
Abstract
Purpose
The goals in this study were several-fold. First, to optimize the in vivo phage display methodology by incorporating phage pharmacokinetic properties, to isolate peptides that target the brain microvasculature, and then to build focused libraries to obtain structure activity relationship information in vivo to identify the optimal targeting motif.
Materials and Methods
The blood pharmacokinetics of filamentous and T7 phage were evaluated to choose the optimal platform. A randomized peptide library with a motif CX10C was constructed in T7 phage and used for in vivo panning. Focused peptide libraries around each structural element of the brain-specific peptide were constructed to perform kinetic structure activity relationship (kSAR) analysis in vivo. To determine potential function, sepsis was induced in mice by LPS administration and four hours later the effect of GST-peptide on adhesion of rhodamine-labelled lymphocytes or CFDA-labelled platelets to pial microvasculature was observed by intravital microscopy.
Results
The blood phamacokinetics of T7 was rapid (half-life of 12 min) which aids the clearance of non-specific phage. In vivo panning in brain enriched for isolates expressing the motif CAGALCY. Kinetic analysis of focused libraries built around each structural element of the peptide provided for rapid pharmacophore mapping. The computer modeling data suggested the peptide showed similarities to peptide mimetics of adhesion molecule ligands. GST-CAGALCY but not GST control protein was able to inhibit the rolling and adhesion of labeled platelets to inflamed pial vasculature. GST-CAGALCY had no effect on lymphocyte adhesion.
Conclusions
Incorporating normal blood phamacokinetics of T7 phage into in vivo phage display improves the ability to recover targeting peptide motifs and allows effective lead optimization by kSAR. This approach led to the isolation of a brain-specific peptide, CAGALCY, which appears to function as an effective antagonist of platelet adhesion to activated pial microvasculature.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
INTRODUCTION
Several lines of evidence suggest that molecular diversity exists within the vasculature. Different molecular recognition systems must exist to account for the specific cellular trafficking that occurs within the body. For example, leukocyte homing to sites of inflammation is facilitated by pro-inflammatory upregulation of specific leukocyte cell adhesion molecules in the target endothelium (1–8). Pro-coagulatory endothelial responses to systemic inflammatory diseases exhibit regional differences in the extent various pro-coagulant factors are upregulated, which underlie the focal response observed in different systemic disorders (9–14). However, to date, the understanding of how specific trafficking pathways are achieved is limited due to the difficulties of recapitulating vascular diversity in vitro.
One approach to overcome this issue has been the use of in vivo panning with phage displayed peptide libraries to identify peptide ligands that specifically home to a given vascular bed to attempt to map the vascular diversity that exists in situ (15–18). However, the original methods had certain drawbacks since phage within various vascular beds were analyzed shortly after IV administration (i.e., ∼Cmax) without taking into adequate consideration the differences in hemodynamics and vascularity of different organs or the normal pharmacokinetic clearance rate of the phage from blood by the phagocytic monophagocytic system (MPS) of liver, kidney and spleen, which is affected by particulate biophysical and surface biochemical characteristics (19). Therefore, we set out to develop a more robust screening method that would take into account well defined parameters for assessing whether a given ligand displays true targeting activity, namely specific retention of a given ligand at the target site compared to clearance from non target organs (e.g., as reviewed by Nilsson et al., 20).
Taking these issues into consideration, an improved in vivo phage display methodology was developed that incorporated the pharmacokinetic behavior of phage particles. This approach led to the identification of the brain-targeting peptide, CAGALCY. By building focused libraries around each structural element of the lead peptide, we developed a novel, rapid and effective means to perform kinetic SAR (kSAR) in vivo. In silico analysis of the pharmacophore indicated some similarities to known ligand mimetics of cell adhesion molecules expressed on blood cells. By observing the behavior of differentially labeled blood cell types under activating conditions (systemic LPS), within the pial microvasculature using intravital microscopy, we were able to demonstrate that the CAGALCY cyclic peptide expressed as a GST-fusion protein was an effective inhibitor of platelet adhesion to brain microvasculature.
MATERIALS AND METHODS
Materials
T7 phage vector, sequencing primers and the BLT 5615 bacterial host were purchased from Novagen (Madison, WI). Synthetic oligonucleotides were custom synthesized by Retrogen (San Diego CA) or Sigma-Aldrich (St. Louis MO). Lipopolysaccharide (LPS) was obtained from Sigma-Aldrich. pGEX-5x-3 vector and glutathione Sepharose 4B were from Pharmacia Biotech, now GE Healthcare (Piscataway, NJ). Limulus endotoxin Kinetic-QCL assay kits were obtained from BioWhittaker (Walkersville, MD). carboxyfluorescein diacetate succinimidyl ester (CFDA) and rhodamine G were obtained from Molecular Probes (Eugene, OR).
Animals
Female BALB/c, FVB/N, and C57BL mice between the ages of 7–12 weeks and Sprague Dawley rats at 8-weeks of age were purchased from Harlan (Indianapolis, IN) and acclimated for at least 3 days before an experiment. They were housed in a fully-accredited AALAC vivarium and all protocols were pre-approved by the Institutional Animal Care and Use Committee.
