
Purpose
We investigated whether dipyridamole and various calcium channel blockers are inhibitors and/or substrates of breast cancer resistance protein (BCRP).
Methods
The effect of dipyridamole and the calcium channel blockers on mitoxantrone efflux by BCRP-overexpressing human embryonic kidney (HEK) cells was determined by flow cytometry. The ability of some of these compounds to reverse BCRP-mediated mitoxantrone resistance was measured by cytotoxicity assays. Transport studies were performed using radiolabeled compounds.
Results
Dipyridamole, nicardipine, nitrendipine, and nimodipine effectively inhibited BCRP-mediated mitoxantrone efflux; however, bepridil, diltiazem, and verapamil had no significant effect. Nifedipine is a much weaker BCRP inhibitor compared with other dihydropyridines tested. Nicardipine and dipyridamole were the most potent BCRP inhibitors among the compounds tested with IC50 values of 4.8 ± 1.3 and 6.4 ± 0.9 μM, respectively. Nicardipine and dipyridamole also effectively reversed BCRP-mediated mitoxantrone resistance in HEK cells. [3H]Nitrendipine was found not to be transported by BCRP. However, the transport of [3H]dipyridamole by BCRP was observed in both HEK and Madin–Darby canine kidney cells stably expressing the transporter, and this transport was completely abolished by fumitremorgin C, a known BCRP inhibitor.
Conclusions
Dipyridamole and several dihydropyridines are effective BCRP inhibitors, but bepridil, diltiazem, and verapamil are not. We also identified a new BCRP substrate, dipyridamole.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The human breast cancer resistance protein (BCRP/ABCG2) is an efflux drug transporter belonging to the subfamily G of the large ABC transporter superfamily (1–4). BCRP confers resistance to a large number of chemotherapeutic agents including topotecan, mitoxantrone, and flavopiridol by enhanced drug efflux (1,5–7). In addition, BCRP is also an active transporter for conjugated or unconjugated organic anions such as estrone-3-sulfate, 17β-estradiol, 17-(β-d-glucuronide), and methotrexate (8–11). Recent studies have demonstrated that amino acid at position 482 in BCRP is an important determinant for activity and substrate specificity of this transporter (11–14).
BCRP has been detected in many types of solid and hematological tumors (1). A potential link between BCRP expression and clinical drug resistance has been implicated in several recent clinical studies (15–17). BCRP is also expressed in many normal tissues. For instance, BCRP is highly expressed in the epithelium of the small intestine, in the liver canalicular membrane, and in the apical membrane of placental syncytiotrophoblasts (18). Consistent with this pattern of tissue localization, a growing number of studies have demonstrated the importance of BCRP in absorption, elimination, and distribution of drugs or xenobiotics/toxins that are substrates of this transporter (19–24).
Calcium channel blockers were first introduced in the 1980s for the treatment of hypertension. Since then, their use has been extended to many other conditions such as angina pectoris, heart failure, myocardial ischemia, and paroxysmal supraventricular tachyarrhythmias (25). In addition to their use in these diseases, calcium channel blockers such as verapamil and dihydropyridines are effective inhibitors of P-gp and multidrug resistance protein 1 (MRP1) (26–34). Some of these compounds have been tested to determine if they can reverse multidrug resistance in cancer patients (35,36). Calcium channel blockers remain one of the most extensively characterized classes of modulating agents for ABC transporters.
In the present study, we investigated the potential interaction of various calcium channel blockers of diverse chemical structures with BCRP. We demonstrated that dipyridamole and several dihydropyridines are effective BCRP inhibitors, but other calcium channel blockers, such as bepridil, diltiazem, and verapamil, are not. These studies also identified a new BCRP substrate, dipyridamole. In the course of preparing this manuscript, a study by Zhou et al. (37) was accepted for publication. These authors also showed that various pyridines and dihydropyridines, including several dihydropyridines synthesized by the authors, were potent BCRP inhibitors. Further studies with Sprague–Dawley rats found that one of the dihydropyridines could inhibit BCRP in vivo.
Materials and Methods
Materials
Nimodipine, nicardipine hydrochloride, nitrendipine, nifedipine, dipyridamole, diltiazem, verapamil hydrochloride, bepridil hydrochloride, and mitoxantrone (MX) hydrochloride were purchased from Sigma (St. Louis, MO, USA). Fumitremorgin C (FTC) was a gift from Dr. Susan E. Bates [National Cancer Institute (NCI), Bethesda, MD, USA]. [3H]Dipyridamole (30 Ci/mmol) was from Moravek Biochemicals (Brea, CA, USA), and [3H]nitrendipine (87 Ci/mmol) was from Perkin-Elmer Life Sciences (Boston, MA, USA). High-performance liquid chromatography grade dimethyl sulfoxide (DMSO) was obtained from Fisher Scientific (Pittsburgh, PA, USA) and was used as the solvent to dissolve the above drugs and FTC. Eagle's minimal essential medium (MEM), penicillin–streptomycin–glutamine solution, and Gibco Opti-MEM were purchased from ATCC (Manassas, VA, USA). Dulbecco's modified Eagle's phenol-free low-glucose medium (DMEM), phosphate-buffered saline (PBS), trypsin–EDTA solution, and fetal bovine serum (FBS) were from Invitrogen (Carlsbad, CA, USA). The mAb BXP-21 was from Kamiya Biomedical Co. (Seattle, WA, USA).
Cell Culture
HEK293 cells stably transfected with the pcDNA empty vector [human embryonic kidney (HEK)/vector] and cDNA coding for wild-type BCRP (HEK/482R) were obtained from Dr. Susan Bates (38). The cell lines were grown and maintained in MEM supplemented with 10% FBS, 2 mM l-glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin, and 0.5 mg/ml G418 (Mediatech, Herndon, VA, USA) at 37°C in a 5% CO2 incubator. Cells were grown to 80–90% confluence and harvested by treating with trypsin–EDTA for subculturing or functional assays. Only cells within six to seven passages were used in subsequent transport experiments.
