y. Zhang and W. M. Pardridge. Mediated efflux of IgG molecules from brain to blood across the blood–brain barrier. J. Neuroimmunol. 114:168–172 (2001).
H. F. Cserr, D. N. Cooper, P. K. Suri, and C. S. Patlak. Efflux of radiolabeled polyethylene glycols and albumin from rat brain. Am. J. Physiol. 240:F319–F328 (1981).
F. Schlachetzki, C. Zhu, and W. M. Pardridge. Expression of the neonatal Fc receptor (FcRn) at the blood–brain barrier. J. Neurochem. 81:203–206 (2002).
D. Triguero, J. Buciak, and W. M. Pardridge. Capillary depletion method for quantification of blood–brain barrier transport of circulating peptides and plasma proteins. J. Neurochem. 54:1882–1888 (1990).
H. J. Lee, B. Engelhardt, J. Lesley, U. Bickel, and W. M. Pardridge. Targeting rat anti-mouse transferrin receptor monoclonal antibodies through blood–brain barrier in mouse. J. Pharmacol. Exp. Ther. 292:1048–1052 (2000).
W. M. Pardridge, Y. S. Kang, J. L. Buciak, and J. Yang. Human insulin receptor monoclonal antibody undergoes high affinity binding to human brain capillaries in vitro and rapid transcytosis through the blood–brain barrier in vivo in the primate. Pharm. Res. 12:807–816 (1995).
D. Wu, J. Yang, and W. M. Pardridge. Drug targeting of a peptide radiopharmaceutical through the primate blood–brain barrier in vivo with a monoclonal antibody to the human insulin receptor. J. Clin. Invest. 100:1804–1812 (1997).
T. S. Salahuddin, B. B. Johansson, H. Kalimo, and Y. Olsson. Structural changes in the rat brain after carotid infusions of hyperosmolar solutions. An electron microscopic study. Acta Neuropathol. (Berlin). 77:5–13 (1988).
A. S. Lossinsky, A. W. Vorbrodt, and H. M. Wisniewski. Scanning and transmission electron microscopic studies of microvascular pathology in the osmotically impaired blood–brain barrier. J. Neurocytol. 24:795–806 (1995).
E. A. Neuwelt and S. I. Rapoport. Modification of the blood–brain barrier in the chemotherapy of malignant brain tumors. Fed. Proc. 43:214–219 (1984).
L. I. Larsson, J. Fahrenkrug, O. Schaffalitzky De Muckadell, F. Sundler, R. Hakanson, and J. R. Rehfeld. Localization of vasoactive intestinal polypeptide (VIP) to central and peripheral neurons. Proc. Natl. Acad. Sci. U. S. A. 73:3197–3200 (1976).
J. McCulloch and L. Edvinsson. Cerebral circulatory and metabolic effects of vasoactive intestinal polypeptide. Am. J. Physiol. 238:H449–H456 (1980).
D. Wu and W. M. Pardridge. Central nervous system pharmacologic effect in conscious rats after intravenous injection of a biotinylated vasoactive intestinal peptide analog coupled to a blood–brain barrier drug delivery system. J. Pharmacol. Exp. Ther. 279:77–83 (1996).
D. Wu and W. M. Pardridge. Neuroprotection with noninvasive neurotrophin delivery to the brain. Proc. Natl. Acad. Sci. U. S. A. 96:254–259 (1999).
T. Sakane and W. M. Pardridge. Carboxyl-directed pegylation of brain-derived neurotrophic factor markedly reduces systemic clearance with minimal loss of biologic activity. Pharm. Res. 14:1085–1091 (1997).
Y. Zhang and W. M. Pardridge. Conjugation of brain-derived neurotrophic factor to a blood–brain barrier drug targeting system enables neuroprotection in regional brain ischemia following intravenous injection of the neurotrophin. Brain Res. 889:49–56 (2001).