Construction of a Random Peptide-expressing T7 Phage Library
A randomized CX10C peptide library was constructed using synthetic oligonucleotides encoding two cysteines separated by ten randomized amino acids. Double-stranded oligonucleotides were synthesized by annealing of the 2nd strand primer with a nucleotide sequence of 5′-TCGAGTGCGGCCGCAAGC-3′ to the single-strand oligonucleotide and extended by Klenow fragment. After digestion with EcoR I and Hind III, the double-strand oligonucleotides were gel-purified and ligated to a T7 (415-1) (Novagen, Madison, WI) vector that had also been digested with EcoR I and Hind III (close to the codon for amino acid 348 of the 10B capsid protein) in a 50 μl reaction volume. The reaction products were mixed with T7 packaging extract in nine reactions and pooled. The titer of the library was determined by plaque assay to be 6.2 × 108 pfu. This library was then amplified in the BLT 5615 bacterial host strain in the presence of 1 mM IPTG. All phage variants were constructed using annealed oligonucleotides, which were then cloned into the EcoR I and Hind III sites of the T7 vector.
In Vivo Phage Selection
The T7 phage was purified by PEG precipitation followed by CsCl gradient centrifugation (20–62.5%, 35,000 rpm for 1 h with Beckman table-top ultracentrifuge and TLS55 rotor). In each round of selection, 300 μl purified phage (1011 pfu) in phosphate-buffered saline (PBS), pH 7.2, was injected (i.v.) into each FVB mouse via the tail vein, 60 min post injection, mice were anesthetized with Avertin and 100 μl of blood was collected from each mouse and mixed with 100 μl of 100 mM sodium citrate. The mouse was euthanized by cervical dislocation and perfused through the heart with 10 ml of ice-cold PBS containing 1% bovine serum albumin (BSA). Brains were harvested, rinsed with cold PBS and homogenized in 500 μl of PBS containing 0.5% NP-40, 1% BSA, 0.5 mM PMSF, 1 μg/ml aprotinin, 1 μg/ml leupeptin and 1 μg/ml pepstatin with a Dounce homogenizer. 1.5 ml of PBS was then added to bring the total volume up to 2 ml. Aliquots of the homogenates were diluted in PBS (1:100) and used to determine phage content by plaque assay. For the plaque assay, 900 μl of a BLT5616 bacterial culture was added to 100 μl of each lysate. After incubating for 10 min at room temperature, duplicate 1, 10 and 100 μl aliquots were added to 3 ml of melted LB agar and plated out onto LB/carbenicillin plates. These were incubated overnight at room temperature and then the numbers of plaques counted. The remaining homogenates were used for amplification in the BLT 5615 host strain in the presence of 1 mM IPTG. The amplified phage were then purified and used for the next round of panning. The blood samples were centrifuged at 4,000 rpm for 10 min at 4°C. The supernatants (plasma) were saved for plaque assay analysis. A total of five rounds of selections were performed.
For panning of focused libraries, equal numbers of each phage variant was mixed with the parental phage (5% for each variant) and injected into 6 Balb/C mice. The brains and blood samples were harvested from three mice at 30 min and 4 h post phage injection. Inserts of approximately 200 plaques of the mixture to be administered were sequenced to determine the relative frequency of each variant. Similar numbers were sequenced from the phage recovered at 30 min and 4 h post-administration.
Sequencing of Phage Isolates
The inserts of individual phage plaques were amplified by PCR using an in-house designed primer, T7 super-up primer, 5′-AGCGGACCAGATTATCGC-3′ (Retrogen), with the manufacturer’s T7Select®DOWN Primer, 5′-AACCCCTCAAGACCCGTTTA-3′ following the manufacturer’s protocol (Novagen, Madison WI). In brief, 2 μl of each primer were added to 5 μl phage suspension (from one plaque resuspended in 50 μl PBS) together with 0.5 μl Taq DNA polymerase and 2 mM NTPs in a final volume of 50 μl. The cycle conditions were 94°C for 2 min, followed by 30 cycles of 94°C for 30 s, 55°C for 30 s and 72°C for 20 s, followed by 1 cycle of 72°C for 5 min. An aliquot of the reaction mixture was then used for sequencing using dye terminator cycle sequencing (DTCS) kits according to the manufacturer’s protocol (Beckman Coulter, Fullerton, CA). The sequences were analyzed using a CEQ™ 8000 Genetic Analysis System (Beckman Coulter).
Pharmacokinetic Analysis of Phage Particles
1 × 1010 pfu each of test phage and T7-lacZ phage (a T7 phage carrying the lacZ gene controlled by the lac promoter in its genome, this phage forms blue plaque in the presence of X-gal and serves as an internal control) were co-injected into FVB female mice. The brain, lung, liver, kidney, blood and spleen were harvested at 2 min, 10 min, 30 min, 1 h, 2 h, 4 h, 6 h, 12 h, and 24 h. The phage contents in each organ were determined by plaque assay in the presence of X-gal in the top agar.
Construction and Purification of GST Fusion Protein
DNA fragments containing sequences for the E tag (5′-GGTGCGCCGGTGCCGTA TCCGGATCCGCTGGAACCGCGTTAA-3′) and E tag-targeting peptide (5′-GGTGCGCCGGTGCCGTATCCGGATCCGCTGGAACCGCGTTGTGCAGGTGCACTCTGTTACTAA-3′) were cloned into the EcoR I and Not I sites of pGEX-5x-3 vector (GE Healthcare, Piscataway, NJ). GST fusion protein expression was induced with 1 mM IPTG and purified using a Glutathione Sepharose 4B gel column according to the manufacturer’s instructions (GE Healthcare), except the wash was performed with ten bed volumes of PBS containing 1% Triton X-100, followed by 20 bed volumes of PBS before eluting the GST protein with 10 mM reduced glutathione, pH 8.0. The eluted fusion proteins were then dialyzed in PBS, pH 7.6, overnight at 4°C. The fusion proteins purified in this way were essentially free of endotoxin as determined by the Limulus endotoxin Kinetic-QCL assay.