Flow Cytometric Efflux Assays
The flow cytometric efflux assays with MX as a model BCRP substrate were performed essentially the same as described previously (39,40) with minor modifications. HEK/482R or HEK/vector control cells were harvested and suspended in incubation buffer [DMEM supplemented with 5 mM 4-(2-Hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) buffer] at a cell concentration of approximately 106 cells in 1 ml of volume. In the accumulation phase, 1 ml of cell suspension was incubated with 10 μM MX and various concentrations of calcium channel blockers and dipyridamole or 10 μM FTC as a positive control at 37°C for 30 min. Cells were then washed once with ice-cold PBS and resuspended in 1 ml of incubation buffer containing the calcium channel blockers and dipyridamole or FTC at the respective concentrations as in the accumulation phase. The cell suspensions were then incubated at 37°C for another 1 h to allow efflux of MX. Cells were then washed once and finally suspended in 500 μl of ice-cold PBS. Intracellular MX fluorescence was measured with a BD FACSCAN flow cytometer equipped with a 488-nm argon laser and a 650-nm band-pass filter as previously described (39). Preliminary studies showed that all the compounds used in this study are not fluorescent compounds as determined using the same instrument setting for MX (data not shown). No more than 1% (v/v) DMSO was used in all experiments. No effects of the vehicle on MX efflux were observed at this concentration. Cells in medium containing MX alone yielded the control histograms. Cells in medium containing MX and the calcium channel blockers or FTC yielded the calcium channel blocker and dipyridamole or FTC histograms, respectively. The percentage of the median fluorescence of the calcium channel blocker and dipyridamole or FTC histograms to the median fluorescence of the control histogram was used to express relative inhibition of BCRP-mediated MX efflux by calcium channel blockers and dipyridamole or FTC, respectively.
IC50 values representing the inhibitory effectiveness of the calcium channel blockers and dipyridamole on BCRP-mediated MX efflux were calculated by fitting the following model to the data (Fig. 3) using nonlinear regression (WinNonLin version 3.2, Pharsight, Mountain View, CA, USA):
where I and I max are the relative inhibition and the maximal relative inhibition, respectively. I o is the I value at zero concentration of an inhibitor, which is generated by nonlinear regression using the above model and close to 100%. C is the concentration of the calcium channel blockers and dipyridamole, γ is the Hill coefficient, and IC50 is the concentration of the calcium channel blockers and dipyridamole leading to half-maximal inhibition of BCRP-mediated MX efflux.
Mitoxantrone Cytotoxicity Assays
Cytotoxicity assays were carried out as follows. HEK/482R or HEK/vector cells were seeded in a 96-well plate at a density of 4000–5000 cells/well in 200 μl of MEM supplemented with 10% FBS. After 24 h, when the cells reached approximately 30–40% confluence, the medium was replaced with fresh medium containing 5% FBS. MX, nicardipine, and dipyridamole were then added to the medium at various concentrations, and the cells were further incubated for 72 h. The medium was then removed, and 200 μl of fresh medium without FBS was immediately added to each well, followed by the addition of 50 μl of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-tetrazolium bromide (Sigma) dissolved in PBS at a concentration of 2.5 mg/ml. The plates were then incubated at 37°C for 4 h in the dark. The medium was then removed, and 200 μl of DMSO was added to each well, followed by the addition of 25 μl of Sorensen's glycine buffer (0.1 M glycine, 0.1 M NaCl equilibrated to pH 10.5 with 0.1 M NaOH). The optical density of the plates was read at 560 nm using a microplate reader (Molecular Devices Thermomax, Ramsey, MN, USA). Cells untreated with MX were used as controls (100% cell survival). In the reversal experiments, cells untreated with MX in the presence of a reversal agent were used as controls (100% cell survival). EC50 values representing MX toxicity in the presence or absence of nicardipine and dipyridamole were estimated by fitting the following model to the data (Fig. 4) using nonlinear regression (WinNonLin version 3.2):
where S is the cell survival as percentage of the optical density of the control cells, S max is the maximal cell survival, S o is the lowest residual cell survival at the high drug concentrations, C is the MX concentration, γ is the Hill coefficient, and EC50 is the MX concentration leading to half-maximal cell survival. Four determinations were carried out within each experiment, and three independent experiments were performed. The concentration of DMSO used as the solvent to dissolve drugs was less than 0.1% (v/v) in all experiments. No cytotoxicity of the vehicle was observed at this concentration.
Efflux of [3H]Dipyridamole and [3H]Nitrendipine
To determine if the calcium channel blockers and dipyridamole are substrates of BCRP, efflux assays using [3H]dipyridamole and [3H]nitrendipine were carried out. HEK/482R and HEK/vector cells at a concentration of approximately 106 cells/ml were incubated at 37°C with the radiolabeled compounds in 0.5 ml of incubation buffer (DMEM supplemented with 5 mM HEPES buffer) in the presence or absence of 10 μM FTC for 30 min to allow accumulation of [3H]dipyridamole or [3H]nitrendipine. The concentrations of [3H]dipyridamole and [3H]nitrendipine used in the assays were 25 and 10 nM, respectively. The cells were then transferred on ice and washed twice with ice-cold PBS. The cell pellets were resuspended in 0.5 ml of incubation buffer in the presence or absence of 10 μM FTC, and incubation was continued for another 1 h at 37°C. The efflux reactions were then stopped by adding 0.95 ml of ice-cold PBS. After washing cells once with PBS, the cell pellets were dissolved in 1 ml of 1% sodium dodecyl sulfate (SDS), and the cell lysates were subjected to counting in a scintillation counter. The intracellular concentrations of [3H]dipyridamole or [3H]nitrendipine were calculated based on radioactivity associated with the cell pellets and presented as femtomoles of drugs per 0.5 × 106 cells.
Stable Expression of BCRP in MDCK Cells
The pcDNA-482R plasmid containing the full length of wild-type human BCRP cDNA, a kind gift from Dr. Susan E. Bates (NCI), and the empty vector were stably transfected into the Madin–Darby canine kidney (MDCK) cells as follows. MDCK cells were maintained in MEM with 10% FBS at 37°C in a 5% CO2 incubator. For stable transfection, approximately 4 × 105 cells were seeded in each well of 12-well plates, and 24 h later, cells were transfected with 2 μg of DNA in 100 μl Opti-MEM (Gibco) at a 1:3 ratio of plasmid DNA to Lipofectamine 2000 (Invitrogen). After 12 h, cells were incubated for an additional 48 h in fresh medium containing 10% FBS before adding 1 mg/ml G418 (Mediatech). Cell culture was continued in 1 mg/ml G418 for 3 days. Then the cells were pooled and subcultured in serial dilutions with frequent removal of dead cells by changing the medium until resistant colonies appeared. After 2 weeks of selection in G418, cell colonies were removed individually using trypsinized sterile cloning disks (Corning Glassworks, Corning, NY, USA) and subcultured in the presence of 1 mg/ml G418. The level of BCRP expression in G418-resistant cell populations was determined by immunoblotting as described below. The cell line expressing the highest level of BCRP was used in the subsequent Transwell transport assays. Cells were grown and maintained in MEM supplemented with 10% FBS and 1 mg/ml G418 at 37°C in a 5% CO2 incubator. Cells were grown to 80–90% confluent and treated with trypsin–EDTA prior to harvesting for subculturing, immunoblotting, confocal microscopy, or Transwell transport experiments. Only cells within six to seven passages were used in subsequent transport experiments.
