
Abstract
Purpose
Glabridin is a major active constituent of Glycyrrhiza glabra which is commonly used in the treatment of cardiovascular and central nervous system (CNS) diseases. Recently, we have found that glabridin is a substrate of P-glycoprotein (PgP/MDR1). This study aimed to investigate the role of PgP in glabridin penetration across the blood–brain barrier (BBB) using several in vitro and in vivo models.
Materials and Methods
Cultured primary rat brain microvascular endothelial cells (RBMVECs) were used in the uptake, efflux and transcellular transport studies. A rat bilateral in situ brain perfusion model was used to investigate the brain distribution of glabridin. The brain and tissue distribution of glabridin in rats with or without coadministered verapamil or quinidine were examined with correction for the tissue residual blood. In addition, the brain distribution of glabridin in mdr1a(−/−) mice was compared with the wild-type mice. Glabridin in various biological matrices was determined by a validated liquid chromatography mass spectrometric method.
Results
The uptake and efflux of glabridin in cultured RBMVECs were ATP-dependent and significantly altered in the presence of a PgP or multi-drug resistance protein (Mrp1/2) inhibitor (e.g. verapamil or MK-571). A polarized transport of glabridin was found in RBMVEC monolayers with facilitated efflux from the abluminal (BL) to luminal (AP) side. Addition of a PgP or Mrp1/2 inhibitor in both luminal and abluminal sides attenuated the polarized transport across RBMVECs. In a bilateral in situ brain perfusion model, the uptake of glabridin into the cerebrum increased from 0.42 ± 0.09% at 1 min to 9.27 ± 1.69% (ml/100 g tissue) at 30 min and was significantly greater than that for sucrose. Co-perfusion of a PgP or Mrp1/2 inhibitor significantly increased the brain distribution of glabridin by 33.6−142.9%. The rat brain levels of glabridin were only about 27% of plasma levels when corrected by tissue residual blood and it was increased to up to 44% when verapamil or quinidine was coadministered. The area under the brain concentration-time curve (AUC) of glabridin in mdr1a(−/−) mice was 6.0-fold higher than the wild-type mice.
Conclusions
These findings indicate that PgP limits the brain penetration of glabridin through the BBB and PgP may cause drug resistance to glabridin (licorice) therapy for CNS diseases and potential drug-glabridin interactions. However, further studies are needed to explore the role of other drug transporters (e.g. Mrp1-4) in restricting the brain penetration of glabridin.
Similar content being viewed by others
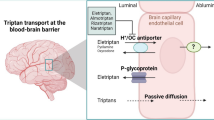

Avoid common mistakes on your manuscript.
INTRODUCTION
The blood-brain barrier (BBB) is formed by tight junctions that connect the brain endothelial cells, thus restricting entry of compounds from circulating blood to the central nervous system (CNS) via paracellular and transcellular routes (1–3). BBB acts as a physical and anatomical barrier notably due to the presence of tight junctions and a multitude of ATP-binding cassette (ABC) transporters such as P-glycoprotein (PgP/MDR1) and multi-drug resistance protein 1 and 2 (MRP1/2) (2,4–6). PgP is localized in the apical membrane of brain capillary endothelial cells and transports substrates toward the blood compartment. As such, the BBB contributes to brain homeostasis by protecting the brain from potentially harmful endogenous and exogenous substances (7). The ABC drug transporter members located in the BBB are increasingly recognized as important determinants of drug distribution to, and elimination from the CNS (2,8,9). Because the majority of drugs do not cross the BBB, CNS disorders remain difficult to treat. The drug delivery challenge posed by the BBB is compelling, particularly as the population ages and the incidence of neurodegenerative diseases such as stroke, Alzheimer’s disease, and Parkinson’s disease increase in prevalence. It has been reported that the BBB blocks delivery of more than 98% of CNS-acting drugs (10), and thus a successful brain penetration is a prerequisite for the design of chemical lead substances for CNS-acting drugs. PgP and other ABC transporters can limit the penetration of drugs into the brain and thus modulate the effectiveness and CNS toxicity of numerous drugs (8). To restrict CNS adverse effects, brain penetration properties are also important for the development of non-CNS acting drugs. Therefore, it is important to determine the BBB penetration of new drugs and existing drugs.
The widely used traditional Chinese herbal medicine, the root of G. glabra (licorice), is one of the most popular herbal medicines in the world due to its exceptional pharmacological properties recognized and claimed by the Traditional Chinese Medicine (11). Licorice has been used as an antidote, demulcent, antioxidant, immuno-modulator, expectorant, and a remedy for inflammation, as well as a flavoring and sweetening agent in Asian countries and less commonly in Western countries (11–15). Licorice extracts contain more than two dozens of ingredients, including glycyrrhizin, oleane triterpenoids, flavonoids and glucose. Glabridin (Fig. 1) is an isoflavan compound, one of the major active flavonoids in licorice. Isoflavans are a subclass of the flavonoid compounds, containing ring A fused to ring C, which is connected to ring B through carbon 3. Glabridin has been found to possess antioxidant, antimicrobial, antiatherosclerotic, hypolipidemic, anti-inflammatory, anti-obesity, hepato-protective, anti-tumor, cytotoxic, apoptosis-inducing, hypoglycemic, cardiovascular-protective, estrogen-like, antinephritic and radical-scavenging activities (16–23). Because of its similar structure to estradiol and similar lipophilicity (LogP ≈ 3.25), glabridin is also used as a phytoestrogen (19,24). Because of a broad spectrum of pharmacological activities, licorice is widely used as a single herbal preparation, or more frequently, in combination with other herbs to treat various diseases in respiratory, digestive, endocrine, cardiovascular, and central nervous system (25–27).

Chemical structures of glabridin and mefenamic acid (used as an internal standard).
Despite the wide use of licorice, data on the pharmacokinetic properties of glabridin are scant in animals and humans. Recently, we have reported that glabridin is a substrate for PgP with a K m of 6.6 μM (28). Following intravenous (i.v.) injection of glabridin at 5 mg/kg in rats, the drug was rapidly distributed in the body, with an elimination half life \( {\left( {t_{{1 \mathord{\left/ {\vphantom {1 2}} \right. \kern-\nulldelimiterspace} 2\beta }} } \right)} \) of 1.92 hr and a plasma clearance of 59.0 ml/min/kg (28). The oral bioavailability of glabridin is about 7.5%. In this study, we hypothesize that the brain penetration of glabridin is restricted by the BBB due to the contribution of PgP expressed in the brain capillary endothelial cells. To test this hypothesis, we examined the transport of glabridin across BBB using several in vitro and in vivo models. The uptake, efflux and transcellular transport of glabridin were examined in the primary rat brain microvascular endothelial cells (RBMVECs). A rat bilateral in situ brain perfusion model was used to investigate the brain distribution of glabridin. We also investigated the brain distribution of glabridin in rats and the effects of coadministered PgP inhibitors (verapamil or quinidine) on its brain distribution. In addition, the effect of PgP on brain penetration of glabridin was examined by comparing the brain distribution of glabridin in mdr1a(−/−) mice and the wild-type mice.
MATERIALS AND METHODS
Chemicals and Reagents
The purified glabridin [(R)-4-(3,4-Dihydro-8,8-dimethyl)-2H,8H-benzo[1,2-b:3,4-b′]dipyran-3yl)-1,3-benzenediol], extracted from the root of G. glabra, was purchased from the National Institute for the Control of Pharmaceutical and Biological Products (Beijing, China). Glabridin was dissolved in dimethyl sulfoxide (DMSO), and freshly prepared for all in vivo and in vitro experiments. The compound has a purity >99.0%, as determined by high performance liquid chromatography (HPLC) with ultraviolet detection. Its chemical structure was identified and confirmed by liquid chromatography mass spectrometry (LC-MS) and 1H-nuclear magnetic resonance (NMR) analysis. Fetal bovine serum, 0.05% trypsin-ethylenediaminetetraacetic acid (EDTA), penicillin-streptomycin, non-essential amino acids, sterilized Hank’s balanced salt solution (HBSS) at pH 7.4 containing 25 mM N-[2-hydroxyethyl] piperazine-N9-[4-butanesulfonic acid] (HEPES) and 25 mM glucose were obtained from Invitrogen (Carlsbad, CA). Trypan blue, propranolol, DMSO, celecoxib, sodium azide, 2, 4-dinitrophenol, verapamil hydrochloride, nifedipine, probenecid, and quinidine sulfate dehydrate were all purchased from Sigma-Aldrich Chemical Co. (St. Louis, MO). The leukotriene D4 receptor antagonist, 3-[[[3-[2-(7-chloro-2-quinolinyl)-(E)-ethenyl]phenyl][[3-(dimethylamino)-3-oxopropyl]thio]methyl]thio]propionic acid (MK-571 or L-660,711) was a gift from Dr Ford Hutchinson (Merck Frosst Canada, Inc., Kirkland, Quebec, Canada). Tissue culture plastics and 0.4 μm pore-size Transwell polycarbonate inserts with a 12 mm inner diameter were from Corning Co. (Corning, NY). Mouse monoclonal antibodies to MDR1 (C219) and to MRP1 were purchased from Abcam Co. (Cambridge, UK). The secondary antibody used was anti-mouse IgG from rat (GE Healthcare, Buckinghamshire, UK). The water used was purified by a Milli-Q purification system (Millipore, Bedford, MA). All other chemicals and reagents were of analytical or HPLC grade as appropriate.
Animals
Male healthy Sprague-Dawley rats (200−260 g) were kept in a room under controlled temperature (22 ± 1°C) and automatic day–night rhythm (12 h-cycle) and housed on wire-bottom cages with paper underneath. The ethical approval of this study was obtained from the Institutional Ethics Committee. The animal experiments were performed in accordance with the Guide for the Care and Use of Laboratory Animals as adopted and promulgated by the National Institutes of Health of the USA (NIH publication No. 85-23, 1985).
Cell Culture
The cryopreserved primary rat brain microvascular endothelial cells (RBMVECs) (catalog no. R840-05) were purchased from Cell Applications Inc. (San Diego, CA). This is a pure endothelial cell population without contamination by pericytes (29). RBMVECs were cultured in a special growth medium (catalog no. R811-500 provided by Cell Applications Inc., Toyoba, Japan) fully supplemented with essential and nonessential amino acids, vitamins, inorganic salts, organic compounds, trace elements, 100 U/ml penicillin and gentamicin, 10% fetal bovine serum, and growth factors including endothelial cell growth factor (1 ng/ml), transferrin (5 μg/ml), and insulin (5 μg/ml) (Cell Applications, Inc., Toyoba, Japan) at 37°C in a 5% CO2, 95% air atmosphere, and were maintained by serial passages in 75-cm plastic culture flasks. These cells can be cultured and propagated to passage 5 and beyond without losing their morphological and phenotypic properties. Cells were used in passages 3–5 only in this study.
Cytotoxicity Assay
The cytotoxic effect of glabridin on RBMVECs examined was determined using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazonium bromide (MTT) assay as described (30). The drug was dissolved in DMSO and then diluted in culture medium and added to the cultures 24 h after cell seeding. Cells were exposed to the drug at different concentrations (0.1−100 μM) in culture medium for 48 h, after which 0.05 mg MTT was added to each well, and the plates were further incubated for 4 h. Thereafter, supernatants were removed and the purple precipitate was dissolved in 100 μl DMSO. The absorbance of formazan, a metabolite of MTT, was measured at a wavelength of 595 nm using a microplate reader (Tecan Instrument Inc., Research Triangle Park, NC).