Y. Zhang and W. M. Pardridge. Neuroprotection in transient focal brain ischemia after delayed intravenous administration of brain-derived neurotrophic factor conjugated to a blood–brain barrier drug targeting system. Stroke. 32:1378–1384 (2001).
B. W. Song, H. V. Vinters, D. Wu, and W. M. Pardridge. Enhanced neuroprotective effects of basic fibroblast growth factor in regional brain ischemia after conjugation to a blood–brain barrier delivery vector. J. Pharmacol. Exp. Ther. 301:605–610 (2002).
L. Belayev, R. Busto, W. Zhao, and M. D. Ginsberg. Quantitative evaluation of blood–brain barrier permeability following middle cerebral artery occlusion in rats. Brain Res. 739:88–96 (1996).
A. Kurihara and W. M. Pardridge. Imaging brain tumors by targeting peptide radiopharmaceuticals through the blood–brain barrier. Cancer Res. 59:6159–6163 (1999).
A. Kurihara, Y. Deguchi, and W. M. Pardridge. Epidermal growth factor radiopharmaceuticals: 111In chelation, conjugation to a blood–brain barrier delivery vector via a biotin-polyethylene linker, pharmacokinetics, and in vivo imaging of experimental brain tumors. Bioconjug. Chem. 10:502–511 (1999).
H. J. Lee, Y. Zhang, C. Zhu, K. Duff, and W. M. Pardridge. Imaging brain amyloid of Alzheimer disease in vivo in transgenic mice with an Abeta peptide radiopharmaceutical. J. Cereb. Blood Flow Metab. 22:223–231 (2002).
Y. Saito, J. Buciak, J. Yang, and W. M. Pardridge. Vector-mediated delivery of 125I-labeled beta-amyloid peptide A beta 1–40 through the blood–brain barrier and binding to Alzheimer disease amyloid of the A beta 1–40/vector complex. Proc. Natl. Acad. Sci. U. S. A. 92:10227–10231 (1995).
T. Suzuki, D. Wu, F. Schlachetzki, J. Y. Li, R. J. Boado, and W. M. Pardridge. Imaging endogenous gene expression in brain cancer in vivo with 111In-peptide nucleic acid antisense radiopharmaceuticals and brain drug-targeting technology. J. Nucl. Med. 45:1766–1775 (2004).
T. Suzuki, Y. Zhang, Y. F. Zhang, F. Schlachetzki, and W. M. Pardridge. Imaging gene expression in regional brain ischemia in vivo with a targeted [111in]-antisense radiopharmaceutical. Mol. Imaging 3:356–363 (2004).
W. M. Pardridge, R. J. Boado, and Y. S. Kang. Vector-mediated delivery of a polyamide (“peptide”) nucleic acid analogue through the blood–brain barrier in vivo. Proc. Natl. Acad. Sci. U. S. A. 92:5592–5596 (1995).
Y. Zhang and W. M. Pardridge. Delivery of beta-galactosidase to mouse brain via the blood–brain barrier transferrin receptor. J. Pharmacol. Exp. Ther. 313:1075–1081 (2005).
M. J. Coloma, H. J. Lee, A. Kurihara, E. M. Landaw, R. J. Boado, S. L. Morrison, and W. M. Pardridge. Transport across the primate blood–brain barrier of a genetically engineered chimeric monoclonal antibody to the human insulin receptor. Pharm. Res. 17:266–274 (2000).
R. J. Boado, Y. Zhang, and W. M. Pardridge. Humanization of anti-human insulin receptor antibody for drug targeting across the human blood–brain barrier. Biotechnol. Bioeng. 96:381–391 (2007).
R. J. Boado, Y. Zhang, and W. M. Pardridge. Genetic engineering, expression, and activity of a fusion protein of a human neurotrophin and a molecular Trojan horse for delivery across the human blood–brain barrier. Biotechnol. Bioeng. in press (2007).