SDS–Polyacrylamide Gel Electrophoresis, Immunoblotting, and Confocal Microscopy
Whole cell lysates from MDCK cell lines were prepared as previously described (39). Plasma membrane vesicles isolated from HEK/482R cells were prepared as previously described (41). Protein concentrations were determined by the Bio-Rad Dc protein assay kit (Bio-Rad, Hercules, CA, USA) using bovine serum albumin as standard. The whole cell lysates and plasma membrane preparations were then subjected to immunoblotting using BXP-21, a BCRP-specific mAb (Kamiya Biomedical) as previously described (39). For detection of β-actin, a mAb specific for human β-actin (Sigma) was used as primary antibody at 1:50,000 dilution. The plasma membrane localization of BCRP in MDCK cells was analyzed by confocal microscopy using mAb BXP-21 essentially the same as previously described (41).
Transwell Transport of [3H]Dipyridamole
Transwell transport assays were performed on microporous polycarbonate membrane filters (3.0-μm pore size, 24-mm diameter, Costar, Corning, NY, USA). Briefly, MDCK cells transfected with BCRP cDNA (MDCK/482R) and the empty vector (MDCK/vector) were seeded onto filter membranes at a density of 1 × 106 cells per well and grown for 7–8 days with replacement of medium every day. To ensure that the cells were polarized and formed a tight junction, transport assays were conducted when the average transepithelial electrical resistance in each well reached 180–200 Ω, measured with a Millicell-ERS (Millipore, Billeria, MA, USA). The MDCK cell monolayer grown under similar culture conditions has been shown to form a tight junction suitable for Transwell transport studies (42). One hour before the experiment, medium was replaced in both compartments with Opti-MEM containing inhibitors (5 μM FTC or 20 μM veparamil or both). The loading volume is 1.5 ml in the apical compartment and 2.5 ml in the basolateral compartment. The experiments were started by replacing the medium with fresh Opti-MEM containing 500 nM dipyridamole traced with [3H]dipyridamole in the presence or absence of the inhibitors in the appropriate compartment. Transport of dipyridamole was tested in two directions (apical-to-basolateral, A → B; and basolateral-to-apical, B → A), every 1 h up to 3 h, by taking aliquots (100 μl) from the opposite compartment. After each sampling, 100 μl of medium with or without inhibitors were added back to the sampling compartment to keep the volume consistent. The amounts of dipyridamole in the aliquots were measured by scintillation counting. The percentage of initially added radioactivity appearing in the opposite compartment was calculated. The transport ratio R was calculated by dividing the basolateral-to-apical transport by the apical-to-basolateral transport at the end of 3 h. The studies were performed in triplicate at 37°C in a humidified incubator.
Statistical Analysis
Data were analyzed for statistical significance using Student's t test. Differences with p values of <0.05 were considered statistically significant.
Results
Inhibition of BCRP-Mediated MX Efflux by Dipyridamole and Calcium Channel Blockers
To investigate the effects of dipyridamole and calcium channel blockers on BCRP efflux activity, MX was used as a model substrate. We measured intracellular MX fluorescence in HEK/482R and HEK/vector cells in the presence or absence of these compounds by flow cytometry. Flow cytometric efflux assays using MX as a model BCRP substrate under similar conditions have been widely used to determine BCRP transport activity and screen BCRP inhibitors (39,40,43). We tested seven calcium channel blockers and dipyridamole with diverse chemical structures (Fig. 1). FTC, a known BCRP inhibitor, was used as a positive control. The intracellular MX fluorescence (arbitrary unit) in HEK/482R and HEK/vector cells in the absence of any test compounds, measured at the end of the efflux assay, was approximately 38 and 140, respectively (data not shown). Thus, the intracellular MX level in HEK/vector control cells is approximately 3.7 times greater than that in HEK/482R cells, indicating that MX is transported out of the cells by BCRP. The addition of 10 μM FTC significantly increased intracellular MX fluorescence in HEK/482R cells approximately 3.5-fold but had no effects on the intracellular MX fluorescence in vector control cells (Fig. 2). These data confirm that FTC is a BCRP inhibitor and validate the efflux assay. Of the eight compounds tested, nicardipine, nitrendipine, nimodipine, and dipyridamole at 10 and 50 μM significantly increased the intracellular MX level approximately 2- to 8-fold in HEK/482R cells; however, no more than 2-fold increase of intracellular MX fluorescence by these compounds was observed in the vector control cells (Fig. 2). These results clearly indicate that nicardipine, nitrendipine, nimodipine, and dipyridamole are effective inhibitors of BCRP. Among the above compounds tested, nicardipine was the most potent BCRP inhibitor, which produced an approximately 5-fold increase of intracellular MX fluorescence in HEK/482R cells at 10 μM, followed by dipyridamole, nitrendipine, and nimodipine. Nifedipine at 10 μM did not significantly inhibit BCRP efflux activity; however, at 50 μM, it increased the intracellular MX level approximately 3-fold, suggesting that nifedipine is a relatively weak BCRP inhibitor (Fig. 2). Diltiazem and verapamil at 10 μM slightly increased intracellular MX fluorescence (less than 2-fold) in the vector control cells but had no significant effects on MX efflux by HEK/482R cells. At 50 μM, diltiazem and verapamil slightly increased the intracellular MX level in both HEK/482R and HEK/vector cells (less than 2-fold). These results suggest that diltiazem and verapamil are not effective BCRP inhibitors. In the vector control cells, bepridil exhibited a minor inhibition on MX efflux at 10 μM; however, its effect at 50 μM could not be measured, possibly because of cytotoxicity. Although intracellular MX fluorescence in HEK/482R cells could be measured in the presence of bepridil at 10 and 50 μM, this compound did not significantly alter MX efflux by HEK/482R cells, suggesting that bepridil is not a BCRP inhibitor (Fig. 2).

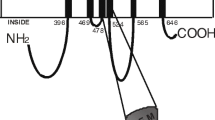
The chemical structure of dipyridamole, bepridil, diltiazem, verapamil, nicardipine, nitrendipine, nimodipine, and nifedipine.