Uptake and Efflux Assay of Glabridin by RBMVECs
The uptake of glabridin by RBMVECs was examined in confluent cell cultures grown on 60-mm plastic culture dishes (Corning Co., Corning, NY) as previously described (31,32). Briefly, exponentially growing cells were exposed to 0−100 μM glabridin over 120 min at 37°C. The medium was aspirated off at indicated times, and the dishes were rapidly rinsed five times with 50 ml of ice-cold phosphate buffered saline (PBS). HPLC analysis of the final washes ensured that they contained no residual glabridin. After five washes using ice-cold PBS, the cells were harvested and each cell pellet was suspended in 200 μl extraction solution (acetonitrile: methanol = 1:1, v/v, with 0.01N HCl) with the addition of 10 μl 1.0 mg/ml mefenamic acid (used as an internal standard). Subsequently, the mixture was sonicated, vortexed and centrifuged. The supernatant was dried under nitrogen gas using a Savant Speedvac (TeleChem International Inc., Sunnyvale, CA) and the residues reconstituted with the mobile phase (50 μl). An aliquot (10−20 μl) was injected into LC-MS for glabridin concentration determination. The cellular uptake of glabridin was corrected by subtraction of extracellular adsorption values of [14C]-sucrose.
Control uptake assays were also performed using confluent monolayers of RBMVECs 3−5 days after seeding. Cells were washed twice using warm HBSS with 25 mM HEPES and incubated over 120 min with 0.3 μCi/well of the extracellular marker [14C]-sucrose (565 mCi/mmol, from GE Healthcare, Buckinghamshire, UK) together with [3H]-propranolol (GE Healthcare, Buckinghamshire, UK). At specified time points, cells were washed three times with warm HBSS. The cell monolayers were solubilized in 0.8% triton-X for 20 min and the solutions were transferred to scintillation vials. The radioactivity in cellular lysates was determined by an LC-6000 liquid scintillation counter (Beckman Instruments, Fullerton, CA). In addition, the uptake of daunomycin (a known PgP substrate) was performed with or without 100 μM verapamil as described above. Preliminary experiments showed that daunomycin uptake was at equilibrium after 60−90 min of incubation with RBMVECs. Therefore, cells were incubated for 30 min with 1.0 μM daunomycin (0.3 μCi/well [3H]-daunomycin (Perkin Elmer, Boston, MA) and unlabelled daunomycin with or without verapamil at 100 μM. Cells were then washed three times with warm HBSS and further processed as described above.
The efflux of glabridin from RBMVECs was examined as described previously (32). Briefly, glabridin (0.1−100 μM) was added to confluent cell cultures grown on 60-mm plastic culture dishes (Corning Co., Corning, NY) after three to five washes of the cells with warm HBSS and incubated over 120 min. At pre-determined time points, an aliquot (200 μl) of culture medium was collected to monitor the drug concentrations in the culture medium. Three volumes of methanol containing the internal standard were added to the sample to precipitate protein and to extract glabridin. In addition, the intracellular level of glabridin was determined over 120 min when glabridin effluxed from RBMVEC cells pre-treated with glabridin at a loading concentration of 1.0 μM and incubated for 120 min to achieve maximum drug uptake. The cells were then washed five times with ice-cold PBS, harvested and lysed as describe above in the uptake assay for glabridin. The concentrations of glabridin in the culture medium and cellular lysates were determined by LC-MS. The efflux kinetic parameters and half-life of drug efflux were calculated using standard formulae with WinNonlin program (Pharsight Co., Moutain View, CA). In addition, a parallel control experiment was conducted using [3H]-vinblastine (GE Healthcare, Buckinghamshire, UK) as a probe substrate, and the radioactivity was determined by an LC-6000 liquid scintillation counter (Beckman Instruments, Fullerton, CA).
The effects of various ATP inhibitors (sodium azide at 10 mM and 2, 4-dinitrophenol at 5 mM), PgP and Mrps inhibitors, including verapamil (100 μM), nifedipine (100 μM), quinidine (50 μM, all PgP inhibitors), MK-571 (a MRP1/2 inhibitor, 100 μM), probenecid (a MRP1/2 inhibitor, 200 μM), and celecoxib (a MRP4 inhibitor, 100 μM), on glabridin cellular uptake and efflux were also investigated in RBMEVEC cells when two substrate concentrations (0.1 and 1.0 μM, for the efflux inhibition assays, these two concentrations were the initial loading ones for drug uptake by the cells) were studied. Because these two concentrations are the low and high values of free blood concentrations of glabridin in rats following a commonly used dosage (5 mg/kg) (28), both have been included in the inhibition studies. All these compounds are known inhibitors for rat P-gp or Mrp1-4 (33). All the inhibitors were freshly prepared by dissolving in DMSO and diluted by HBSS with 25 mM HEPES. The final concentration of DMSO was 0.2% (v/v). These inhibitors at indicated concentrations showed little cytotoxicity (<6.0%) to the cells tested when incubated for 4 h. For uptake inhibition assay, all inhibitors were pre-incubated with cells for 2 hr and co-incubated further for 30 min in the presence of glabridin (33). Thereafter, cells were washed five times with cold PBS. The cells were then harvested, lysed by sonication and extracted using ice-cold acetonitrile: methanol mixture (1:1, v/v, with 0.01N HCl) as described above. The supernatant was dried under nitrogen gas and reconstituted with the mobile phase and an aliquot (10−20 μl) was injected into LC-MS for the determination of glabridin concentrations. For the efflux inhibition assay, glabridin was loaded at 0.1 or 1.0 μM and incubated for 120 min to achieve maximum drug uptake. Thereafter, after five washes with 4°C PBS of the cells to eliminate the extracellular drugs and stop any transporter-mediated drug flux, cells were incubated in culture medium for 30 min at 37°C with the inhibitor. All uptake and efflux assays in the absence and presence of inhibitor were studied in at least three independent experiments. DMSO (0.2% in the culture buffer), used to dissolve the all inhibitors, did not change the accumulation or efflux of glabridin in RBMVECs (data not shown).
Transport of Glabridin in Rat Brain Microvascular Endothelial Monolayers
RBMVECs were seeded at a density of 2.5 × 105/well onto rat tail collagen-precoated (10 μg/cm2) polycarbonate membrane Transwell inserts (Corning Co., Corning, NY) in a 12-well plate as described previously (34). The culture medium (0.5 ml in the luminal donor chamber of the insert and 1.5 ml in the abluminal well chamber) was replaced every 24 h. The effective transepithelial electric resistance (TEER) of the monolayers was examined routinely before and after the experiment using the Millicell-ERS apparatus (Millipore Co., Billerica, MA). Transport experiments of glabridin in RBMVEC monolayers were performed when the cells had reached integrated confluence 6–10 days after seeding. Cells were rinsed twice with HBSS buffered with 25 mM HEPES (pH 7.4) before transport study. A pH of 7.4 for the transport study was chosen as it was close to the brain pH value and this pH resulted in maximum luminal (AP) to abluminal (BL) and BL-AP transport of glabridin. After two washes with warm HBSS, the plates were incubated at 37°C for 15 min, and the TEER was monitored and inserts distributed evenly between treatments based on the measured TEER values. Glabridin (0.1−100 μM) was loaded to the AP or BL side, and an aliquot (50 μl) of sample was collected from the receiving side at predetermined times (5, 10, 15, 20, 25, 30, 35, 40, 45, 50, 55 and 60 min) over 60 min. After each sampling, 50 μl of HBSS with 25 mM HEPES were added to the receiving side to maintain a constant volume. Glabridin solutions were freshly prepared by dissolving in DMSO, and then diluted by the transport buffer HBSS. The final concentration of DMSO in incubations was 0.2% (v/v). All experiments were conducted in triplicate to nine times.
The effects of pH, Na+, temperature and ATP on glabridin transport across RBMVEC monolayers were also investigated. The effect of AP or BL pH (5.5−7.4) on the AP to BL and BL to AP fluxes of glabridin at 0.1 and 1.0 μM was examined at pH 7.4 for the receiving side. The pH was altered by substituting appropriate amounts of HEPES in the incubation medium by equimolar (25 mM) 2-[N-morpholino]ethanesulfonic acid. In experiments to investigate the effect of Na+ on the fluxes of glabridin at 0.1 and 1.0 μM across the RBMVEC monolayers, the sodium chloride in the HBSS was replaced by equimolar amounts (140 mM) of potassium chloride. The permeability of glabridin from the AP to BL and BL to AP side was measured after incubation for 30 min at 4 or 37°C. To determine energy dependency of glabridin fluxes in RBMVEC mohnolayers, the transport medium depleted in glucose was used in both sides of the cell monolayers. Sodium azide (10 mM) or 2, 4-dinitrophenol (5 mM) (both ATPase inhibitors) was added to both AP and BL sides and the monolayers were incubated for 30 min at 37°C. In addition, [3H]-inulin (paracellular marker) and [3H]-propranolol (transcellular marker) were used as model compounds to confirm monolayer integrity.
The apparent permeability coefficient (P app) in RBMVEC cellular monolayers is expressed in cm/sec, and calculated as following equation:
where ΔQ/Δt is the permeability rate (μg/sec); A is the surface area of the membrane (cm2); and C0 is the initial drug concentration in the donor chamber (μg/ml). Samples from 30 min point were used for P app calculations as at this time steady state has achieved.
Bilateral in situ Brain Perfusion in Rats
Adult male Sprague-Dawley rat brain perfusion (n = 8) was performed using the method reported previously (35). Briefly, the common carotid arteries were catheterized and connected to a perfusion system and the jugular veins were sectioned at the start of perfusion. Glabridin was introduced into the perfusion medium at 0.5 ml/min and achieved a final concentration of 0.5 μM. The rat was decapitated and its brain was removed at indicated time points, cerebrospinal fluid (CSF) was collected, and the cerebral hemispheres and cerebellum were weighed, rinsed, homogenized and centrifuged. The samples were then processed as above in the uptake, efflux and transport studies. In a separate series of single-time point (10 min) experiments, the brain uptake of glabridin at 0.5 μM was examined in the presence of verapamil (100 μM), nifedipine (100 μM), or quinidine (50 μM), MK-571 (100 μM), or probenecid (200 μM). In vivo BBB permeability (K in, μl/min/g of brain) was calculated using Eq. 2 by single-time point analysis as described previously (36) after correcting for the remaining intravascular glabridin, estimated from the apparent brain uptake of [3H]-sucrose
where T is the length of perfusion in minutes, and R brain is the amount of glabridin in the brain over that in the perfusate.