Computer Modeling
The Tripos software package (37) was utilized for visualizing and generating energy minimizing structures. The Sybyl molecular mechanics force field was used for molecular dynamics and minimization studies. First, the linear sequence was generated and during a constrained minimization step, the peptide was cyclized via SS bonding. Minimum-energy conformers of the CAGALCY peptide were generated using molecular dynamics at high temperatures. Fifteen hundred structures were generated by molecular dynamics at 700 K, capturing snapshots every 2 ps. We used a time step of 1 fs under controlled temperature. Each conformer was then submitted to 2 ps of molecular dynamics at 298 K. Then, the 200 lowest energy structures were energy minimized using the steepest descent, conjugate gradient and Newton–Rahpson methodologies until the gradient was less than 0.001 kcal·mol−1·Å−1. The final ten structures were then submitted to minimization using semi-empirical molecular orbital methodology (PM3/MOPAC). Then, a representative structure was chosen as a peptide model for the construction of the pharmacophore model.
Intravital Microscopy Study
The mice were anesthetized by injection (i.p.) of ketamine (200 mg/kg body weight) plus xylazine (10 mg/kg). The jugular vein was cannulated with polyethylene tubing (PE-10 or PE-50) to administer rhodamine 6G, platelets, or KCl for euthanasia. An incision was made over the skull, which was removed by creating a circular opening with a handheld surgical drill. The mouse was then placed on the stage of a Leica upright epi-fluorescence microscope (DM-FSA) with an intensified camera (Cascade, Roper) and the 10 × images recorded on videotape for off-line analysis. At least five non-overlapping regions were analyzed for each mouse and each region was videotaped for about 2 min (approximately 25 min total observation period per mouse).
Mouse platelets were obtained from blood isolated via a cannula implanted into the jugular vein, which was centrifuged at 120 × g for 10 min to generate platelet rich plasma (PRP; the supernatant). Human PRP was obtained from the San Diego Blood Bank and platelets were fluorescently labeled with carboxyfluorescein diacetate succinimidyl ester (CFDA).
Mice were injected (i.p.) with 100 μg of LPS (Sigma Aldrich, St. Louis, MO) to induce endotoxemia. Three and a half hours later, the mice were anesthetized, cannulated, and a brain window developed in the skull. About 4 h after LPS injection, test compounds were injected in a blinded fashion via the jugular cannula and then 1 × 108 CFDA-labeled platelets were injected. The number of platelets detected in a 100 μm section of vessel was recorded and the vessel diameter was measured from the screen by an independent blinded scorer. The average number of platelets per unit area was calculated for each mouse and then the blind was broken, individual mice assigned to their relevant group and groups compared with each other using the StatView program.
RESULTS
In Vivo Screening for Brain-targeting Phage
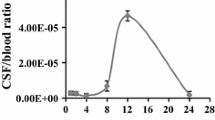
The original in vivo phage display methodology is limited by very high background. Before screening for brain-specific ligands, we first assessed the pharmacokinetic properties of the phage particles themselves to investigate whether the natural clearance mechanism of particulates from the circulation by the MPS could be exploited to improve the yield of positive hits by removing non-binding phage, i.e., reduce noise. The plasma pharmacokinetics of M13 and fUSE5 filamentous phage as well as T7 phage were assessed following intravenous administration of a 1010 pfu dose. Preliminary studies revealed that anesthetizing the mice with Avertin before IV administration of phage resulted in a slower and altered pattern of blood pharmacokinetics over the first hour (data not shown). Therefore, for all subsequent studies, phage were administered by IV tail injection to non-anesthetized mice and Avertin was only administered 5 min before euthanasia. The plasma pharmacokinetics of filamentous phage (fUSE5 and M13) were slow (7 and 9 h, respectively; data not shown), similar to published studies (21). In contrast, the non-filamentous phage, T7, showed a half-life in blood of 12 min, which resulted in a one log decrease in the phage content of blood by 1 h (similar to the results shown in Fig. 1a). This indicated that the natural clearance mechanisms for T7 phage could be exploited to increase the signal to noise ratio in target tissues. Furthermore, choosing these longer time points for harvesting and analyzing individual phage at a given site would select for phage displaying peptides that confer slow off-rates and thereby improve retention at the target site, desired features for a targeting peptide.

Panels a–c: Pharmacokinetic profiles of T7-CAGALCY and T7-lacZ phage in the blood (a), brain (b), and spleen (c). 1 × 1010 pfu of T7-CAGALCY and T7-lacZ phage were co-injected into FVB female mice. At the indicated time intervals, the brain, blood, and spleen were harvested. The numbers of phage particles were determined by plaque assay, and expressed as yield (numbers of phage recovered divided by that of injected)/g. d: Selectivity index of CAGALCY phage in the brain and reference organs. Selectivity index is defined as the ratio of T7-CAGALCY and T7-lacZ phage recovered from the same organ. Equal numbers of CAGALCY and T7-lacZ phage were co-injected into 3 Balb/c mice via the tail vein. Organs were harvested two hours post injection. The numbers of T7-CAGALCY and T7-lacZ phage were determined by plaque assay and the selectivity indices were calculated for each organ of individual mice. The bar in each column represents the mean selectivity index.