Effects of dipyridamole and the calcium channel blockers on mitoxantrone (MX) efflux in human embryonic kidney (HEK) cells. Intracellular MX fluorescence in the presence of 10 μM (A) and 50 μM (B) dipyridamole or the calcium channel blockers was measured as described in Materials and Methods. The solid bars represent the intracellular MX levels in HEK/vector control cells. The open bars represent the intracellular MX levels in HEK/482R cells. The intracellular MX levels in the absence of test compounds in both cell lines were set as controls (100%). Relative MX levels in the presence of the test compounds as compared with the controls are shown. The data shown are mean ± SD from three independent experiments. The differences in MX levels are statistically significant: *p < 0.05; **p < 0.01; ***p < 0.001 as compared with the controls.
Concentration-Dependent Inhibition of BCRP-Mediated MX Efflux by Dipyridamole and Calcium Channel Blockers
To further evaluate the relative inhibitory potency of dipyridamole and the calcium channel blockers for BCRP, concentration-dependent effects of nicardipine, dipyridamole, and nimodipine on MX efflux by HEK/482R cells were measured by flow cytometry. Figure 3 illustrates that the intracellular MX level in HEK/482R cells was increased by addition of nicardipine, dipyridamole, and nimodipine in a concentration-dependent manner. The IC50 values for inhibition of BCRP-mediated MX efflux were then calculated to be 4.8 ± 1.3, 6.4 ± 0.9, and 13.7 ± 3.8 μM for nicardipine, dipyridamole, and nimodipine, respectively. Therefore, nicardipine seems to be the most potent BCRP inhibitor, followed by dipyridamole and nimodipine. This pattern of relative inhibitory potency is consistent with the data shown in Fig. 2. Although not determined, the IC50 value of nitrendipine would be expected to be in between the IC50 values of dipyridamole and nimodipine based on the data shown in Fig. 2.

Concentration-dependent effects of nicardipine (A), dipyridamole (B), and nimodipine (C) on MX efflux by HEK/482R cells. Intracellular MX fluorescence in the presence of the test compounds at a range of concentrations (0–20 μM for nicardipine and dipyridamole; 0–50 μM for nimodipine) was measured as described in Materials and Methods. Intracellular MX levels in the absence of any test compounds were set as controls (100%). Relative intracellular MX levels in the presence of the test compounds as compared with the controls are shown. The data are expressed as mean ± SD from three independent experiments.
Reversal of MX Resistance by Dipyridamole and Nicardipine
To further confirm the inhibitory effects of dipyridamole and the calcium channel blockers on BCRP and investigate the potential of using these compounds as modulators of BCRP-mediated drug resistance, we examined the capability of nicardipine and dipyridamole to reverse MX resistance of HEK cells conferred by BCRP. Because nicardipine and dipyridamole are the two most potent BCRP inhibitors found in the above studies, they were used as representatives in the cytotoxicity studies. To determine whether nicardipine or dipyridamole itself is toxic to HEK cells, both HEK/482R and HEK/vector control cells were incubated with dipyridamole or nicardipine alone for 72 h at 5 and 10 μM. Neither nicardipine nor dipyridamole showed significant toxicity to HEK cells at concentrations up to 10 μM (data not shown).
Therefore, we first examined the effects of nicardipine and dipyridamole on MX toxicity to HEK/vector control cells. As shown in Fig. 4B and D and in Table I, in the absence of any test compounds, the EC50 value of MX in HEK/vector cells was 0.92 ± 0.19 nM. The EC50 values of MX in the vector control cells were not significantly changed by the addition of 5 or 10 μM nicardipine. The addition of 10 μM dipyridamole also had no apparent effect on the MX toxicity profile to the vector control cells; however, 5 μM dipyridamole decreased the EC50 value to 0.3 ± 0.1 μM. Because we found that dipyridamole at 5 μM had no apparent toxicity to HEK cells, at this point, we do not have an explanation for this observation. Thus, nicardipine and dipyridamole do not seem to have significant effects on the MX toxicity profile of the HEK/vector control cells.

Effects of nicardipine and dipyridamole on MX toxicity in HEK cells. (A) The MX toxicity in HEK/vector cells (□) and HEK/482R cells (▪) in the absence of nicardipine, and in HEK/482R cells in the presence of nicardipine (•, 5 μM; ▵, 10 μM). (B) The MX toxicity in HEK/vector cells in the absence (□) and presence of nicardipine (•, 5 μM; ▵, 10 μM). (C) The MX toxicity in HEK/vector cells (□) and HEK/482R cells (▪) in the absence of dipyridamole, and in HEK/482R cells in the presence of dipyridamole (•, 5 μM; ▵, 10 μM). (D) The MX toxicity in HEK/vector cells in the absence (□) and presence of dipyridamole (•, 5 μM; ▵, 10 μM). Results shown are mean ± SD from four determinations in a typical experiment. Similar results were obtained in two additional independent experiments.
Further analysis revealed that BCRP confers an approximately 36-fold resistance to MX in HEK/482R cells with an EC50 value of 33.2 ± 13.0 nM (Fig. 4A, C and Table I). We then examined whether nicardipine and dipyridamole are able to restore the sensitivity of HEK/482R cells to MX. Upon the addition of 5 μM nicardipine, the MX toxicity profile of HEK/482R cells was shifted toward that of HEK/vector cells with a decrease in EC50 value to 14.4 ± 10.1 nM (Fig. 4A and Table I). When 10 μM nicardipine was used, the EC50 value was further decreased to 4.4 ± 1.4 nM (Fig. 4A and Table I). Thus, nicardipine at 10 μM effectively reversed MX resistance in HEK/482R cells with a significant decrease in relative resistance (RR) from 36.0 to 6.0. Similarly, dipyridamole also drastically and effectively reversed MX resistance. Upon the addition of 5 and 10 μM dipyridamole, the RR values were decreased to 5.3 and 0.5, respectively (Fig. 4B and Table I). Taken together, the MX toxicity studies confirmed that nicardipine and dipyridamole are indeed BCRP inhibitors and are able to reverse BCRP-mediated resistance to MX in vitro in HEK cells.