Brain and Tissue Distribution Study of Glabridin in Rats
Healthy male Sprague Dawley rats (200−250 g, n = 6 per group per time point) were treated orally by gavage with glabridin dissolved in DMSO (final concentration 0.2%, v/v) at 5 mg/kg. At pre-determined time points (15 min, 30 min, 1, 2, 4, 6, 8, 12 and 24 h), the rats were sacrificed under ether anesthesia, and blood, the brain, liver, heart, spleen, lung and kidney were immediately collected for drug analysis. Plasma samples were immediately separated by centrifugation at 3,000 × g for 10 min at 4°C. Brain and other tissues were quickly excised, rinsed with ice-cold PBS, weighed, and homogenized with 1–2 volumes of 4°C PBS using a Heidolph homogenizer (Heidolph Instruments GmbH & Co. KG, Germany). Samples were kept frozen at −80°C until analysis. Plasma, the brain and other tissue samples were extracted with three volumes of ethanol containing the internal standard mefenamic acid and then centrifuged at 10,000 × g for 10 min to obtain the supernatants. The samples were evaporated to dryness under nitrogen gas using a Savant SPD111V SpeedVac concentrator (TeleChem International, Inc., Sunnyvale, CA) and reconstituted with 50 μl mobile phase. An aliquot (10−20 μl) was injected onto the LC-MS system for glabridin concentration determination. To determine the tissue distribution extent compared to the levels in plasma (partition coefficient, R i), we calculated the R i value using the following equation:
The levels in tissue and plasma mainly refer to the maximum concentration (Cmax) and area under the concentration-time curve (AUC). Since the total drug concentration in a tissue is primarily a result of the drug found in the capillaries and thus the R i value is an overestimate of the real distribution coefficient, it is necessary to correct for the presence of drug in the residual blood of the tissue (\( R^{\prime }_{{\text{i}}} \)) using the following equation (37):
where (V F)B is the volume fraction of residual blood in individual tissues in rats and mice (rats and mice have similar (V F)B values: liver: 0.06; heart: 0.13; kidney: 0.10; lung: 0.13; brain: 0.02; and spleen: 0.25) (37,38).
Effects of Coadministered Verapamil or Quinidine on the Brain and Tissue Distribution of Glabridin in Rats
In separate kinetic experiments, we examined the effects of coadministered verapamil at 50 or 100 mg/kg and quinidine at 10 or 50 mg/kg dissolved in water on the brain and tissue distribution of glabridin of glabridin in healthy male Sprague Dawley rats. The dose of verapamil and quinidine selected was approximately the maximum tolerated dose, as assessed in pilot experiments in male Sprague Dawley rats. Rats were randomized to receive the following different treatment (n = 6 per time point): glabridin at 5 mg/kg by gavage plus water (0.3 ml, control vehicle); glabridin at 5 mg/kg by gavage in combination with verapamil at 50 or 100 mg/kg by oral gavage dissolved in water; and glabridin at 5 mg/kg by gavage in combination with quinidine at 10 or 50 mg/kg by oral gavage dissolved in water. The inhibitor was administered 2 h before glabridin dosing. Rats were sacrificed at pre-determined time points after glabridin dosing and the blood, brain, liver, heart, lung, spleen and kidney tissues were collected processed as described above. The concentrations of glabridin in plasma, the brain and other tissues were determined by LC-MS. To examine the specificity of the effect of verapamil and quinidine, the brain uptake and the R i and \( R^{\prime }_{{\text{i}}} \) (concentration of brain:plasma) values of the paracellular transport marker, [3H]-mannitol, at 5 and 15 min after i.v. administration (1 μCi/rat) was also determined according to the method described above.
Brain and Tissue Distribution of Glabridin in mdr1a(−/−) Mice
FVB/NJ (20−35 g) and mdr1a gene-deficient mice (25−30 g) were purchased from Jackson Laboratories (Bar Harbor, ME) and Taconic Farms, Inc. (Germantown, NY), respectively. The mice were treated with oral glabridin at 5 mg/kg by gavage (n = 4 per time points). At pre-determined time points, the mice were sacrificed by neck dislocation. Blood, the brain, heart, liver, lung, spleen and kidney were immediately collected, rinsed with saline, and weighed. Plasma was obtained by centrifugation at 5,000 × g for 8 min at 4°C and all tissues were process as described above. The concentration of glabridin in plasma and tissues was determined by an LC-MS method.
LC-MS and HPLC Analysis
The concentrations of glabridin in rat and mouse plasma, tissue homogenates, transport medium in cellular monolayers and cellular lysates were determined by an LC-MS system equipped with an Agilent 1100 LC connected to an Applied Biosystems Q-Trap 4000 mass spectrometer through an electrospray ionization source. Chromatographic separation was achieved using a C18 Hyperclon ODS column (200 mm × 4.6 mm i.d.) (Phenomenex, Torrance, CA) preceded by a Phenomenex C18 guard cartridge at room temperature (22°C). The mobile phase was methanol/0.1% (v/v) formic acid (85:15, v/v) at a flow rate of 0.2 ml/min. The injection volume was 10−20 μl of a sample kept in an autosampler set at 10°C. Air was used as heater gas set at a temperature of 600°C and with a setting of 70 psi, and nebulizing gas with a setting of 40 psi. The capillary temperature was at 450°C and the spray voltage was set at 4,000 V. The product ion was recorded using multiple reaction monitoring negative ion detection mode. The monitor ion and collision energy were m/z 323.1→201.3 and −32 eV for glabridin and m/z 240.1→196.1 and −24 eV for the internal standard. The lower limit of quantitation of glabridin was 0.025–0.05 ng/ml in rat and mouse plasma, tissue homogenates and other matrices used in this study. Glabridin was recovered >96.0%, and was stable when kept at 10°C for 72 h, at −20°C for 4 months, and after five to eight freeze–thaw cycles.
Western Blotting Analysis
The expression levels of PgP and Mrp1 in RBMVEC cells were monitored every passage by Western blotting analysis as described previously (39). The cells were homogenized in a buffer containing 10 mM Tris-Cl at pH 7.4, 10 mM KCl, 5 mM EDTA, 1.5 mM MgCl2, 0.5% sodium dodecyl sulphate (SDS, w/v) and a cocktail of protease inhibitors (#P-8340, Sigma-Aldrich Chemical Co., St. Louis, MO). Cellular lysates were centrifuged at 12,000 × g at 4°C for 10 min. Total protein content in the extracts was determined using the Lowry method (40). Ten micrograms of protein were separated by 7.5% SDS-polyacrylamide gel [with 0.1% SDS (w/v)] electrophoresis, and transferred onto nitrocellulose membranes (GE Healthcare, Buckinghamshire, UK). The membranes were blotted with antibody against MDR1 or MRP1 (Abcam Co., Cambridge, UK). To assure equivalent protein loading, the membranes were also incubated with mouse anti-β-actin monoclonal antibodies (1:2,000 in Tris-buffered saline/Tween), and subsequently with a corresponding horseradish peroxidase-conjugated second antibody IgG (GE Healthcare, Buckinghamshire, UK) and developed using Chemiluminescence Reagent Plus (Biorad, Hercules, CA). β-Actin was used as the control for loading quantity of the samples. The densitometric analysis was carried out using CHEMDOC image scanner (Biorad, Hercules, CA).
Pharmacokinetic Calculation
The plasma/tissue concentration-time curves of glabridin were obtained by plotting the mean plasma/tissue concentrations of glabridin versus time on a semi-logarithmic scale. Pharmacokinetics parameters were calculated by standard model-independent pharmacokinetic formulae using WinNonlin program (Pharsight Co., Mountain View, CA). The \( t_{{1 \mathord{\left/ {\vphantom {1 2}} \right. \kern-\nulldelimiterspace} 2\beta }} \) value was calculated as 0.693/β, where β is the elimination rate constant calculated from the terminal linear portion of the log plasma concentration-time curve. The total areas under plasma concentration-time curve from time zero to the last quantifiable time point (AUC0–t) and from time zero to infinity \( {\left( {{\text{AUC}}_{{0 - \infty }} } \right)} \) were calculated using the log trapezoidal rule. The \( {\left( {{\text{AUC}}_{{0 \to \infty }} } \right)} \) was calculated as following equation:
where Ct is the last measurable plasma concentration. The maximum plasma concentration (Cmax) for glabridin was obtained by visual inspection of the plasma concentration-time curve, whereas the initial drug concentration (the extrapolated concentration at zero time) of the drug following i.v injection was calculated by back extrapolation of the plasma concentration-time curve to y-axis.
Data Analysis
Data are presented as mean ± SD. The initial statistical analysis to evaluate the differences in the mean values among different groups was carried out by a one-way analysis of variance (ANOVA) followed with a post-hoc test (Dunnett’s multiple comparison test). The Student’s t-test was conducted for the between-group comparisons with a significance level of p < 0.05. Several models to describe the kinetics of the transport of glabridin (single and two binding sites with and without a passive component, substrate inhibition, and the sigmoid models) were fitted using the Prism 3.0 program (Graphpad Software, San Diego, CA).
where v is the rate of glabridin uptake, efflux or transcellular transport; Vmax is the maximum velocity; K m is Michaelis-Menten constant; [S] is the substrate concentration; PD, the passive diffusion component; K is, the substrate inhibition constant; h′, the Hill coefficient for cooperative substrate binding; and subscripts 1 and 2 represent the first and the second type of binding sites. The choice of model was confirmed by F-test and comparison of Akaike’s information criterion values (41).
RESULTS
Cytotoxicity and Metabolism of Glabridin in Primary Rat Brain Microvascular Endothelial Cells
Glabridin at 0.1−100 μM did not show significant cytotoxicity (<10%) to RBMVEC cells when incubated for up to 48 h as determined by the MTT assay. No detectable oxidative metabolites and conjugates were observed when glabridin at 0.1−100 μM was incubated with primary rat brain microvascular endothelial cells for 2−48 h as determined by HPLC and LC-MS analysis.
Expression of PgP and Mrp1 in Rat Brain Microvascular Endothelial Cells
Western blot analysis using the PgP monoclonal antibody C219 and antibody against Mrp1 detected a marked single band at approximately 170 ∼ 180 kDa and a weak band (about 1/10 density of PgP) at about 190 kDa in RBMVEC cells (Fig. 2). The stable overexpression of PgP in RBMVECs over passages 2 to 7 was maintained. The marked expression of PgP and minor to moderate expression of Mrp1 detected by Western blotting analysis in RBMVECs indicates that this cell culture model exhibits important functional features of in vitro BBB model.

Protein expression of rat P-glycoprotein and multidrug resistance associated protein 1 (Mrp1) in primary rat brain microvascular endothelial cells at passages 2−7, as determined by the Western blotting analysis. Ten micrograms of the cellular protein were separated by 7.5% SDS-polyacrylamide gel electrophoresis, and transferred onto nitrocellulose membranes. The membranes were blotted with antibody against human MDR1/PgP or MRP1 which can cross-react with rat PgP or Mrp1, respectively. β-Actin was used as the control for loading quantity of the protein samples.