R. J. Boado, Y. Zhang, C. F. Xia, and W. M. Pardridge. Fusion antibody for Alzheimer’s disease with bidirectional transport across the blood–brain barrier and abeta fibril disaggregation. Bioconjug Chem. 18:447–455 (2007).
J. Norman, W. Denham, D. Denham, J. Yang, G. Carter, A. Abouhamze, C. L. Tannahill, S. L. MacKay, and L. L. Moldawer. Liposome-mediated, nonviral gene transfer induces a systemic inflammatory response which can exacerbate pre-existing inflammation. Gene Ther. 7:1425–1430 (2000).
D. Simberg, S. Weisman, Y. Talmon, A. Faerman, T. Shoshani, and Y. Barenholz. The role of organ vascularization and lipoplex-serum initial contact in intravenous murine lipofection. J. Biol. Chem. 278:39858–39865 (2003).
N. Shi and W. M. Pardridge. Noninvasive gene targeting to the brain. Proc. Natl. Acad. Sci. U. S. A. 97:7567–7572 (2000).
Y. Zhang, F. Schlachetzki, and W. M. Pardridge. Global non-viral gene transfer to the primate brain following intravenous administration. Mol. Ther. 7:11–18 (2003).
N. Shi, Y. Zhang, C. Zhu, R. J. Boado, and W. M. Pardridge. Brain-specific expression of an exogenous gene after i.v. administration. Proc. Natl. Acad. Sci. U. S. A. 98:12754–12759 (2001).
N. Shi, R. J. Boado, and W. M. Pardridge. Receptor-mediated gene targeting to tissues in vivo following intravenous administration of pegylated immunoliposomes. Pharm. Res. 18:1091–1095 (2001).
Y. Zhang, F. Schlachetzki, J. Y. Li, R. J. Boado, and W. M. Pardridge. Organ-specific gene expression in the rhesus monkey eye following intravenous non-viral gene transfer. Mol. Vis. 9:465–472 (2003).
D. J. Zack, J. Bennett, Y. Wang, C. Davenport, B. Klaunberg, J. Gearhart, and J. Nathans. Unusual topography of bovine rhodopsin promoter-lacZ fusion gene expression in transgenic mouse retinas. Neuron. 6:187–199 (1991).
C. Zhu, Y. Zhang, Y. F. Zhang, J. Yi Li, R. J. Boado, and W. M. Pardridge. Organ-specific expression of the lacZ gene controlled by the opsin promoter after intravenous gene administration in adult mice. J. Gene Med. 6:906–912 (2004).
Y. Zhang, F. Schlachetzki, Y. F. Zhang, R. J. Boado, and W. M. Pardridge. Normalization of striatal tyrosine hydroxylase and reversal of motor impairment in experimental parkinsonism with intravenous nonviral gene therapy and a brain-specific promoter. Hum. Gene Ther. 15:339–350 (2004).
Y. Zhang, C. Zhu, and W. M. Pardridge. Antisense gene therapy of brain cancer with an artificial virus gene delivery system. Mol. Ther. 6:67–72 (2002).
Y. Zhang, Y. F. Zhang, J. Bryant, A. Charles, R. J. Boado, and W. M. Pardridge. Intravenous RNA interference gene therapy targeting the human epidermal growth factor receptor prolongs survival in intracranial brain cancer. Clin. Cancer Res. 10:3667–3677 (2004).
W. M. Pardridge. Recent advances in blood–brain barrier transport. Annu. Rev. Pharmacol. Toxicol. 28:25–39 (1988).
Y. F. Zhang, R. J. Boado, and W. M. Pardridge. Absence of toxicity of chronic weekly intravenous gene therapy with pegylated immunoliposomes. Pharm. Res. 20:1779–1785 (2003).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Pardridge, W.M. Drug Targeting to the Brain. Pharm Res 24, 1733–1744 (2007). https://doi.org/10.1007/s11095-007-9324-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11095-007-9324-2