Efflux of [3H]Nitrendipine and [3H]Dipyridamole
To further examine whether dipyridamole and the calcium channel blockers are substrates of BCRP, direct efflux assays using radiolabeled compounds were carried out. We examined [3H]nitrendipine and [3H]dipyridamole because they are the only two drugs commercially available in a radiolabeled form among the BCRP inhibitors found in this study. As shown in Fig. 5A, the efflux of [3H]nitrendipine was not significantly different in HEK/482R and HEK/vector control cells. Also, the addition of 10 μM FTC did not affect [3H]nitrendipine efflux in either cell line. These data suggest that [3H]nitrendipine is not a BCRP substrate, although it is an effective inhibitor for the transporter. On the other hand, the intracellular levels of [3H]dipyridamole in HEK/482R cells were significantly reduced to approximately half of those in HEK/vector control cells (Fig. 5B). Furthermore, this reduction of [3H]dipyridamole levels in HEK/482R cells was completely abrogated by the addition of 10 μM FTC; however, the addition of FTC had no significant influence on the [3H]dipyridamole levels in HEK/vector cells. These results indicate that dipyridamole is a BCRP substrate.

Efflux of [3H]nitrendipine (A) and [3H]dipyridamole (B) in HEK cells. Efflux of [3H]nitrendipine (10 nM) and [3H]dipyridamole (25 nM) in HEK/vector and HEK/482R cells was determined as described in Materials and Methods. The open bars represent intracellular drug levels in the absence of FTC. The solid bars represent intracellular drug levels in the presence of 10 μM FTC. Results shown are mean ± SD of triplicate determinations in a typical experiment. Similar results were obtained from two additional independent experiments. The differences in intracellular drug levels are statistically significant: *p < 0.05; **p < 0.01.
Transwell Transport of Dipyridamole
Further evidence that dipyridamole is a BCRP substrate was obtained from Transwell transport studies. To perform Transwell transport studies, we developed a stable expression system for human BCRP in MDCK cells. Stable transfection of the plasmid pcDNA-482R containing the full length of wild-type human BCRP cDNA into MDCK cells yielded a clone (MDCK/482R) with substantial expression of BCRP as determined by immunoblotting (Fig. 6A, lane 4). The apparent molecular weight (approximately 75 kDa) of BCRP expressed in MDCK cells (Fig. 6A, lane 4) is the same as the BCRP produced in HEK cells (Fig. 6A, lane 1). No BCRP expression was detected in the MDCK/vector control cells (Fig. 6A, lanes 2 and 3). Moreover, MX efflux by the MDCK/482R cells was completely inhibited by FTC (data not shown). The immunofluorescent experiment using mAb BXP-21 revealed strong plasma membrane staining, indicating that BCRP was predominantly expressed in the plasma membrane of the MDCK cells (Fig. 6B). These results confirmed that BCRP was functionally expressed and properly routed to the plasma membrane of the MDCK cells.

Stable expression of breast cancer resistance protein (BCRP) in Madin–Darby canine kidney (MDCK) cells. (A) Immunoblotting of BCRP using mAb BXP-21 (dilution 1:500) (40 μg of total protein in each lane). Lane 1, plasma membrane vesicles isolated from HEK/482R cells; lanes 2 and 3, whole cell lysates prepared from MDCK/vector control cells; lane 4, whole cell lysates prepared from MDCK/482R cells. BCRP and β-actin are indicated by arrows. (B) Confocal microscopy of MDCK/482R cells. BCRP was detected using mAb BXP-21 as described under Materials and Methods and is indicated in green. No green fluorescence was detected in MDCK/vector cells (data not shown). A selected area of MDCK/482R cells is shown.
In the MDCK/vector cells, the B → A transport of dipyridamole was found to be significantly greater than the A → B transport, and this Transwell transport of dipyridamole was completely inhibited by the addition of 20 μM verapamil; however, the addition of 5 μM FTC showed no significant effect (data not shown). These data suggest that there is an endogenous efflux transporter in MDCK/vector control cells, which can transport dipyridamole and can be inhibited by verapamil. This endogenous efflux transporter is unlikely to be BCRP, as we have already shown that verapamil at concentrations up to 50 μM did not inhibit BCRP, and that FTC is a specific BCRP inhibitor (Fig. 2). This endogenous efflux transporter is most likely to be P-gp because the parental MDCK cells have been shown to express substantial amounts of endogenous P-gp [44]. We also found similar P-gp levels in the MDCK/vector and MDCK/482R cells detected by immunoblotting (data not shown). Because 20 μM verapamil completely inhibited the dipyridamole transport in MDCK/vector cells, to eliminate the contribution of this endogenous transporter on dipyridamole transport, we included 20 μM verapamil in all subsequent Transwell transport experiments. Indeed, when 20 μM verapamil was added, comparable B → A and A → B transports of dipyridamole were observed in MDCK/vector cells (Fig. 7C), and the addition of 5 μM FTC had no effect on both the B → A and A → B transports (Fig. 7D). In contrast, in the presence of 20 μM verapamil, the B → A transport of dipyridamole in MDCK/482R cells was significantly greater than the A → B transport with a transport ratio of approximately 2.5 (Fig. A). The addition of 5 μM FTC completely abolished the difference between the B → A and A → B translocations of dipyridamole (Fig. 7B). These data indicate that BCRP mediates the apically directed transport of dipyridamole in MDCK/482R cells.

Transwell transport of dipyridamole. Transepithelial transport of dipyridamole (490 nM cold dipyridamole plus 10 nM [3H]dipyridamole) across MDCK/482R (A and B) and MDCK/vector (C and D) cell monolayers in the absence (A and C) or the presence (B and D) of 5 μM FTC. The experiments were started by adding dipyridamole to one compartment. Verapamil (20 μM) was included in all the experiments. The radioactivity appearing in the opposite compartment was measured, and the molar amounts of dipyridamole were calculated. The percentage of the amount of initially applied dipyridamole appearing in the opposite compartment was plotted. The results are presented as mean ± SD from three independent experiments. Translocation from the basolateral compartment to the apical compartment (▪, □); translocation from the apical compartment to the basolateral compartment (▴, ▵). R values are the transport ratios of the basolateral-to-apical transport to the apical-to-basolateral transport at the end of the experiments (3 h).