Intracellular Accumulation and Efflux of Glabridin in Rat Brain Microvascular Endothelial Cells
The effects of incubation time and substrate concentration intracellular accumulation of glabridin in RBMVECs were examined (Fig. 3). The accumulation amounts of glabridin in RBMVECs were linear up to 30 min with a maximum uptake rate (12.5 ± 1.3 ng/mg cellular protein), and gradually declined thereafter until 120 min. The pattern of glabridin uptake kinetics by RBMVECs is remarkably different from those compounds studied in brain endothelial cells and other types of cells where there is a typical stead-state plateau after achieving the maximum uptake (42). This could be due to the involvement of multiple transporters with differential affinity to glabridin in its uptake by RBMVECs and a rapid intracellular distribution of the drug molecules by binding to its target organelles and proteins. The uptake of glabridin by RBMVECs was about 0.5−7.5% of the loading dose. The intracellular accumulation of glabridin in RBMVEC cells also increased with increasing substrate concentrations, and followed Michaelis-Menten kinetics with one-binding site model without a passive diffusion component being the best fit. The estimated K m and Vmax for glabridin uptake by RBMVECs was 10.87 ± 1.46 μM and 4.81 ± 0.20 ng/min/mg cellular protein. The best fit of one binding-site model without incorporation of a passive diffusion component for the accumulation of glabridin in RBMVECs suggested the involvement of one predominant transporter or multiple transporters with similar affinity to the substrate, but passive diffusion may also play a minor role in its intracellular accumulation in RBMVECs. The exclusion of a passive diffusion component in the best fit model could be due to our limited data points and a minor role of passive diffusion in the transport of glabridin. Uptake of the probe markers sucrose and propranolol into RBMVECs was determined at various time points up to 120 min. Propranolol penetrated into rat brain capillary endothelial cells to a low degree (3.2−5.1% of added dose), and diffusion of sucrose into the cells was very minimal (0.04−0.06% of added dose). The uptake of sucrose and propranolol did not increase with incubation time in RBMVECs. In the additional experiment using daunomycin as a model PgP substrate, its uptake by RBMVECs was significantly increased by 65.3 ± 8.3% when co-incubated with 100 μM verapamil (data not shown). We did not estimate the non-specific binding of glabridin to RBMVEC cells because the radio-labeled glabridin is not available and the LC/MS method for glabridin determination has a limited sensitivity when measuring very low levels of glabridin.

Effects of incubation time (a and c, substrate concentration: 1.0 μM) and substrate concentration (b and d, incubation time: 30 min) on the intracellular accumulation and efflux of glabridin in primary rat brain microvascular endothelial cells cultured in HBSS. The curves in plots b and d represent the best fit of one binding-site model without a passive diffusion component. Plot e shows the efflux profile of glabridin from RBMVEC cells when preloaded at 1.0 μM and incubated for 120 min to achieve maximum drug uptake. Cells were washed with cold PBS and then incubated at 37°C over 120 min. The intracellular concentrations of glabridin were monitored by LC-MS. The data are the mean ± SD of at least 6–9 determinations.
The efflux of glabridin was measured at 0.1−100 μM when incubated for up to 120 min in RBMVEC cells. The effects of substrate concentration and incubation time on the efflux rate of glabridin (appearance rate in culture medium) are shown in Fig. 3. The efflux of glabridin from RBMVECs was dependent on the incubation time, linear up to 60 min, and then slightly declined thereafter up to 120 min. Like its uptake by RBMVECs, the pattern of glabridin efflux kinetics from RBMVECs is remarkably different from those compounds studied in brain endothelial cells and other types of cells where there is a typical stead-state plateau after achieving the maximum efflux (42). The reason for this is unknown, but involvement of multiple transporters with differential affinity to glabridin in its efflux from RBMVECs and other factors are implied. The efflux of glabridin from RBMVECs also increased depending on the substrate concentration and followed Michaelis-Menten kinetics with one-binding site model without a passive diffusion component being the best fit. The exclusion of a passive diffusion component in the best fit model could be due to our limited data points and a minor role of passive diffusion in the efflux of glabridin from RBMVECs. The estimated K m and Vmax for glabridin efflux from RBMVEC cells was 15.48 ± 2.94 μM and 1.32 ± 0.08 pg/min/mg cellular protein. The efflux of the model substrate vinblastine was also dependent on the substrate concentration and incubation time, and its cellular efflux was also characterized by a bi-exponential kinetics with the half-lives were 11.2 ± 1.4 min and 2.18 ± 0.31 h, respectively (data not shown). The K m1 and K m2 for vinblastine were 1.25 ± 0.3 and 23.2 ± 5.3 μM, respectively. The Vmax1 and Vmax2 for vinblastine uptake by RBMVECs were 7.37 ± 1.16 and 3.11 ± 0.21 ng/min/mg cellular protein, respectively. These results are comparable to those reported in the literature (43,44). We also monitored the intracellular concentration of glabridin at 1.0 μM upon incubation over 120 min (Fig. 3). It appeared that the efflux of glabridin from RBMVECs was characterized by a bi-exponential kinetics with half-life values of 2.55 ± 0.37 and 14.62 ± 1.53 min, respectively, indicating a rapid exit of the drug from RBMVECs in the initial 5 min and followed by a relatively rapid exit from RBMVECs. The two-phase exponential efflux of glabridin from RBMVECs suggested the involvement of multiple transporters with differential affinity to the substrate, but passive diffusion may also play a minor role in its efflux. Another explanation for the biphasic kinetics is a PgP-mediated initial rapid efflux phase followed by a relatively slower passive efflux.
The effects of 2-h pre-incubation and 30-min co-incubation of various ATP and transporter inhibitors, including sodium azide, 2,4-dinitrophenol, verapamil, nifedipine, quinidine, probenecid, MK-571, and celecoxib on the uptake of glabridin by RBMVEC cells are shown in Fig. 4. In the presence of sodium azide or 2,4-dinitrophenol, the uptake of glabridin at 0.1 and 1.0 μM was significantly (p < 0.05) increased by 52.1 ± 5.8% and 57.3 ± 6.5%; and 62.8 ± 7.8% and 64.5 ± 7.6%, respectively. When verapamil, nifedipine (both at 100 μM), or quinidine (50 μM) was pre-incubated for 2 h and co-incubated further for 30 min with RBMVEC cells, the uptake of glabridin at 0.1 μM was significantly (p < 0.05 or 0.01) increased by 78.3 ± 8.9%, 87.2 ± 9.4%, and 91.4 ± 11.2%, respectively. Similar results were found when the concentration of glabridin was increased to 1.0 μM. MK-571 (100 μM) and probenecid (200 μM) also significantly increased glabridin uptake in RBMVECs (p < 0.05). However, celecoxib (100 μM) did not significantly alter the uptake of glabridin. These findings demonstrated that the uptake of glabridin by RBMVECs was ATP-dependent, and pre-incubation with PgP or Mrp1/2 inhibitors such as verapamil and MK-571 significantly increased the uptake of glabridin in RBMVECs. These data provided further evidence indicating that glabridin is a substrate for PgP and possibly for Mrp1/2, but not for Mrp4.

Effects of sodium azide, 2, 4-dinitrophenol, verapamil, nifedipine, quinidine, probenecid, MK-571 and celecoxib on the uptake (a and b) and efflux (c and d) of glabridin at 0.1 (a and c) and 1.0 (b and d) μM in primary rat brain microvascular endothelial cells. For uptake inhibition assays, the inhibitors added to both luminal (AP0) and abluminal (BL) sides were pre-incubated with the cells for 2 h, and co-incubated further for 30 min in the presence of glabridin. For efflux inhibition assays, glabridin at 0.1 or 1.0 μM was added to the cell cultures and incubated for 120 min to achieve maximum drug uptake. Cells were washed with ice-cold PBS and then incubated in the presence of an inhibitor for 30 min. The data are the mean ± SD of at least 6–9 determinations. *p < 0.05; **p < 0.01.
The effects of 2-h pre-incubation and 30-min co-incubation of various ATP and transporter inhibitors, including sodium azide, 2,4-dinitrophenol, verapamil, nifedipine, quinidine, probenecid, MK-571, and celecoxib on the efflux of glabridin (monitored by measuring appearance of substrate in culture medium) in RBMVECs are shown in Fig. 4. In the presence of sodium azide or 2,4-dinitrophenol, the efflux of glabridin at 0.1 and 1.0 μM from RBMVECs was significantly (p < 0.05) decreased by 45.4 ± 5.2% and 47.3 ± 5.4%; and 44.5 ± 5.8% and 46.1 ± 5.7%, respectively. When verapamil, nifedipine (both at 100 μM), or quinidine (50 μM), was pre-incubated for 2 h and co-incubated further for 30 min with the cells, the efflux of glabridin from RBMVECs when the initial loading concentration of glabridin was 0.1 μM was significantly (p < 0.05 or 0.01) decreased by 50.7 ± 6.1%, 51.8 ± 6.3% and 55.0 ± 6.2%, respectively. MK-571 (100 μM) and probenecid (200 μM) also significantly decreased glabridin efflux in RBMVECs (p < 0.05). In contrast, celecoxib (100 μM) did not significantly affect glabridin efflux. Similar results were observed when the initial glabridin loading concentration was increased to 1.0 μM. These findings demonstrated that pre-incubation of the cells with a PgP or Mrp1/2 inhibitor such as verapamil, or MK-571, significantly decreased glabridin efflux from RBMVECs.
Transport of Glabridin in Rat Brain Microvascular Endothelial Monolayers
Inulin was used as a functional indicator of the integrity of the tight junctions in RBMVEC monolayers, and an endothelial permeability coefficient for inulin P app higher than 6.67 × 10−6 cm/s was indicative of a leaky BBB based on literature data (45,46). In our in vitro rat BBB model, the P app of inulin across the RBMVEC monolayer from the AP to BL direction was 2.25 ± 0.23 × 10−6 cm/sec under control conditions. For propranolol, the P app was 3.5 ± 0.4 × 10−6 cm/s. Moreover, the initial integrity of RBMVEC monolayers before each experiment was monitored and confirmed by measuring the TEER values; the mean value was 643.8 ± 62.3 Ω•cm2. All these data indicate the integrity of the RBMVEC monolayers used in our transport experiments. This is largely consistent with the inulin permeability data. However, RBMVECs appeared to form tight junctions at a moderate level, because its ability to differentiate high permeable compounds (e.g. inulin) from low permeable compounds such as propranolol. Coculture with astrocytes might be a useful alternative approach to improve the predictability of in vitro brain endothelial cell model, but coculture with astrocytes may complicate the binding, uptake, and transcellular transport of drugs and cell–cell interaction may occur that may influence drug transport behavior in RBMVECs.
After incubation of glabridin at 0.1–100 μM loaded at the AP or BL side, the sample was collected from the receiving side for LC-MS analysis. No detectable metabolites were observed when glabridin was loaded on luminal or abluminal side at all concentrations over 60 min. The time course and concentration effect of glabridin flux from the AP to BL or BL to AP side has been examined and the results are shown in Fig. 5. After a luminal or abluminal loading, glabridin appeared on the receiving side by 5 min. The flux rate (nmol/min/cm2) of glabridin from the AP to BL side was largely proportional to glabridin concentrations over 0.1−100 μM and was linear up to 60 min of incubation time. The transport rate of glabridin across RBMVEC monolayers from the BL to AP side was linear up to 60 min of incubation time. The transport rate of glabridin across RBMVEC monolayers from the BL to AP side was significantly (2- to 3-fold) higher than that from the AP to BL side. The P app of glabridin from the BL to AP side (1.28−5.93 × 10−6 cm/s) was about 2- to 3-fold higher than those from the AP to BL side (0.51−2.26 × 10−6 cm/s) with a marked decrease in P app values for both directional flux at increasing glabridin concentration (Fig. 5). The BL to AP efflux rate of glabridin increased with increasing glabridin concentrations over 0.1−100 μM but appeared saturable when glabridin concentration was ≥25 μM as indicated by a non-proportional increase in the efflux (Fig. 6). Consistently, there was a significant decrease in P app values for the BL to AP fluxes at glabridin concentrations ≥25 μM (p < 0.01).