To screen for brain vascular-specific peptides, a CX10C peptide phage library was constructed and the endotoxin levels determined. PEG precipitation (used in the original studies for filamentous phage (15,18) failed to adequately reduce the endotoxin level to physiologically acceptable levels and injecting phage resulted in rapid complement activation (data not shown). Cesium chloride gradient centrifugation reduced endotoxin levels by 15,000-fold to approximately 15 U/mL, well within a physiologically acceptable range (22); this method was used throughout this study. A dose of 1011 pfu in 300 μL PBS was injected intravenously into each mouse and the phage allowed to circulate for one hour before harvesting the target organ, brain. Five rounds of selection were performed, and the phage numbers in the brain and in blood were determined by plaque assay. The ratio of phage numbers in the brain to the numbers in blood increased after each round of selection. After the 5th round of selection, the inserts downstream from codon 348 of the 10B capsid protein gene in the DNA isolated from individual plaques were PCR amplified and then sequenced. Of the first 48 sequenced, 47 gave readable sequences for the inserted peptide beginning with the first cysteine (C1). As shown in Table 1, 64% of the recovered sequences encoded the peptide sequence, CAGALCY, with a termination codon immediately after the tyrosine, instead of the expected CX10C motif inserted between the EcoR I and Hind III sites (codons 351 and 359) of the 10B capsid protein. This phage was designated T7-CAGALCY.
To characterize the pharmacokinetics of the T7-CAGALCY phage, it was co-injected in equal numbers into mice together with a control reporter phage, T7-lacZ, which was constructed to carry the lacZ gene controlled by the lac promoter in its genome but with normal T7 coat protein (Fan et al., unpublished data). Groups of mice were euthanized at various time intervals and tissues harvested and analyzed. Both T7-CAGALCY and T7-lacZ phage exhibited similar rapid plasma half-lives of 8.9 and 14.7 min, respectively, over the first 4 h, followed by a slower beta phase of clearance (Fig. 1a), which indicates that both phage have similar blood pharmacokinetics. In the brain, however, whereas the control phage declined rapidly with kinetics similar to the clearance from blood, the T7-CAGALCY phage appeared to accumulate over the first 2 h, then decline slowly thereafter (Fig. 1b). In contrast T7-CAGALCY phage was rapidly cleared from the reference organs studied. In the spleen, a major site for phage clearance, T7-CAGALCYphage was cleared slightly more slowly than T7-lacZ phage, but more rapidly than its clearance from brain (Fig. 1c). These pharmacokinetic data suggest that T7-CAGALCY indeed selectively targets brain microvasculature, while it is rapidly cleared from the blood and other organs.
To confirm selective targeting, the ratio of the numbers of the targeting phage to control phage in the same organ (defined as the selectivity index) was determined by comparing the number of clear (test phage) to blue (T7-lacZ phage) plaques (pfu) in each recovered organ and in blood 2 h after phage administration. The selectivity indices for each organ indicate that T7-CAGALCY phage specifically targets the brain vasculature with a selectivity index around 1,000, but exhibits low specificity for the vascular beds of the other organs (Fig. 1d).
To confirm that the CAGALCY peptide governed the observed phage targeting activity to brain, E-tagged GST fusion proteins with and without the CAGALCY peptide were evaluated for their ability to compete phage targeting in vivo. GST-E-tag-CAGALCY dose-dependently inhibited T7-CAGALCY phage targeting, but had no effect on T7-lacZ control phage levels (Fig. 2a). Control GST-E-tag protein did not affect the accumulation of T7-CAGALCY phage in the brain even at the highest dose tested (100 μg/mouse), confirming that T7-CAGALCY targeting to brain is solely mediated by the peptide moiety, CAGALCY.

GST-E-tag-CAGALCY dose-dependently inhibited T7-CAGALCY phage targeting to the brain. The numbers of phage particles were determined by plaque assay, and expressed as yield/g. GST-E-tag-CAGALCY at a dose of 25 μg/mouse or higher significantly inhibited T7-CAGALCY (a), but not T7-lacZ (b), phage accumulation in the brain (*P < 0.05, **P < 0.01), while GST-E-tag even at a dose as high as 100 μg/mouse had no effect.
In Vivo Kinetic Structure Activity Relationship Analysis
The most notable structural features of the brain-specific peptide were the bulky hydrophobic core formed by the four residues bounded by the two cysteines, the apparent cyclic nature of the peptide and the extracyclic tyrosine at the carboxy terminus. We decided to perform a type of structural activity relationship (SAR) analysis in vivo by building focused libraries around each of these features, to use the relative rates of clearance to rank preferred amino acids at each position, a method we called kinetic SAR or kSAR.
First, we determined whether brain-targeting activity required the peptide to be cyclized through cysteine disulphide bonding. A phage variant carrying a peptide with cysteine-6 mutated to serine was constructed and evaluated by injecting equal numbers of the C-6 and S-6 phage species and determining which was preferentially retained within the brain over time. The substitution of cysteine with serine completely abolished the targeting activity in vivo (data not shown). Since the only difference between serine and cysteine is that the former has an OH instead of an SH group, these results suggest that a cyclic ring formed by disulphide bonding is required for activity.