Discussion
In the present study, we investigated the interactions of BCRP with cardiovascular drugs including dipyridamole and seven calcium channel blockers with different chemical structures (Fig. 1). For example, verapamil belongs to the class of phenylalkylamines, diltiazem to the class of benzothiazepines, and nicardipine to the class of dihydropyridines. We found that the dihydropyridines (nicardipine, nitrendipine, and nimodipine) and dipyridamole can effectively inhibit BCRP-mediated MX efflux in HEK/482R cells (Figs. 2 and 3), indicating that these compounds are highly efficient BCRP inhibitors. We also noticed a small but statistically significant increase of intracellular MX fluorescence in HEK/vector control cells by the above compounds (Fig. 2). These results suggest that there is an endogenous efflux transporter(s) for MX in HEK cells that can be inhibited by the dihydropyridines and dipyridamole. MX is also a substrate of P-gp or MRP1 (45,46). We have previously shown that the HEK cells express little endogenous P-gp, MRP1, or MRP2 (39). Thus, it seems unlikely that any of these pumps is the endogenous transporter for MX that can be inhibited by the dihydropyridines and dipyridamole. Nicardipine is the most potent inhibitor among the compounds tested, followed by dipyridamole, nitrendipine, and nimodipine (Fig. 2). Consistent with this trend, the IC50 value of nimodipine was approximately three times greater than those of nicardipine and dipyridamole (Table I). Both nicardipine and dipyridamole could effectively inhibit MX efflux in HEK/482R cells at concentrations below 5 μM (Fig. 3). Nifedipine is a significantly weaker inhibitor, which only mildly inhibited BCRP at 50 μM (Fig. 2). Very recently, Zhou et al. (37) also reported effective inhibition of BCRP-mediated MX efflux by various dihydropyridines in BCRP-expressing human breast cancer MCF-7/MX100 cells and human large lung cancer H640/MX20 cells. Although different expression systems were used, similar results as obtained in this study with respect to inhibition of BCRP by dihydropyridines were reported. Both studies showed that nicardipine is the most potent BCRP inhibitor among the commercially available dihydropyridines with IC50 value around 5 μM. Both studies also found that nifedipine is a much weaker BCRP inhibitor compared with other dihydropyridines tested. Zhou et al. (37) also found that various pyridine and dihydropyridine analogs they synthesized were effective BCRP inhibitors.
We report here for the first time that three other calcium channel blockers, bepridil, diltiazem, and verapamil, only slightly influenced MX efflux in both HEK/482R and HEK/vector control cells (Fig. 2). Therefore, bepridil, diltiazem, and verapamil are not BCRP inhibitors. It should be noted that verapamil is an efficient inhibitor of P-gp. Therefore, verapamil is an ideal inhibitor that can be used in transport studies to differentiate P-gp and BCRP. Interestingly, bepridil at 50 μM in the presence of 10 μM MX enhanced MX toxicity to HEK/vector control cells, leading to cell lysis, and hence, intracellular MX accumulation in HEK/vector cells could not be measured. However, bepridil and MX at the same concentrations did not display similar toxicity to HEK/482R cells. One possible explanation is that bepridil can inhibit an endogenous efflux transporter for MX other than BCRP and thus further increases intracellular MX concentration in HEK/vector cells to a high level that is sufficient to produce toxicity to HEK cells. This, however, does not occur in HEK/482R cells because of efficient MX efflux by BCRP. Another possibility is that bepridil itself is toxic to HEK cells and is a BCRP substrate. However, we found that BCRP did not confer resistance to bepridil toxicity in HEK/482R cells (data not shown).
Additional evidence that dipyridamole and the dihydropyridines are effective BCRP inhibitors was obtained from in vitro drug resistance studies. Both nicardipine and dipyridamole at 5 and 10 μM effectively reversed resistance of HEK cells to MX conferred by BCRP (Fig. 4 and Table I). Because nicardipine or dipyridamole alone at 5 and 10 μM did not display significant toxicity to HEK cells (data not shown), the reversal effects of nicardipine and dipyridamole are most likely because of inhibition of BCRP-mediated MX efflux. Dipyridamole has been shown to significantly inhibit secretion of nitrofurantoin into milk in rats (47). BCRP has been identified to be the transporter that is responsible for secreting nitrofurantoin into milk (48). Our data therefore support the notion that dipyridamole reduces milk secretion of nitrofurantoin by inhibiting BCRP.
Upon analyzing the chemical structures of the compounds tested (Fig. 1), we noticed that the BCRP inhibitors identified in this study all contain either a single-ring pyridine (the dihydropyridines) or a double-ring pteridine (dipyridamole); however, such single or multiple nitrogen-containing aromatic rings do not exist in bepridil, diltiazem, and verapamil, which are not BCRP inhibitors. Therefore, the nitrogen-containing heterocyclic compounds are likely to constitute one class of effective BCRP inhibitors. This finding is consistent with the observation of Zhou et al. (37) that the pyridine compounds are also effective BCRP inhibitors. The size of side chain groups attached to the heterocyclic ring seems to be critical in determining inhibition potency. Among the dihydropyridines, nicardipine, the most potent BCRP inhibitor, contains the largest group at position R1 and the smallest group at position R2 attached to pyridine (Fig. 1). In contrast, nifedipine, the least potent BCRP inhibitor, contains the smallest group at position R1 and the largest group at position R2. Nitrendipine and nimodipine, with inhibitory potency in between that of nicardipine and nifedipine, coincidentally contain side chains attached to pyridine that are of intermediate size. The molecular mechanism by which these nitrogen-containing heterocyclic compounds inhibit BCRP is currently unknown. These compounds may either directly interact with BCRP at the substrate-binding sites to compete with substrates for binding on BCRP or inhibit BCRP by modulating its ATPase activity. Both types of interaction have been reported between the dihydropyridines and P-gp (49,50).
To investigate whether the inhibitors identified are also BCRP substrates, we performed direct transport studies using radiolabeled compounds. We found that [3H]nitrendipine is not a BCRP substrate, although it is an effective BCRP inhibitor (Fig. A). Likewise, the anti-HIV protease inhibitors (39) and the glucocorticoid drugs (51) have also been shown to be effective inhibitors but not substrates of BCRP. On the other hand, [3H]dipyridamole was found to be actively transported by BCRP with two different assays: the efflux assay using HEK/482R cells in suspension (Fig. 5) and the Transwell transport assay using MDCK/482R cells (Fig. 7). Dipyridamole is highly lipophilic with a logP value 3.92 (n-octanol/0.1 N NaOH), but does not cross the blood–brain barrier (52). Because BCRP is highly expressed at the blood–brain barrier, our data suggest that BCRP may play a role in limiting brain penetration of the drug. As BCRP is highly expressed in the apical membrane of the epithelium in the small intestine, our data that dipyridamole is a BCRP substrate may, in part, explain the relatively low and variable bioavailability (11–44%) typically associated with the drug (53).