Transport of glabridin across rat brain microvascular endothelial monolayers. Plot a illustrates the effect of concentration of glabridin (0.1−100 μM) on the permeability (P app) of glabridin from the luminal (AP) to abluminal (BL) and BL to AP side when incubated for 30 min. Plot b shows the effect of incubation time (0−60 min) on the permeability (P app) of glabridin at 1.0 μM in the AP to BL and BL-AP directions. Plots c and d show the effect of various ATP and ABC transporter inhibitors, on the AP to BL and BL to AP transport of glabridin at 0.1 μM (c) or 1.0 μM (d) in RBMVEC monolayers. The inhibitors added to both AP and BL sides were pre-incubated with the cells for 2 h. Glabridin (0.1 or 1.0 μM) was then loaded on either luminal or abluminal side and co-incubated further for 30 min at 37°C. Samples from receiving side were collected and glabridin concentrations were determined by an LC-MS method. Data are the mean ± SD from at least 6–9 determinations. *p < 0.05; **p < 0.01.

Effects of concentration on the transport rate of glabridin (0.1−100 μM) from the luminal (AP) to abluminal (BL) side and from BL to AP side when incubated for 30 min (plot a: AP-BL); plot b: BL-AP). Glabridin was loaded on either AP or BL side and incubated for 30 min at 37°C. Samples from receiving side were collected and glabridin was determined by an LC/MS method. Data are expressed as the mean ± SD from 3–9 determinations.
The effects of ATP inhibitors and various ABC transporter inhibitors on the transport of glabridin (0.1 and 1.0 μM) in RBMVEC monolayers were also investigated. Addition of the transport buffer at both sides with sodium azide (10 mM), 2, 4-dinitrophenol (5 mM), verapamil (100 μM), nifedipine (100 μM), or quinidine (50 μM) significantly (p < 0.05 or 0.01) increased the AB-BL fluxes of glabridin at 0.1 μM by 46.1 ± 6.5%, 47.8 ± 6.7%, 55.4 ± 6.8%, 56.9 ± 6.9%, and 66.3 ± 7.2%, respectively (p < 0.05) (Fig. 5). In contrast, these compounds caused a significant (p < 0.05 or 0.01) decrease in the BL to AP fluxes of glabridin at 0.1 μM by 30.2 ± 3.4%, 33.2 ± 4.2%, 45.6 ± 4.8%, 56.35 ± 6.3%, and 45.5 ± 5.2%, respectively. Probenecid and MK-571, but not celecoxib, also significantly increased the AP-BL fluxes and decreased the BL-AP fluxes of glabridin (p < 0.05). Similar results were observed when the concentration of glabridin was increased to 1.0 μM in the presence of above inhibitors. These results indicated the involvement of PgP in the transport of RBMVECs, and Mrp1-2, but not Mrp3-4, may be involved.
Permeability of Glabridin in Bilateral Perfused Brain in Rats
Entry of glabridin into the CNS was examined by means of the in situ brain perfusion technique in rats and compared with results for the plasma space marker molecule sucrose (Fig. 7). The uptake of glabridin into the cerebrum increased from 0.42 ± 0.09% at 1 min to 9.27 ± 1.69% (ml/100 g tissue) at 30 min and was significantly greater than that measured for sucrose at all measured time points. The values for cerebellum were 2.02 ± 0.41% at 1 min and 6.43 ± 1.29%, respectively. Lower values of glabridin were observed in CSF, which were 1.10 ± 0.23% and 3.95 ± 0.86%, respectively. The rank order of glabridin distribution in rat CNS was cerebrum > cerebellum > CSF.

Distribution of glabridin at 0.1−100 μM in cerebrum (a and d), cerebellum (b and e), and cerebrospinal fluid (CSF, c and f) when perfused for 1−30 min in a bilateral in situ rat brain perfusion model. Plots g, h, and i show the effects of various inhibitors on the uptake of glabridin at 0.5 μM after 10 min perfusion in cerebrum, cerebellum and CSF, respectively. The uptake of glabridin in cerebrum and cerebellum is expressed as a percentage ratio of tissue to plasma concentrations (R brain %; ml/100 g, for CSF, ml/100 ml). Each point represents the mean ± SD of eight animals. *p < 0.05; **p < 0.01 (for Plots a–e, glabridin vs sucrose; for Plots g–i, treatment with an inhibitor vs control).
The K in of glabridin in cerebrum, cerebellum and CSF significantly decreased when the substrate concentration was ≥1 μM (Fig. 7), suggesting the involvement of a saturable mechanism for glabridin uptake by CNS. The glabridin-specific R brain values at 0.1, 0.25, 0.5, 1, 2.5, 10, 25, 50 and 100 μM substrate concentration were 8.86, 7.11, 5.12, 4.06, 3.62, 3.28, 2.64, 2.44, and 2.13 ml/min/g of cerebrum tissue after subtraction of the value of [3H]-sucrose, respectively, with significant concentration-dependent decrease. Lower values were observed for cerebellum and CSF but significant concentration-dependent decrease remained. In addition, the effect of verapamil, nifedipine, quinidine, MK-571, probenecid, or celecoxib on the K in of glabridin in cerebrum, cerebellum and CSF at 0.5 μM after 10 min perfusion is shown Fig. 7. Verapamil, nifedipine, quinidine, MK-571, probenecid all significantly increased the R cerebrum, R cerebellum and R CSF values by 33.6−142.9%.
Brain and Tissue Distribution of Glabridin in Rats
We have reported the plasma pharmacokinetics of glabridin in rats (28). To examine the tissue distribution of glabridin in rats, blood and tissue levels of glabridin were determined in rats.
Fig. 8 shows representative plasma/tissue concentrations-time profiles over 24 h after treatment of oral glabridin at 5 mg/kg in rats. The R i values of AUC0–24 hr of glabridin for brain, lung, liver, kidney, spleen and heart over that in plasma were 0.19, 2.22, 1.59, 2.01, 1.70, and 1.54, respectively (Table I). The corrected R i values based on volume fraction of residual blood (\( R^{\prime }_{{\text{i}}} \)) were 0.27, 2.15, 1.57, 1.96, 1.70, 1.60 and 1.54, respectively. Both R i and \( R^{\prime }_{{\text{i}}} \) had similar values except for the brain where a 42.1% error was observed (R i vs \( R^{\prime }_{{\text{i}}} \): 0.19 vs 0.27). Thus, the rank order of glabridin tissue distribution as evidenced by AUC0–24 hr, R i, or \( R^{\prime }_{{\text{i}}} \) was lung > kidney > spleen > liver > heart > brain. The most apparent result is the limited brain distribution as indicated by its lowest \( R^{\prime }_{{\text{i}}} \). In contrast, glabridin considerably accumulated in the lung, kidney, heart, liver and spleen. In terms of the rate of glabridin tissue distribution, the Tmax ranged from 2.59 (heart) to 4.95 (spleen) h, with the highest Cmax values observed in the lung (33.4 ng/g tissue), followed by the heart (25.8 ng/g tissue), kidney (25.4 ng/g tissue), liver (18.7 ng/g tissue), spleen (19.9 ng/g tissue) and brain (2.8 ng/g tissue). In addition, the \( t_{{1 \mathord{\left/ {\vphantom {1 2}} \right. \kern-\nulldelimiterspace} 2\beta }} \) values for glabridin in rat plasma, brain, lung, liver, kidney, spleen and heart were 3.20 ± 0.93, 3.56 ± 1.08, 3.73 ± 1.12, 4.93 ± 1.14, 3.90 ± 1.07, 4.95 ± 1.33, and 2.59 ± 0.82 h, respectively.

The concentration-time profiles of glabridin in rat plasma (a), brain (b), liver (c), kidney (d), spleen (e), heart (f) and lung (g). Rats were treated with glabridin at 5 mg/kg by gavage, in combination with control vehicle, 25 m/kg or 100 mg/kg verapamil by gavage. Samples from the plasma, brain, lung, heart, liver, spleen and kidney over 24 h were collected, and processed as described in “Materials and Methods,” and drug concentrations were determined by LC-MS analysis. Data are the mean ± SD of six rats for each time point. Symbols: ■, glabridin at 5 mg/kg with control vehicle; ▲, glabridin at 5 mg/kg with combined 25 mg/kg verapamil; and ▼, glabridin at 5 mg/kg with combined 100 mg/kg verapamil. The data for the plasma in control rats are from Cao et al. (28).
Effects of Coadministered Verapamil or Quinidine on the Brain and Tissue Distribution of Glabridin in Rats
We have reported that coadministered verapamil significantly increased the plasma concentrations and AUCs of glabridin in rats (28). The plasma/tissue concentration-time profiles when glabridin was coadministered with verapamil at 25 or 100 mg/kg are shown in Fig. 8 and the pharmacokinetic parameter changes are shown in Table I. Combined verapamil significantly (p < 0.05) increased the brain Cmax and AUC0–24 h of glabridin in a dose-dependent manner. Co-administered verapamil at 25 and 100 mg/kg caused a significant increase (p < 0.05) in brain Cmax [from 2.8 ± 0.8 (control) to 4.5 ± 1.3 and 9.5 ± 2.8 ng/ml, respectively] and AUC0–24 h [from 18.3 ± 5.9 (control) to 23.7 ± 7.2 and 54.8 ± 17.6 ng h/ml, respectively] (Fig. 8 and Table I). The R i of AUC0–24 h for the brain was increased from 0.19 (control rats) to 0.20 and 0.32, respectively, when in combination with 25 and 100 mg/kg verapamil. The \( R^{\prime }_{{\text{i}}} \) of AUC0–24 h for the brain was increased from 0.27 (control rats) to 0.28 and 0.36, respectively. In addition, a similar magnitude in the increase of AUC0–24 h and Cmax for the lung, liver, kidney, spleen and heart was observed in a dose-dependent manner, when verapamil was coadministered at 25 and 100 mg/kg in rats. The R i and \( R^{\prime }_{{\text{i}}} \) values were largely unchanged for the lung, liver, kidney, spleen and heart at a 25 mg/kg of verapamil, but these values were significantly decreased when a higher dose (100 mg/kg) of verapamil was coadministered to the rat. This could be due to the effect of verapamil on PgP-mediated transport of glabridin in these tissues, but changes in the protein binding, metabolism, and clearance may also contribute.
The plasma/tissue concentration-time profiles when glabridin was coadministered with quinidine at 10 or 50 mg/kg are shown in Fig. 9 and the pharmacokinetic parameters are shown in Table II. Coadministered quinidine at 10 and 50 mg/kg caused a significant increase (p < 0.05) in rat brain Cmax [from 2.8 ± 0.8 (control) to 4.3 ± 1.3 and 11.6 ± 3.6 ng/ml, respectively] and AUC0–24 h [from 18.3 ± 5.9 (control) to 22.3 ± 6.5 and 64.2 ± 20.1 ng hr/ml, respectively] of glabridin in a dose-dependent manner (Fig. 9 and Table II). Consistently, the R i of AUC0–24 h for the brain was increased from 0.19 (control rats) to 0.21 and 0.41, respectively, when in combination with 10 and 50 mg/kg quinidine. Similarly, the \( R^{\prime }_{{\text{i}}} \) for the brain was increased from 0.27 (control rats) to 0.28 and 0.44, respectively. In addition, a similar magnitude in the increase of AUC0–24 hr and Cmax of glabridin for the lung, liver, kidney, spleen and heart was observed in a dose-dependent pattern, when quinidine was coadministered at 10 and 50 mg/kg in rats. Interestingly, the R i and \( R^{\prime }_{{\text{i}}} \) values were largely unchanged for the liver, kidney, and spleen by combined quinidine, but these values were significantly decreased in the lung but increased in the heart when quinidine was coadministered to the rat. This suggests that quinidine exhibited tissue-specific effect on glabridin distribution.