Next, the role of the terminal tyrosine with a free carboxy terminus was assessed. Phage variants carrying peptides with an additional alanine residue before or after the tyrosine of CAGALCY or lacking the terminal tyrosine altogether exhibited no brain targeting activity. To determine whether a phenyl character with a carboxyl residue at a fixed distance from the hydrophobic ring was required or whether the key element was simply having a free carboxyl residue at a fixed distance from the ring we built a focused library containing all 20 amino acid variants at the Y7 position and performed a kSAR analysis by comparing the frequencies of the injected dose with the relative frequencies in blood and brain at 30 min and 4 h after administration. The actual percentage of each phage variant in the injected dose was determined by sequencing the inserts of 184 phage randomly picked from 184 individual plaques grown from the starting mixture; the frequency ranged from 0–12% with the majority of variants falling between 2 and 7% (Fig. 3a). However, within as short a circulation time as 30 minutes in mice, a selective accumulation of some variants over others was evident. Most notably, the T7–CAGALCY phage, increased markedly from 2.1% in the input mixture to 24% of the recovered pool in perfused brain at 30 min (Fig. 3b). At 4 hours post injection, only six phage variants were detected; T7-CAGALCY accounted for 65.5% and the phage variant carrying a peptide ending with a phenylalanine residue accounted for 24.14% (Fig. 3c) of the recovered phage pool, indicating that a phenyl ring carrying a free carboxylic acid is important for the observed brain targeting activity.

Kinetic structure activity relationship analysis in vivo (kSAR). Frequencies of the 20 variants of CAGALCX in the input phage pool administered (a) and the phage recovered from brains at 30 min (b) and 4 h (c) post injection were determined by growing up individual phage plaques from the pool recovered from the target site, brain, and sequencing the inserts.
In the 3rd set of studies, the nature of the hydrophobic core was assessed. First, each of the non-glycine residues within the two cysteines was mutated to the amino acid, glycine (A2G, A4G and L5G) to determine whether the overall size of the hydrophobic cyclized core was important for brain targeting activity. Each variant was evaluated in vivo in a head-to-head comparison with the lacZ control phage. Mutation of any one of these residues resulted in selectivity indices of less than 1 for these variants, in contrast to the high selectivity index of 700 exhibited by the parent phage (Fig. 4a). The loss of targeting activity was not due to shorter half-lives of these variants, because the ratios of these variants to T7-lacZ control phage recovered from plasma were similar to the parental phage (Fig. 4b). The analysis of the intracyclic residues was expanded by generating phage variants carrying peptides with one of all 20 amino acids at the L5 position. Each phage variant was carefully titered and mixed at equal concentrations (5% for each variant) and a 109 pfu dose of the mixture injected per mouse (three per group). One group was sacrificed at 30 min and one at 4 h and the levels of each phage variant in blood and brain determined. Even within 30 min after injection, a shift in frequency was evident; by 4 h however, only three variants, N5, M5, and L5 were detected. The results suggested that the overall size, hydrophobic nature and nature of the side chain at the five position were all important elements of the pharmacophore and that N or M may be more preferred at the five position (Fig. 4c and d).

Panels a–b: Selectivity indices of T7-CAGALCY and its G-derivatives for the brain (a) and blood (b) in FVB mice. Panels c–d: Complete kSAR analysis of the CAGAXCY library, where a mixture containing approximately 5% of each amino acid variant at the L5 position was injected and phage content in blood isolated 30 min (c) and 4 h (d) after IV administration was analyzed by seeding individual plaques in wells of a 96-well plate (one plate per time point) and sequencing the inserts to quantify the frequency of each variant.
The kSAR analysis and the molecular modeling studies of the CAGALCY peptide and its variants yielded the three dimensional structure shown in Fig. 5a. This structure is representative of a cluster of low energy conformers obtained using the approach described in the Computer Modeling Methods section. It is reasonable to postulate that the bioactive conformation of CAGALCY is found within this cluster of low energy conformers and therefore we proposed a 3D pharmacophore model based on the chemical and structural features of this peptide. Fig. 5b displays a cartoon of the proposed pharmacophore; five pharmacophoric elements were defined from the seven residues of the peptide model: the carboxylic group, benzyl group (Y7) representing an aromatic hydrophobic point, the isobutyl group (L5) and the side chains of A2 and A4 representing aliphatic hydrophobic points. We have also included another optional pharmacophoric element represented by the OH group (Y7) since our analysis indicates that its presence may increase targeting activity. The carboxylic group is regarded as a key pharmacophore element with a specific position within the 3D shape of CAGALCY. It can be assumed that an acid group at this position is essential for interactions with basic regions of the receptor. A pi–pi interaction between the aromatic ring and another aromatic system from the receptor is very plausible since the orientation of this system with respect to the other pharmacophoric points is well defined, our internal data suggests that any deviation of this spatial orientation will result in lost of targeting activity, as in the case of mutating Leucine with Proline (L5P) which produces a kink on the cyclic system resulting in a perturbation of the pharmacophore. The importance of the alkyl chains (A2, A4) indicates that there are two small pockets in the active site of the receptor; the size and hydrophobic properties tolerate no changes. The region occupied by L5 seems to be more flexible since it can accommodate N and M residues; it is not clear from our data what the function of this pharmacophoric element should be.

a. Minimized 3-D structure of cyclic MeCONH-CAGALCY-COOH using molecular mechanics (Sybyl force field) and molecular orbital semi-empirical methodology (PM3). The rendering was done using ArgusLab 4.01 (39), omitting H atoms for clarity. The atoms are represented by the following colors: carbon=gray, nitrogen = blue, oxygen = red and sulfur = yellow. b: A cartoon representing the 3-D pharmacophore of a representative structure of the CALGACY peptide. The pharmacophore elements are represented by the following colors: light green is an aromatic hydrophobic; dark green represents aliphatic hydrophobic; cyan corresponds to the carboxylic acid and red is the OH group. The distances are measured in Angstroms.