Nicardipine and dipyridamole inhibit BCRP with IC50 values at low micromolar concentrations, which could be achieved in the small intestine after oral administration of these drugs. For example, the current recommended dose of nicardipine is 30 mg three times a day. Assuming 100% dissolution of the drug in the intestine, the estimated intestinal luminal concentration of nicardipine would be approximately 40–120 μM. Dipyridamole is orally administered to patients at doses around 100–400 mg daily. At such doses, the estimated intestinal luminal concentrations of dipyridamole would be around 130–530 μM. These concentrations far exceed the IC50 values of nicardipine and dipyridamole for BCRP. Thus, inhibition of intestinal BCRP by the orally administered nicardipine or dipyridamole is expected. Our finding that dipyridamole and several dihydropyridines are effective BCRP inhibitors is therefore clinically significant with respect to drug–drug interactions. Zhou et al. (37) have demonstrated that coadministration of one of the dihydropyridines they synthesized with topotecan produced a 2-fold increase in oral bioavailability and the peak plasma concentration of topotecan in female Sprague–Dawley rats. Because topotecan is a high-affinity BCRP substrate and does not undergo extensive metabolism in rats, the authors concluded that this increase in topotecan exposure was a result of inhibition of BCRP. Thus, our data and those reported by Zhou et al. (37) predict that coadministration of oral nicardipine or dipyridamole and topotecan would result in a pharmacokinetic drug interaction.
In conclusion, the data reported in this study and by Zhou et al. (37) suggest that the dihydropyridines and dipyridamole are effective BCRP inhibitors. Drug–drug interactions with the dihydropyridines and dipyridamole may occur in patients taking these drugs simultaneously with BCRP substrate drugs. These BCRP inhibitors may be used as lead compounds for further development of clinically useful BCRP inhibitors to be used as reversal agents for tumors overexpressing BCRP. There are not many drugs, other than chemotherapeutic agents, to be identified as BCRP substrates. Our studies found a new BCRP substrate, dipyridamole. BCRP is likely to play a role in in vivo disposition of the drug. We also developed an expression system for human BCRP in MDCK cells, which is useful for investigating the interaction of drugs with BCRP.
Abbreviations
- ABC:
-
ATP-binding cassette
- BCRP:
-
breast cancer resistance protein
- DMSO:
-
dimethyl sulfoxide
- FTC:
-
fumitremorgin C
- HEK:
-
human embryonic kidney cells
- mAb:
-
monoclonal antibody
- MDCK:
-
Madin–Darby canine kidney
- MRP1:
-
multidrug resistance protein 1
- MRP2:
-
multidrug resistance protein 2
- MX:
-
mitoxantrone
- P-gp:
-
P-glycoprotein
References
L. A. Doyle D. D. Ross (2003) ArticleTitleMultidrug resistance mediated by the breast cancer resistance protein BCRP (ABCG2) Oncogene 22 7340–7358 Occurrence Handle14576842
L. A. Doyle W. Yang L. V. Abruzzo T. Krogmann Y. Gao A. K. Rishi D. D. Ross (1998) ArticleTitleA multidrug resistance transporter from human MCF-7 breast cancer cells Proc. Natl. Acad. Sci. USA 95 15665–15670 Occurrence Handle9861027 Occurrence Handle10.1073/pnas.95.26.15665 Occurrence Handle1:CAS:528:DyaK1MXhvFeitQ%3D%3D
K. Miyake L. Mickley T. Litman Z. Zhan R. Robey B. Cristensen M. Brangi L. Greenberger M. Dean T. Fojo S. E. Bates (1999) ArticleTitleMolecular cloning of cDNAs which are highly overexpressed in mitoxantrone-resistant cells: demonstration of homology to ABC transport genes Cancer Res. 59 8–13 Occurrence Handle9892175 Occurrence Handle1:CAS:528:DyaK1MXmsFKqsQ%3D%3D
R. Allikmets L. M. Schriml A. Hutchinson V. Romano-Spica M. Dean (1998) ArticleTitleA human placenta-specific ATP-binding cassette gene (ABCP) on chromosome 4q22 that is involved in multidrug resistance Cancer Res. 58 5337–5339 Occurrence Handle9850061 Occurrence Handle1:CAS:528:DyaK1cXotVSkt7Y%3D
T. Litman T. E. Druley W. D. Stein S. E. Bates (2001) ArticleTitleFrom MDR to MXR: new understanding of multidrug resistance systems, their properties and clinical significance Cell. Mol. Life Sci. 58 931–959 Occurrence Handle11497241 Occurrence Handle1:CAS:528:DC%2BD3MXlvFSms74%3D
A. Haimeur G. Conseil R. G. Deeley S. P. Cole (2004) ArticleTitleThe MRP-related and BCRP/ABCG2 multidrug resistance proteins: biology, substrate specificity and regulation Curr. Drug Metab. 5 21–53 Occurrence Handle14965249 Occurrence Handle10.2174/1389200043489199 Occurrence Handle1:CAS:528:DC%2BD2cXhvVCisLo%3D
S. E. Bates R. Robey K. Miyake K. Rao D. D. Ross T. Litman (2001) ArticleTitleThe role of half-transporters in multidrug resistance J. Bioenerg. Biomembranes 33 503–511 Occurrence Handle1:CAS:528:DC%2BD38XlsVyltw%3D%3D
Y. Imai S. Asada S. Tsukahara E. Ishikawa T. Tsuruo Y. Sugimoto (2003) ArticleTitleBreast cancer resistance protein exports sulfated estrogens but not free estrogens Mol. Pharmacol. 64 610–618 Occurrence Handle12920197 Occurrence Handle10.1124/mol.64.3.610 Occurrence Handle1:CAS:528:DC%2BD3sXmvFaiu7g%3D
M. Suzuki H. Suzuki Y. Sugimoto Y. Sugiyama (2003) ArticleTitleABCG2 transports sulfated conjugates of steroids and xenobiotics J. Biol. Chem. 278 22644–22649 Occurrence Handle12682043 Occurrence Handle1:CAS:528:DC%2BD3sXksF2it7w%3D