The concentration-time profiles of glabridin in rat plasma (a), brain (b), liver (c), kidney (d), spleen (e), heart (f) and lung (g). Rats were treated with glabridin at 5 mg/kg by gavage, in combination with control vehicle, 10 m/kg or 50 mg/kg quinidine by gavage. Samples from the plasma, brain, lung, heart, liver, spleen and kidney over 24 h were collected, and processed as described in “Materials and Methods,” and drug concentrations were determined by LC-MS analysis. Data are the mean ± SD of six rats for each time point. Symbols: ■, glabridin at 5 mg/kg with control vehicle; ▲, glabridin at 5 mg/kg with coadministered 10 mg/kg quinidine; and ▼, glabridin at 5 mg/kg with coadministered 50 mg/kg quinidine. The data for the plasma in control rats are from Cao et al. (28).
The brain uptake, R i and \( R^{\prime }_{{\text{i}}} \) values of the paracellular transport marker [3H]-mannitol at 5 or 15 min post-doing was not affected by coadministered verapamil at 50 or 100 mg/kg or quinidine at 10 or 50 mg/kg (data not shown). This indicated the functional integrity of rat BBB in our study and both verapamil and quinidine would not cause injuries and marked toxicities of the BBB and consequently increase glabridin brain penetration. PgP inhibition in the BBB is thus considered the major mechanism for increased brain penetration of glabridin by coadministered verapamil and quinidine. However, other factors, such as protein binding and metabolism, should also be considered.
Brain and Tissue Distribution of Glabridin in mdr1a Knockout Mice
We have reported the plasma pharmacokinetics of glabridin in mdr1a(−/−) in comparison with the wild-type mice and found that the plasma AUC0–24 h of glabridin in mdr1a(−/−) mice was 3.8-fold higher than that in the wild-type mice (28). We have also investigated the pharmacokinetics of glabridin in the brain and other various tissues in mdr1a(−/−) mice and wild-type mice. The results are shown in Fig. 10 and Table III. In wild-type mice, the R i values of AUC0–24 h of glabridin for the brain, lung, liver, kidney, spleen and heart over that in plasma were 0.20, 2.42, 2.05, 2.05, 1.70, and 1.36, respectively (Table III). The \( R^{\prime }_{{\text{i}}} \) values for the brain, lung, liver, kidney, spleen and heart were 0.28, 2.34, 2.02, 2.00, 1.60, and 1.33, respectively. Both R i and \( R^{\prime }_{{\text{i}}} \) had similar values except for the brain where a 40% error was observed (R i vs \( R^{\prime }_{{\text{i}}} \): 0.20 vs 0.28). These values were comparable to those observed in the rat. Thus, the rank order of glabridin tissue distribution as evidenced by AUC0–24 h, R i, or \( R^{\prime }_{{\text{i}}} \) was lung > liver > kidney > spleen > heart > brain. The most apparent result is the limited brain distribution \( {\left( {R^{\prime }_{{\text{i}}} = 0.28} \right)} \) in mice which was close to that in rats, suggesting that PgP in BBB efficiently restricts the brain penetration of glabridin in wild-type mice. In addition, the Tmax ranged from 2.00 (plasma) to 4.25 (liver) h in the wild-type mice.

The concentration-time profiles of glabridin in the plasma (a), brain (b), liver (c), kidney (d), spleen (e), heart (f), lung (g) from mdr1a(−/−) and wild-type mice. Mice were treated with glabridin at 5 mg/kg by gavage and samples from the plasma, brain, liver, kidney, spleen, heart, and lung over 24 h were collected, and processed as described in “Materials and Methods.” The concentrations of glabridin were determined by LC-MS analysis. Data are the mean ± SD of four mice per time point. Symbols: ■, mdr1a(−/−) mice; and ▲, wild-type mice. The data for the plasma in wild-type and mdr1a(−/−) mice are from Cao et al. (28).
In contrast, the pharmacokinetic profiles and brain penetration in mdr1a(−/−) mice were significantly different from those in the wild-type mice. The brain AUC0–24 h in mdr1a(−/−) mice were 6.0-fold higher than the wild-type mice, with a significantly increased brain R i and \( R^{\prime }_{{\text{i}}} \) values (0.30 and 0.35, respectively). The R i values of AUC0–24 h of glabridin for the brain, lung, liver, kidney, spleen and heart in mdr1a(−/−) mice were 0.30, 1.27, 1.01, 1.50, 1.09, and 1.07, respectively (Table III). The \( R^{\prime }_{{\text{i}}} \) values were 0.35, 1.24, 1.01, 1.47, 1.07, and 1.06, respectively (Table III). It was clear that the R i and \( R^{\prime }_{{\text{i}}} \) values decreased in all tissues examined except the brain in mdr1a(−/−) mice. Tissue accumulation of glabridin was not marked in mdr1a(−/−) mice. These values were significantly different from those observed in the wild-type mice and in rats. Thus, the rank order of glabridin tissue distribution in mdr1a(−/−) mice as evidenced by AUC0–24 h, Ri or \( R^{\prime }_{{\text{i}}} \) was kidney > lung > spleen > heart > > liver > brain. The Tmax ranged from 1.25 (heart) to 2.50 (brain) h in mdr1a(−/−) mice, with the highest Cmax values observed in the lung (74.5 ng/g tissue), followed by the heart (71.5 ng/g tissue), kidney (64.6 ng/g tissue), spleen (48.8 ng/g tissue), liver (44.6 ng/g tissue) and brain (19.1 ng/g tissue). In addition, the \( t_{{1 \mathord{\left/ {\vphantom {1 2}} \right. \kern-\nulldelimiterspace} 2\beta }} \) values of glabridin for the brain, lung, and heart in mdr1a(−/−) mice were decreased, but these values were increased in the plasma, liver, kidney and spleen compared to the wild-type mice.
DISCUSSION
There is an increasing application of herbal medicines to manage various diseases and to promote health in recent years in Asian and Western countries. The incorporation of herbal therapy into the main stream medical-care system has been encouraged by the World Health Organization and the governments of many Asian countries including China, Japan, Korea and Singapore despite the lack or scanty of evidence for the efficacy and safety data of most commonly herbal medicines. Though there is of importance of licorice and its major active constituent glabridin in human health because of their wide clinical use, little is known about their ability to reach sites of action including CNS. In this study, we evaluated, for the first time, the role of PgP in the brain penetration of glabridin using several in vitro and in vivo rat and mouse models.
In vitro models of the BBB, including animal (mainly bovine, mouse and rat) and human brain vascular endothelial cells, play a major role in the study of BBB permeability of drugs and other xenobiotics. The expression of typical BBB drug transporters, in particular PgP, is mandatory for the usability of these in vitro cell culture systems. The cells we used, RBMVECs, expressed high levels of PgP and minor to moderate levels of Mrp1 (Fig. 2) which ensured its functional features of an in vitro model for the study of drugs across BBB. RBMVECs are specialized endothelial cells forming tight junctions between adjacent cells, which maintain physiological cell morphology and exhibit many of the biochemical and biophysical properties of the intact BBB, providing an useful in vitro model with a restrictive paracellular barrier to solute permeability. Both the uptake and efflux of glabridin was investigated in RBMVECs. The kinetic parameters (Km and Vmax for the uptake and efflux of glabridin were different, indicating the presence of active transport across these cells. The inhibition data provided further evidence that PgP and possibly Mrp1–2 participated in the transport of glabridin in RBMVECs (Fig. 4). The uptake and efflux of glabridin in RBMVECs were ATP-dependent. PgP are highly likely to be involved in the influx and efflux of glabridin in RBMVECs, as indicated by the significant enhanced accumulation and decreased efflux of the substrate in the presence of PgP inhibitors. A number of flavonoids can be readily taken up and pumped out by human epithelial cells and tumor cells through active (may by PgP and MRPs) and/or passive diffusion (42). Because of the concentration gradient across the cellular membranes and high lipophilicity of glabridin (LogP ≈ 3.25), a passive diffusion is likely involved in the uptake and efflux of glabridin. The physico-chemical properties of drugs (e.g., hydrophilicity, lipophilicity, hydrogen bonding potential) largely determine the passive transport of drugs across the BBB. Passive transcellular processes are mainly dependent on the LogP (log-partition coefficient) or LogD (LogP at pH 7.4) values and their hydrogen bonding potential. There is a linear relationship between LogP and BBB transport of drugs in vivo (6). Because glabridin has a low molecular weight (322 Dalton) and high lipophilicity, the most likely mechanism would be active efflux into the systemic circulation across the BBB rather than low BBB permeability or low tissue binding in the brain. The facts that glabridin shares common characteristics (high lipophilicity, low molecular weight and planar structure) as a substrate for PgP indicate that PgP mediates the efflux of glabridin across the BBB, resulting in the restricted entry of the drug into the brain.
Our study demonstrated a polarization in the RBMVEC transport toward glabridin and a significantly higher efflux from the abluminal to luminal side, namely from the brain to systemic circulation (Fig. 5). Co-treatment with PgP inhibitors such as verapamil or Mrp1/2 inhibitors such as MK-571 significantly inhibited the efflux of glabridin from the abluminal to luminal side in RBMVECs (Fig. 5). These results indicated that glabridin was a substrate for PgP and probably for Mrp1/2 and these transporters located in brain capillary endothelial cells could efflux the drug from the brain into the systemic circulation. PgP is able to limit the brain penetration of glabridin and thus greatly diminish its concentrations in the brain and thus may affect its therapeutic efficacy when Licorice or glabridin is used for the treatment of CNS diseases. As control experiments, the inhibitors used above were also incubated on RBMVEC monolayers, and the BBB integrity was preserved as shown by inulin permeability (2.12−2.35 × 10−6 cm/s). This result clearly demonstrates that glabridin transport across RBMVEC monolayers in the luminal to abluminal direction is increased in the presence of ATP inhibitors, PgP inhibitors, or Mrp1/2 inhibitors, not due to reduced or loss of BBB monolayer integrity. The active efflux of glabridin and numerous other compounds by the BBB indicates that the BBB works as a detoxification and protective system in the brain by restricting the entry of exogenous compounds such as glabridin in the circulating blood to the brain.
The results from the bilateral in situ brain perfusion study in rats indicate that the BBB can restrict the entry of glabridin into CNS, probably due to the contribution of PgP and Mrp1/2 to a less extent (Fig. 7). Notably, the brain uptake of glabridin after considering vascular space was significantly greater than uptake into CSF. Although it is appreciated that in in vivo studies the separation of uptake across either the BBB or the blood-CSF barrier is not possible, it would appear unlikely that the blood-CSF route would produce the glabridin levels observed in the brain.