The relative distances of all pharmacophoric elements shown in Fig. 5b can be used to perform a three-dimensional query of small molecules databases that can potentially be used to design non-peptidic ligands based on this pharmacophore model. The distances were obtained from the peptide model using a centroid around each of the pharmacophoric elements.
CAGALCY Inhibits Platelet Adhesion to Endothelium In Vivo
The requirement for a terminal free carboxylic acid on an aromatic amino acid for T7-CAGALCY brain-targeting activity is similar to the tyrosine acid analogue nature of the αIIbβ3 (gpIIb/IIIa) integrin antagonist, Tirofiban® (23,24). Specific orientation of a tyrosine acid relative to a hydrophobe is also a hallmark of mimetics of other integrin binding sites (25–33). To address the working hypothesis that the CAGALCY peptide represents a mimetic of a receptor/ligand binding site expressed on circulating cells that is involved in cell adhesion to the brain endothelium, the ability of GST-CAGALCY to inhibit platelet or leukocyte adhesion to the pial microvasculature activated by endotoxin was measured by intravital microscopy to monitor blood cell adhesion under flow directly in vivo.
First, white blood cells were labeled in situ with rhodamine G and the pial vasculature of mice that had been injected with LPS 4 h earlier was observed before and after administration of 100 μg GST-CAGALCY or control GST protein. While the rolling and adhesion of the white cells to the microvascular endothelium of venules was clearly visible, GST-CAGALCY had no effect on the numbers of rolling or adherent white cells (data not shown). However, when labeled platelets were injected into mice with endotoxemia a marked effect of GST-CAGALCY was observed on platelet adhesion to the pial vasculature. While mouse platelets adhered markedly to small and large venules and large and small arterioles of endotoxic mice injected with PBS or GST control protein, mice injected with GST-CAGALCY had significantly lower numbers of rolling and adherent platelets in all vessel types (Fig. 6).

Inhibition of platelet adhesion to small and large inflamed venules and arterioles by GST-CAGALCY. Groups of LPS-treated mice were injected with fluorescent CFDA-labelled platelets and the pial vasculature image captured at two 5-min intervals before injecting either control GST or GST-CAGALCY protein compounds. Images at 1 and 20 min post administration of test compound were scored for numbers of adherent platelets. Treatment groups are indicated on the x-axis (n is shown in the upper panel) and the mean ± SE of platelet number per unit vessel area for five vessels per animal is plotted on the y-axis (panels a–d). e: fluorescent images prior to and 20 min after administration of GST-CAGALCY fusion protein into endotoxemic mice; the larger fluorescent spots represent mini thrombi.
DISCUSSION
Phage display of peptide libraries has been explored as a means to map the vascular diversity of different organs (15–17). The rationale for this approach is that a single phage with multiple copies of a single peptide could behave much as an activated blood cell, with the multiple copies of a given ligand conferring avidity to complement the low affinity typically associated with short peptide ligands. However, screening in vivo has inherent issues of high background, all the more apparent when a high complexity library is administered. The data presented in this report, indicate that incorporating the natural clearance mechanisms for particulates via the MPS phagocytic system can be harnessed to reduce noise. By incorporating kinetic and selectivity parameters into the screening methods we identified a cyclic peptide, CAGALCY, which was highly selective for brain vasculature.
Having identified a lead, it is standard practice in pharmaceutical research to perform lead optimization and definition of the basic pharmacophore structure by structure-activity relationship (SAR) analysis (35–37). This approach has been demonstrated in a number of instances for peptide ligands. For example, the rational design of RGD peptides that bind selectively to platelet αIIbβ3 or αvβ3 integrin compared to other integrin family members for the purpose of improving thrombotic responses or tumor-targeting properties, respectively, has been described (28–34). Here we describe a novel, rapid and effective means to perform kinetic SAR (kSAR) in vivo using a modification of the phage panning method whereby focused libraries around different structural elements are evaluated over time to determine the relative ranking order for the retention of each variant at the target site. Using this approach, we demonstrated that a key element of the CAGALCY targeting peptide was the free carboxylic acid of the C-terminal tyrosine residue. This terminal tyrosine-carboxylic acid was a fortuitous outcome of the termination codon that occurred immediately after the Y7 codon of the inserted DNA that resulted in a C-terminus immediately after the inserted CAGALCY peptide motif and the deletion of ∼35 amino acids from the C-terminus of the wild-type capsid protein. Analysis of the pharmacophore indicated some similarities to known ligand mimetics of cell adhesion molecules expressed on blood cells. By differential labeling of the different blood cell types under activating conditions (systemic LPS), we were able to demonstrate that the CAGALCY cyclic peptide expressed as a GST-fusion protein was an effective inhibitor of platelet adhesion to brain microvasculature.
Our own unpublished studies using the CAGALCY-expressing phage as a positive control, indicate that there is an upper limit of library diversity of one in 105 pfu, if a hit is to be reliably identified by in vivo phage display. Although Pasqualini and Ruoslahti (15–17) state that in vivo phage display allows panning with libraries of large diversity (up to 1011), in fact their methods cite isolating 300 individual plaques after each round of panning for amplification for the subsequent round of panning. Thus after the first round, the library diversity was automatically reduced to a maximum diversity of 300. Our data indicate that in vivo phage display can be used most effectively when library complexity is relatively low, as is the case with focused libraries around a structural component within a peptide lead. Such kSAR studies provide a rapid means to study peptide structure activity relationship in vivo. This method can be applied to any peptide that can be displayed on phage surface, and allows the optimization of the structure of the peptide ligand for its interaction with its cognate receptor in its physiological environment; therefore it represents a major advancement in peptide ligand SAR research.