The rat brain levels of glabridin were about 27% of that in plasma when corrected by tissue residual blood (Fig. 8 and Table I), indicating that the entry of glabridin to the brain was considerably restricted in rats. The ratios of plasma Cmax and AUC0–24 h of glabridin over those in brain tissue were 4.38−4.85, indicating limited brain distribution of glabridin in rats. Previous studies indicated that the expression of PgP in various tissues such as the brain, heart, kidney, lung and liver minimized the distribution of many drugs which are PgP substrates into these tissues (47,48). The accumulation of glabridin in rat heart, lung and kidney suggested the possible presence of high-affinity binding proteins for glabridin and/or low level expression of PgP expression in these tissues. In addition, the distribution of glabridin was high in these tissues possibly because it is widely distributed to highly perfused organs such as the kidney and lung. The difference in glabridin distribution between these organs and the highly perfused brain is most likely due to high PgP levels at the BBB site. This is also reflected by the brain having the lowest Cmax, R i and \( R^{\prime }_{{\text{i}}} \) values among all tissues examined. The elimination half-lives of glabridin for the plasma, brain, lung and heart varied markedly in a range of 2.59−4.93 h, with the value in the spleen being the highest and the heart the lowest. This difference may reflect the differential distribution, binding, disposition and elimination of glabridin in these different tissues. For example, the liver and the spleen may contain lower levels of PgP or more binding proteins for glabridin than other tissues and thus retain glabridin for a longer time. Thus, after oral glabridin administration, the observed terminal half-lives may reflect drug absorption rather than drug elimination (i.e., a flip-flop kinetic model is operable). There is a possibility that glabridin accumulates in some organs such as the kidney, lung, liver and spleen due to specific binding to tissue proteins and thus glabridin elimination from these organs is delayed.
The brain levels of glabridin could be increased to up to 44% of plasma levels when verapamil or quinidine was co-administered in rats (Figs. 8 and 9, Tables I and II). Our results in rats are consistent with previous reports where the rat brain uptake of colchicine and vinblastine was significantly enhanced (3- to 7-fold) by co-treated verapamil at 1.0 mg/kg by i.v. (49). In the same way, SDZ PSC 833 (valspodar), a potent PgP inhibitor, also significantly increased the brain penetration of cyclosporine and vincristine (both PgP substrates) in rats (50). However, in several other studies using verapamil at 50 mg/kg by i.p. as a PgP inhibitor, it could not enhance the brain penetration of typical PgP substrates such as vinblastine in mice (51). The reason for the different findings in mice and rats in these studies might be due to marked species-dependent differences in PgP expression and activity, elimination and disposition of PgP substrates and inhibitors, and sensitivity of PgP protein to inhibitors. Notably, the increased magnitude of R i and \( R^{\prime }_{{\text{i}}} \) of glabridin for the brain in the presence of PgP inhibitors such as verapamil or quinidine is low. This may suggest the involvement of transporters other than PgP at the BBB to influence the uptake of glabridin and it may also reflect the fact that PgP is a protein that is very difficult to be completely suppressed for its activity by currently available inhibitors in vivo.
Coadministered verapamil increased the \( t_{{1 \mathord{\left/ {\vphantom {1 2}} \right. \kern-\nulldelimiterspace} 2\beta }} \) values of glabridin in the plasma, lung, kidney and heart in a dose-dependent manner, indicating that verapamil slowed down the elimination of glabridin from plasma and these tissues, probably due to inhibition of PgP-mediated hepato-biliary and renal efflux of glabridin. Clearance of PgP substrates through biliary and/or renal secretion can be significantly decreased in the presence of a PgP inhibitor (52). The localization of the protein also suggests that the function of PgP is related to transport mechanism of glabridin in the liver and kidneys because there are high levels of PgP protein in the brush border of renal proximal tubules and biliary surface of hepatocytes. Interestingly, coadministered quinidine significantly (p < 0.05) increased the \( t_{{1 \mathord{\left/ {\vphantom {1 2}} \right. \kern-\nulldelimiterspace} 2\beta }} \) values of glabridin in the plasma, kidney, and heart, but decreased those for the brain, liver, and spleen. Such difference may reflect differential effects of quinidine on the tissue binding, disposition and elimination in different organs. Though both quinidine and verapamil are potent PgP inhibitors with similar K i (1–4 μM), their effects on the cytochrome P450s and their interaction with PgP protein (e.g. ATPase activity) are different. It seemed that the plasma and tissue Tmax values were not significantly altered by combined verapamil or quinidine. This suggests that the intestinal absorption rate and distribution rate to different tissue of glabridin are not affected by verapamil or quinidine.
The present study showed that the brain AUC0–24 h in mdr1a(−/−) mice was 6.0-fold higher than the wild-type mice, with an significantly increased brain penetration in the mdr1a(−/−) mice (Fig. 10 and Table III). In the endothelium of mouse brain capillary, mdr1a/b is exclusively expressed and the mdr1a contributes solely to the efflux of drugs from the brain back into the blood circulation (6). Our results with glabridin as a PgP substrate are consistent with those reported in the literature. The absence of active PgP as observed in mdr1a knockout mice lacking PgP and thus exhibiting unrestricted access of PgP substrates to the brain yields significantly increased central nervous system concentrations often exceeding those observed in wild-type mice by orders of magnitude (53).
Results from the present study suggest that PgP/MDR1 play a major role in the transport of glabridin across the BBB, but the contribution of MDR3 and other ABC transporters such as Mrp1/2 cannot be excluded. Not all these proteins confer resistance to drugs; transfection experiments have shown that the expression of human PgP/MDR1 or rodent mdr1a or mdr1b is sufficient to confer resistance to anticancer agents, whereas MDR3 and mdr2 are involved in the transport of other molecules, such as phospholipids in the bile (4). It would be interesting to investigate the relative contributions of these P-glycoproteins in rats, mice and humans in the transport of glabridin. Moreover, PgP/MDR1-mediated drug-drug interactions have been reported, drug interaction studies of glabridin with PgP substrates/inhibitors should be considered in future clinical trials.
The in vivo results are consistent with our in vitro brain distribution study in rats, where coadministered verapamil or quinidine significantly enhanced the brain penetration of glabridin in rats. In addition, since the search for novel and more potent modulators of PgP is of major importance because most of the PgP inhibitors including verapamil cause side effects in vivo, the in vitro BBB system such as RBMVECs can be used as a efficient screening system for identifying new and potent PgP reversing agents. The safety data on glabridin in humans have not been evaluated in details, but it may become a useful probe for PgP functional studies in vitro and in vivo.
The identification of PgP in limiting the brain entry of glabridin based on in vitro and in vivo rodent studies has important clinical implications, though there is no direct evidence indicating that this important transporter restricts the entry of glabridin into the CNS in humans. The results from the present study using mouse and rat models strongly suggest that human PgP and probably MRP1 may play a role in restricting the brain penetration of glabridin despite the fact that there are species differences in the expression and activity of these transporters between rodents and human. In our study, there were some species differences in the organ distribution of glabridin in rats and mice. Since species differences in the expression and activity, substrate specificity, inhibition, sensitivity to modulator and organ expression profiles of PgP and MRP1-5 in rodents and humans have been reported (54,55), caution should be taken when extrapolating the data from rodent studies to humans.
In conclusion, glabridin was found to have a limited brain penetration in rats but increased when coadministered with PgP inhibitors. Our in vitro studies using RBMVECs and a bilateral in situ brain perfusion model indicated that PgP-mediated active efflux across the BBB restricted the entry of glabridin to the brain. The brain AUC of glabridin in mdr1a(−/−) mice was 6.0-fold higher than the wild-type mice. PgP may cause drug resistance to glabridin (licorice) therapy for CNS diseases and potential drug-glabridin interactions. Based on the inhibition studies, Mrp1/2 were probably involved in the transport of glabridin and limited the brain penetration of glabridin, but to a lesser extent compared to PgP. Further research addressing the role of PgP and other transporters in the disposition and brain distribution of glabridin and possible drug interactions through PgP modulation is warranted.
Abbreviations
- ABC:
-
ATP-binding cassette
- AP:
-
Luminal
- AUC:
-
the area under the plasma concentration-time curve
- BBB:
-
blood-brain barrier
- BL:
-
abluminal
- Cmax :
-
maximum plasma concentration
- CNS:
-
central nervous system
- CSF:
-
cerebrospinal fluid
- DMSO:
-
dimethyl sulfoxide
- EDTA:
-
ethylenediaminetetraacetic acid
- HBSS:
-
Hank’s balanced salt solution
- HEPES:
-
N-[2-hydroxyethyl] piperazine-N9-[4-butanesulfonic acid]
- HPLC:
-
high performance liquid chromatography
- K in :
-
in vivo BBB permeability
- K m :
-
Michaelis-Menten constant
- LC-MS:
-
liquid chromatography mass spectrometry
- MRP (Mrp for rat):
-
multi-drug resistance protein
- MTT:
-
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazonium bromide
- P app :
-
permeability coefficient
- PD :
-
passive diffusion component
- PgP:
-
P-glycoprotein
- R brain :
-
the amount of a drug in the brain over that in the perfusate
- RBMVEC:
-
rat brain microvascular endothelial cells
- SDS:
-
sodium dodecyl sulphate
- \( t_{{1 \mathord{\left/ {\vphantom {1 2}} \right. \kern-\nulldelimiterspace} 2\beta }} \) :
-
elimination half life
- Tmax :
-
time to achieve Cmax
- TEER:
-
transepithelial electric resistance
- (V F)B :
-
volume fraction of residual blood in a tissue
- Vmax :
-
maximum velocity
References
W. A. Banks. Physiology and pathology of the blood-brain barrier: implications for microbial pathogenesis, drug delivery and neurodegenerative disorders. J. Neurovirol. 5:538–555 (1999).
W. Loscher and H. Potschka. Blood-brain barrier active efflux transporters: ATP-binding cassette gene family. NeuroRx 2:86–98 (2005).
P. L. Golden and G. M. Pollack. Blood-brain barrier efflux transport. J. Pharm. Sci. 92:1739–1753 (2003).
P. Borstand and R. O. Elferink. Mammalian ABC transporters in health and disease. Annu. Rev. Biochem. 71:537–592 (2002).
H. Sun, H. Dai, N. Shaik, and W. F. Elmquist. Drug efflux transporters in the CNS. Adv. Drug Deliv. Rev. 55:83–105 (2003).
A. G. de Boer, I. C. van der Sandt, and P. J. Gaillard. The role of drug transporters at the blood-brain barrier. Annu. Rev. Pharmacol. Toxicol. 43:629–656 (2003).
T. Terasaki and S. Ohtsuki. Brain-to-blood transporters for endogenous substrates and xenobiotics at the blood-brain barrier: an overview of biology and methodology. NeuroRx 2:63–72 (2005).
D. J. Begley. ABC transporters and the blood-brain barrier. Curr. Pharm. Des. 10:1295–1312 (2004).
M. Demeule, A. Regina, J. Jodoin, A. Laplante, C. Dagenais, F. Berthelet, A. Moghrabi, and R. Beliveau. Drug transport to the brain: key roles for the efflux pump P-glycoprotein in the blood-brain barrier. Vascul. Pharmacol. 38:339–348 (2002).
E. M. Taylor. The impact of efflux transporters in the brain on the development of drugs for CNS disorders. Clin. Pharmacokinet. 41:81–92 (2002).
Y.-P. Zhu. Chinese materia medica: chemistry, pharmacology and applications, Harwood Academic Publishers, Amsterdam, The Netherlands, 1998.
R. K. Dhiman and Y. K. Chawla. Herbal medicines for liver diseases. Dig. Dis. Sci. 50:1807–1812 (2005).
C. Fiore, M. Eisenhut, E. Ragazzi, G. Zanchin, and D. Armanini. A history of the therapeutic use of liquorice in Europe. J. Ethnopharmacol. 99:317–324 (2005).
L. Bielory. Complementary and alternative interventions in asthma, allergy, and immunology. Ann. Allergy Asthma Immunol. 93:S45–S54 (2004).
W. J. Craig. Health-promoting properties of common herbs. Am. J. Clin. Nutr. 70:491S–499S (1999).
P. A. Belinky, M. Aviram, B. Fuhrman, M. Rosenblat, and J. Vaya. The antioxidative effects of the isoflavan glabridin on endogenous constituents of LDL during its oxidation. Atherosclerosis 137:49–61 (1998).
T. Yokota, H. Nishio, Y. Kubota, and M. Mizoguchi. The inhibitory effect of glabridin from licorice extracts on melanogenesis and inflammation. Pigment Cell Res. 11:355–361 (1998).
M. Rosenblat, P. Belinky, J. Vaya, R. Levy, T. Hayek, R. Coleman, S. Merchav, and M. Aviram. Macrophage enrichment with the isoflavan glabridin inhibits NADPH oxidase-induced cell-mediated oxidation of low density lipoprotein. A possible role for protein kinase C. J. Biol. Chem. 274:13790–13799 (1999).
S. Tamir, M. Eizenberg, D. Somjen, N. Stern, R. Shelach, A. Kaye, and J. Vaya. Estrogenic and antiproliferative properties of glabridin from licorice in human breast cancer cells. Cancer Res. 60:5704–5709 (2000).
F. Aoki, S. Honda, H. Kishida, M. Kitano, N. Arai, H. Tanaka, S. Yokota, K. Nakagawa, T. Asakura, Y. Nakai, and T. Mae. Suppression by licorice flavonoids of abdominal fat accumulation and body weight gain in high-fat diet-induced obese C57BL/6J mice. Biosci. Biotechnol. Biochem. 71:206–214 (2007).
J. S. Kang, Y. D. Yoon, M. H. Han, S. B. Han, K. Lee, K. H. Lee, S. K. Park, and H. M. Kim. Glabridin suppresses intercellular adhesion molecule-1 expression in tumor necrosis factor-alpha-stimulated human umbilical vein endothelial cells by blocking sphingosine kinase pathway: implications of Akt, extracellular signal-regulated kinase, and nuclear factor-kappaB/Rel signaling pathways. Mol. Pharmacol. 69:941–949 (2006).
R. K. Dhiman. Herbal hepatoprotective agents: marketing gimmick or potential therapies? Trop. Gastroenterol. 24:160–162 (2003).
Z. Y. Wang and D. W. Nixon. Licorice and cancer. Nutr. Cancer 39:1–11 (2001).
C. E. Piersen. Phytoestrogens in botanical dietary supplements: implications for cancer. Integr. Cancer Ther. 2:120–138 (2003).
D. Armanini, C. Fiore, M. J. Mattarello, J. Bielenberg, and M. Palermo. History of the endocrine effects of licorice. Exp. Clin. Endocrinol. Diabetes 110:257–261 (2002).
L. Langmead and D. S. Rampton. Review article: herbal treatment in gastrointestinal and liver disease-benefits and dangers. Aliment. Pharmacol. Ther. 15:1239–1252 (2001).
A. Olukoga and D. Donaldson. Liquorice and its health implications. J. R. Soc. Health 120:83–89 (2000).
J. Cao, X. Chen, J. Liang, X. Q. Yu, A. L. Xu, E. Chan, W. Duan, M. Huang, J. Y. Wen, X. Y. Yu, X. T. Li, F. S. Sheu, and S. F. Zhou. Role of P-glycoprotein in the intestinal absorption of glabridin, an active flavonoid from the root of Glycyrrhiza glabra. Drug. Metab. Dispos. 35:539–553 (2007).
F. Roux and P. O. Couraud. Rat brain endothelial cell lines for the study of blood-brain barrier permeability and transport functions. Cell. Mol. Neurobiol. 25:41–58 (2005).
T. Mosmann. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J. Immunol. Methods 65:55–63 (1983).
S. Zhou, X. Feng, P. Kestell, J. W. Paxton, B. C. Baguley, and E. Chan. Transport of the investigational anti-cancer drug 5,6-dimethylxanthenone-4-acetic acid and its acyl glucuronide by human intestinal Caco-2 cells. Eur. J. Pharm. Sci. 24:513–524 (2005).
B. El Hafny, N. Cano, M. Piciotti, A. Regina, J. M. Scherrmann, and F. Roux. Role of P-glycoprotein in colchicine and vinblastine cellular kinetics in an immortalized rat brain microvessel endothelial cell line. Biochem. Pharmacol. 53:1735–1742 (1997).
Q. Tian, J. Zhang, E. Chan, W. Duan, and S. F. Zhou. Multidrug resistance proteins (MRPs) and implication in drug development. Drug Dev. Res. 51:1–18 (2005).
J. Zhang, M. Huang, S. Guan, H. C. Bi, Y. Pan, W. Duan, S. Y. Chan, X. Chen, Y. H. Hong, J. S. Bian, H. Y. Yang, and S. Zhou. A mechanistic study of the intestinal absorption of cryptotanshinone, the major active constituent of Salvia miltiorrhiza. J. Pharmacol. Exp. Ther. 317:1285–1294 (2006).
Y. Takasato, S. I. Rapoport, and Q. R. Smith. An in situ brain perfusion technique to study cerebrovascular transport in the rat. Am. J. Physiol. 247:H484–H493 (1984).
I. Tamai, M. Senmaru, T. Terasaki, and A. Tsuji. Na(+)- and Cl(−)-dependent transport of taurine at the blood-brain barrier. Biochem. Pharmacol. 50:1783–1793 (1995).
S. P. Khor and M. Mayersohn. Potential error in the measurement of tissue to blood distribution coefficients in physiological pharmacokinetic modeling. Residual tissue blood. I. Theoretical considerations. Drug Metab. Dispos. 19:478–485 (1991).
V. C. de Boer, A. A. Dihal, H. van der Woude, I. C. Arts, S. Wolffram, G. M. Alink, I. M. Rietjens, J. Keijer, and P. C. Hollman. Tissue distribution of quercetin in rats and pigs. J. Nutr. 135:1718–1725 (2005).
U. K. Laemmli. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680–685 (1970).
O. H. Lowry, N. J. Rosebrough, A. L. Farr, and R. J. Randall. Protein measurement with the Folin phenol reagent. J. Biol. Chem. 193:265–275 (1951).
K. Yamaoka, T. Nakagawa, and T. Uno. Application of Akaike's information criterion (AIC) in the evaluation of linear pharmacokinetic equations. J. Pharmacokinet. Biopharm. 6:165–167 (1978).
S. Zhou, L. Y. Lim, and B. Chowbay. Herbal modulation of P-glycoprotein. Drug Metab. Rev. 36:57–104 (2004).
T. Miyama, H. Takanaga, H. Matsuo, K. Yamano, K. Yamamoto, T. Iga, M. Naito, T. Tsuruo, H. Ishizuka, Y. Kawahara, and Y. Sawada. P-glycoprotein-mediated transport of itraconazole across the blood-brain barrier. Antimicrob. Agents Chemother. 42:1738–1744 (1998).
S. V. Ambudkar, C. O. Cardarelli, I. Pashinsky, and W. D. Stein. Relation between the turnover number for vinblastine transport and for vinblastine-stimulated ATP hydrolysis by human P-glycoprotein. J. Biol. Chem. 272:21160–21166 (1997).
C. J. Ek, M. D. Habgood, K. M. Dziegielewska, A. Potter, and N. R. Saunders. Permeability and route of entry for lipid-insoluble molecules across brain barriers in developing Monodelphis domestica. J. Physiol. 536:841–853 (2001).
S. Ohtsuki, S. Sato, H. Yamaguchi, M. Kamoi, T. Asashima, and T. Terasaki. Exogenous expression of claudin-5 induces barrier properties in cultured rat brain capillary endothelial cells. J. Cell. Physiol. 210:81–86 (2007).
A. H. Schinkel, J. J. Smit, O. van Tellingen, J. H. Beijnen, E. Wagenaar, L. van Deemter, C. A. Mol, M. A. van der Valk, E. C. Robanus-Maandag, H. P. te Riele, et al. Disruption of the mouse mdr1a P-glycoprotein gene leads to a deficiency in the blood-brain barrier and to increased sensitivity to drugs. Cell 77:491–502 (1994).
J. van Asperen, O. van Tellingen, F. Tijssen, A. H. Schinkel, and J. H. Beijnen. Increased accumulation of doxorubicin and doxorubicinol in cardiac tissue of mice lacking mdr1a P-glycoprotein. Br. J. Cancer 79:108–113 (1999).
N. Drion, M. Lemaire, J. M. Lefauconnier, and J. M. Scherrmann. Role of P-glycoprotein in the blood-brain transport of colchicine and vinblastine. J. Neurochem. 67:1688–1693 (1996).
M. Lemaire, A. Bruelisauer, P. Guntz, and H. Sato. Dose-dependent brain penetration of SDZ PSC 833, a novel multidrug resistance-reversing cyclosporin, in rats. Cancer Chemother. Pharmacol. 38:481–486 (1996).
M. Arboix, O. G. Paz, T. Colombo, and M. D’Incalci. Multidrug resistance-reversing agents increase vinblastine distribution in normal tissues expressing the P-glycoprotein but do not enhance drug penetration in brain and testis. J. Pharmacol. Exp. Ther. 281:1226–1230 (1997).
S. Song, H. Suzuki, R. Kawai, and Y. Sugiyama. Effect of PSC 833, a P-glycoprotein modulator, on the disposition of vincristine and digoxin in rats. Drug Metab. Dispos. 27:689–694 (1999).
A. H. Schinkel, U. Mayer, E. Wagenaar, C. A. Mol, L. van Deemter, J. J. Smit, M. A. van der Valk, A. C. Voordouw, H. Spits, O. van Tellingen, J. M. Zijlmans, W. E. Fibbe, and P. Borst. Normal viability and altered pharmacokinetics in mice lacking mdr1-type (drug-transporting) P-glycoproteins. Proc. Natl. Acad. Sci. U. S. A. 94:4028–4033 (1997).
S. Baltes, A. M. Gastens, M. Fedrowitz, H. Potschka, V. Kaever, and W. Loscher. Differences in the transport of the antiepileptic drugs phenytoin, levetiracetam and carbamazepine by human and mouse P-glycoprotein. Neuropharmacology 52:333–346 (2007).
M. Katoh, N. Suzuyama, T. Takeuchi, S. Yoshitomi, S. Asahi, and T. Yokoi. Kinetic analyses for species differences in P-glycoprotein-mediated drug transport. J. Pharm. Sci. 95:2673–2683 (2006).
Acknowledgements
The authors appreciate the support provided by Guangdong Provincial Cardiovascular Institute (Guangzhou, China), National 973 Grand Project of China (Grant No. 2005CB523305), the Australian Institute of Chinese Medicine, Sydney, Australia (Grant Nos. R-106-00341 & R-106-00382), Singapore Cancer Syndicate Grant SCS PS0023, and the Queensland University of Technology Academic Staff Research Fund (Brisbane, Queensland, Australia).
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article
Cite this article
Yu, XY., Lin, SG., Zhou, ZW. et al. Role of P-glycoprotein in Limiting the Brain Penetration of Glabridin, An Active Isoflavan from the Root of Glycyrrhiza glabra . Pharm Res 24, 1668–1690 (2007). https://doi.org/10.1007/s11095-007-9297-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11095-007-9297-1