CONCLUSION
Incorporating the normal blood phamacokinetics of T7 phage into in vivo phage display methodology improves the ability to recover targeting peptide motifs. Applying kSAR analysis to the targeting peptide allows effective lead optimization and definition of the pharmacophore. This approach led to the isolation of a brain-specific peptide, CAGALCY, which appears to function as an effective antagonist of platelet adhesion to activated pial microvasculature.
Abbreviations
- CFDA:
-
carboxyfluorescein diacetate succinimidyl ester
- GST:
-
glutathione S-transferase
- IPTG:
-
Isopropyl β-D-1-thiogalactopyranoside
- KSAR:
-
kinetic structure activity relationship
- LPS:
-
Lipopolysaccharide
- MPS:
-
monophagocytic system
- PEG:
-
polyethylene glycol
References
P. R. Streeter, E. L. Berg, B. T. N. Rouse, R. F. Bargatze, and E. C. Butcher. A tissue-specific endothelial cell molecule involved in lymphocyte homing. Nature 331:41–46 (1988).
L. A. Lasky. Selectins: interpreters of cell-specific carbohydrate information during inflammation. Science 258:964–969 (1992).
T. M. Carlos and J. M. Harlan. Leukocyte-endothelial adhesion molecules. Blood 84:2068–2101 (1994).
M. Salmi and S. Jalkanen. How do lymphocytes know where to go: current concepts and enigmas of lymphocyte homing. Adv. Immunol. 64:139–218 (1997).
E. C. Butcher and L. J. Picker. Lymphocyte homing and homeostasis. Science 272:60–66 (1996).
R. O. Hynes. Integrins: bidirectional, allosteric signaling machines. Cell 110:673–687 (2002).
P. Kubes and P. A. Ward. Leukocyte recruitment and the acute inflammatory response. Brain Pathol. 10:127–135 (2000).
C. Robert and T. S. Kupper. Inflammatory skin diseases, T cells, and immune surveillance. N. Engl. J. Med. 341:1817–1828 (1999).
T. A. Springer. Traffic signals on endothelium for lymphocyte recirculation and leukocyte emigration. Annu. Rev. Physiol. 57:827–872 (1995).
U. H. von Andrian and C. R. Mackay. T-cell function and migration. Two sides of the same coin. N. Engl. J. Med. 343:1020–1034 (2000).
R. P. McEver. Adhesive interactions of leukocytes, platelets, and the vessel wall during hemostasis and inflammation. Thromb. Haemost. 86:746–756 (2001).
W. C. Aird. The role of the endothelium in severe sepsis and multiple organ dysfunction syndrome. Blood 101:3765–3777 (2003).
J. N. George, J. E. Sadler, and B. Lammle. Platelets: thrombotic thrombocytopenic purpura. Hematology (Am. Soc. Hematol. Educ. Program) 1:315–334 (2002).
J. L. Moake. Thrombotic microangiopathies. N. Engl. J. Med. 347:589–600 (2002).
R. Pasqualini and E. Ruoslahti. Organ targeting in vivo using phage display peptide libraries. Nature 380:364–366 (1996).
D. Rajotte, W. Arap, M. Hagedorn, E. Koivunen, R. Pasqualini, and E. Ruoslahti. Molecular heterogeneity of the vascular endothelium revealed by in vivo phage display. J. Clin. Invest. 102:430–437 (1998).
W. Arap, R. Pasqualini, and E. Ruoslahti. Cancer treatment by targeted drug delivery to tumor vasculature in a mouse model. Science 279:377–380 (1998).
E. Ruoslahti and D. Rajotte. An address system in the vasculature of normal tissues and tumors. Annu. Rev. Immunol. 18:813–827 (2000).
T. M. Allen. Interactions of liposomes and other drug carriers with the mononuclear phagocyte system. In G. Gregoriadis (ed.), Liposomes as Drug Carriers, Recent Trends and Progress, Wiley, Chichester, 1988, pp. 37–50.
F. Nilsson, L. Tarli, F. Viti, and D. Neri. The use of phage display for the development of tumour targeting agents. Adv. Drug. Deliv. Rev. 43:165–196 (2000).
Y. L. Yip, N. J. Hawkins, G. Smith, and R. L. Ward. Biodistribution of filamentous phage-Fab in nude mice. J. Immunol. Methods 225:171–178 (1999).
J. O’Malley, L. E. Matesic, M. C. Zink, J. D. Strandberg, M. L. Mooney, A. De Maio, and R. H. Reeves. Comparison of acute endotoxin-induced lesions in A/J and C57BL/6J mice. J. Heredity 89:525–530 (1998).
D. G. Abraham, E. M. Nutt, R. A. Bednar, B. Bednar, R. J. Gould, and L. T. Duong. Arginine-glycine-aspartic acid mimics can identify a transitional activation state of recombinant alphaIIb beta3 in human embryonic kidney 293 cells. Mol. Pharmacol. 52:227–236 (1997).
B. K. Blackburn, A. Lee, M. Baier, B. Kohl, A. G. Olivero, R. Matamoros, K. D. Robarge, and R. S. McDowell. From peptide to non-peptide. 3. Atropisomeric GPIIbIIIa antagonists containing the 3,4-dihydro-1H-1,4-benzodiazepine-2,5-dione nucleus. J. Med. Chem. 40:717–729 (1997).
N. J. Dubree, D. R. Artis, G. Castanedo, J. Marsters, D. Sutherlin, L. Caris, K. Clark, S. M. Keating, M. H. Beresini, H. Chiu, S. Fong, H. B. Lowman, N. J. Skelton, and D. Y. Jackson. Selective alpha4beta7 integrin antagonists and their potential as anti-inflammatory agents. J. Med. Chem. 45:3451–3457 (2002).
M. S. Egbertson, C. T. Chang, M. E. Duggan, R. J. Gould, W. Halczenko. G. D. Hartman, W. I. Laswell, J. J. Lynch, R. J. Lynch, and P. D. Manno. Non-peptide fibrinogen receptor antagonists. 2. Optimization of a tyrosine template as a mimic for Arg-Gly-Asp. J. Med. Chem. 37:2537–2551 (1994).
T. R. Gadek, D. J. Burdick, R. S. McDowell, M. S. Stanley, J. C. Marsters Jr., K. L. Paris, D. A. Oare, M. E. Reynolds, C. Ladner, K. A. Zioncheck, W. P. Lee, P. Gribling, W. Dennis, L. G. Presta, and S. C. Bodary. Generation of an LFA-1antagonist by the transfer of the ICAM-1 immunoregulatory epitope to a small molecule. Science 295:1086–1089 (2002).
M. Gurrath, G. Muller, H. Kesslar, M. Aumailley, and R. Timpl. Conformation/activity studies of rationally designed potent anti-adhesive RGD peptides. Eur. J. Biochem. 210:911–921 (1992).
R. Haubner, H. J. Wester, F. Burkhart, R. Senekowitsch-Schmidtke, W. Weber, S. L. Goodman, H. Kessler, and M. Schwaiger. Glycosylated RGD-containing peptides: tracer for tumor targeting and angiogenesis imaging with improved biokinetics. J. Nucl. Med. 42:326–336 (2001).
D. Y. Jackson, C. Quan, R. R. Artis, T. Rawson, B. Blackburn, M. Struble, G. Fitzgerald, K. Chan, S. Mullins, J. P. Burnier, W. J. Fairbrother, K. Clark, M. Berisini, H. Chui, M. Renz, S. Jones, and S. Fong. Protein alpha 4 beta 1 peptide antagonists as potential anti-inflammatory agents. J. Med. Chem. 40:3359–3368 (1997).
D. Y. Jackson. Alpha 4 integrin antagonists. Curr. Pharm. Des. 8:1229–1253 (2002).
R. J. Kok, A. J. Schraa, E. J. Bos, H. E. Moorlag, S. A. Asgeirsdottir, M. Everts, D. K. Meijer, and G. Molema. Preparation and functional evaluation of RGD-modified proteins as alpha(v)beta(3) integrin directed therapeutics. Bioconjug. Chem. 13:128–135 (2002).
I. Sircar, K. S. Gudmundsson, R. Martin, J. Liang, S. Nomura, H. Jayakumar, B. R. Teegarden, D. M. Nowlin, P. M. Cardarelli, J. R. Mah, S. Connell, R. C. Griffith, and E. Lazarides. Synthesis and SAR of N-benzoyl-L-bipheylalanine derivatives: discovery of TR-14035, a dual alpha(4)beta(7)/alpha(4)beta(1) integrin antagonist. Bioorg. Med. Chem. 10:2051–2066 (2002).
D. M. Huryn, A. W. Konradi, S. Ashwell, S. B. Freedman, L. J. Lombardo, M. A. Pleiss, E. D. Thorsett, T. Yednock, and J. D. Kennedy. The identification and optimization of orally efficacious, small molecule VLA-4 antagonists. Curr. Top. Med. Chem. 4:1473–1484 (2004).
J. K. Seydel. Sulfonamides, structure-activity relationship, and mode of action. Structural problems of the antibacterial action of 4-aminobenzoic acid (PABA) antagonists. J. Pharm. Sci. 57:1455–1478 (1968).
M. S. Tute. Principles and practice of Hansch analysis: a guide to structure-activity correlation for the medicinal chemist. Adv. Drug Res. 6:1–77 (1971).
Y. C. Martin. Theoretical basis of medicinal chemistry: structure activity relationships and three dimensional structures of small and macromolecules. In Y. C. Martin, V. Austel, and E. Kutter (eds.), Modern Drug Research. Paths to Better and Safer Drugs, Marcel Dekker, New York, 1989, pp 161–216.
Alchemy 2000. Tripos Inc., St Louis, Missouri, http://www.tripos.com.
ArgusLab 4.0.1 Mark A. Thompson, Planaria Software LLC, Seattle, WA http://www.arguslab.com.
Acknowledgments
The authors thank Jie Li and Clara Polizzi for their technical assistance with the intravital microscopy studies and Zhe Li, Shaan Tolani, and Zaid Yusufi for technical assistance with the phage display work. The intravital microscopy studies were supported in part by NIH grant A140667-06 to HvdH.
Author information
Authors and Affiliations
Corresponding author
Additional information
Targeted Molecules Corp. is now a wholly owned subsidiary of Chromos Molecular Systems Inc, 8081 Lougheed Highway, Burnaby, BC, V5A 1W9, Canada.
Rights and permissions
About this article
Cite this article
Fan, X., Venegas, R., Fey, R. et al. An In Vivo Approach to Structure Activity Relationship Analysis of Peptide Ligands. Pharm Res 24, 868–879 (2007). https://doi.org/10.1007/s11095-007-9238-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11095-007-9238-z