E. L. Volk E. Schneider (2003) ArticleTitleWild-type breast cancer resistance protein (BCRP/ABCG2) is a methotrexate polyglutamate transporter Cancer Res. 63 5538–5543 Occurrence Handle14500392 Occurrence Handle1:CAS:528:DC%2BD3sXntlersrs%3D
Z.-S. Chen R. W. Robey M. G. Belinsky I. Shchaveleva X.-Q. Ren Y. Sugimoto D. D. Ross S. E. Bates G. D. Kruh (2003) ArticleTitleTransport of methotrexate, methotrexate polyglutamates, and 17{beta}-estradiol 17-({beta}-d-glucuronide) by ABCG2: effects of acquired mutations at R482 on methotrexate transport Cancer Res. 63 4048–4054 Occurrence Handle12874005 Occurrence Handle1:CAS:528:DC%2BD3sXlsFOju7s%3D
Y. Honjo C. A. Hrycyna Q. W. Yan W. Y. Medina-Perez R. W. Robey A. Laar Particlevan de T. Litman M. Dean S. E. Bates (2001) ArticleTitleAcquired mutations in the MXR/BCRP/ABCP gene alter substrate specificity in MXR/BCRP/ABCP-overexpressing cells Cancer Res. 61 6635–6639 Occurrence Handle11559526 Occurrence Handle1:CAS:528:DC%2BD3MXntFaisr0%3D
J. D. Allen S. C. Jackson A. H. Schinkel (2002) ArticleTitleA mutation hot spot in the Bcrp1 (Abcg2) multidrug transporter in mouse cell lines selected for doxorubicin resistance Cancer Res. 62 2294–2299 Occurrence Handle11956086 Occurrence Handle1:CAS:528:DC%2BD38Xjt1ejtbo%3D
C. Ozvegy-Laczka G. Koblos B. Sarkadi A. Varadi (2005) ArticleTitleSingle amino acid (482) variants of the ABCG2 multidrug transporter: major differences in transport capacity and substrate recognition Biochim. Biophys. Acta 1668 53–63 Occurrence Handle15670731
K. Yoh G. Ishii T. Yokose Y. Minegishi K. Tsuta K. Goto Y. Nishiwaki T. Kodama M. Suga A. Ochiai (2004) ArticleTitleBreast cancer resistance protein impacts clinical outcome in platinum-based chemotherapy for advanced non-small cell lung cancer Clin. Cancer Res. 10 1691–1697 Occurrence Handle15014021 Occurrence Handle1:CAS:528:DC%2BD2cXhvFCltr0%3D
A. Suvannasankha H. Minderman K. L. O'Loughlin T. Nakanishi L. A. Ford W. R. Greco M. Wetzler D. D. Ross M. R. Baer (2004) ArticleTitleBreast cancer resistance protein (BCRP/MXR/ABCG2) in adult acute lymphoblastic leukaemia: frequent expression and possible correlation with shorter disease-free survival Br. J. Haematol. 127 392–398 Occurrence Handle15521915 Occurrence Handle10.1111/j.1365-2141.2004.05211.x Occurrence Handle1:CAS:528:DC%2BD2cXhtVOms7jE
Z. Benderra A. M. Faussat L. Sayada J. Y. Perrot D. Chaoui J. P. Marie O. Legrand (2004) ArticleTitleBreast cancer resistance protein and P-glycoprotein in 149 adult acute myeloid leukemias Clin. Cancer Res. 10 7896–7902 Occurrence Handle15585622 Occurrence Handle1:CAS:528:DC%2BD2cXhtVCrs73O
M. Maliepaard G. L. Scheffer I. F. Faneyte M. A. Gastelenvan A. C. Pijnenborg A. H. Schinkel M. J. Vijvervan ParticleDe R. J. Scheper J. H. Schellens (2001) ArticleTitleSubcellular localization and distribution of the breast cancer resistance protein transporter in normal human tissues Cancer Res. 61 3458–3464 Occurrence Handle11309308 Occurrence Handle1:CAS:528:DC%2BD3MXjtFSju7o%3D
J. W. Jonker J. W. Smit R. F. Brinkhuis M. Maliepaard J. H. Beijnen J. H. Schellens A. H. Schinkel (2000) ArticleTitleRole of breast cancer resistance protein in the bioavailability and fetal penetration of topotecan J. Natl. Cancer Inst. 92 1651–1656 Occurrence Handle11036110 Occurrence Handle10.1093/jnci/92.20.1651 Occurrence Handle1:CAS:528:DC%2BD3cXnvV2mtbw%3D
J. W. Jonker M. Buitelaar E. Wagenaar M. A. Valk ParticleVan Der G. L. Scheffer R. J. Scheper T. Plosch F. Kuipers R. P. Elferink H. Rosing J. H. Beijnen A. H. Schinkel (2002) ArticleTitleThe breast cancer resistance protein protects against a major chlorophyll-derived dietary phototoxin and protoporphyria Proc. Natl. Acad. Sci. USA 99 15649–15654 Occurrence Handle12429862 Occurrence Handle10.1073/pnas.202607599 Occurrence Handle1:CAS:528:DC%2BD3sXjvV2k
C. M. Kruijtzer J. H. Beijnen H. Rosing W. W. Bokkel Huinink M. Schot R. C. Jewell E. M. Paul J. H. Schellens (2002) ArticleTitleIncreased oral bioavailability of topotecan in combination with the breast cancer resistance protein and P-glycoprotein inhibitor GF120918 J. Clin. Oncol. 20 2943–2950 Occurrence Handle12089223 Occurrence Handle1:CAS:528:DC%2BD38Xlslyrt7w%3D
S. Cisternino C. Mercier F. Bourasset F. Roux J. M. Scherrmann (2004) ArticleTitleExpression, up-regulation, and transport activity of the multidrug-resistance protein abcg2 at the mouse blood–brain barrier Cancer Res. 64 3296–3301 Occurrence Handle15126373 Occurrence Handle10.1158/0008-5472.CAN-03-2033 Occurrence Handle1:CAS:528:DC%2BD2cXjs1ehtb8%3D
A. E. Herwaarden J. W. Jonker E. Wagenaar R. F. Brinkhuis J. H. Schellens J. H. Beijnen A. H. Schinkel (2003) ArticleTitleThe breast cancer resistance protein (Bcrp1/Abcg2) restricts exposure to the dietary carcinogen 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine Cancer Res. 63 6447–6452 Occurrence Handle14559835
G. Merino J. W. Jonker E. Wagenaar A. E. Herwaardenvan A. H. Schinkel (2005) ArticleTitleThe breast cancer resistance protein (BCRP/ABCG2) affects pharmacokinetics, hepatobiliary excretion, and milk secretion of the antibiotic nitrofurantoin Mol. Pharmacol. 67 1758–1764 Occurrence Handle15709111 Occurrence Handle1:CAS:528:DC%2BD2MXktFegtLg%3D
E. Grossman F. H. Messerli (2004) ArticleTitleCalcium antagonists Prog. Cardiovasc. Dis. 47 34–57 Occurrence Handle15517514 Occurrence Handle1:CAS:528:DC%2BD2cXhtFWqurvJ Occurrence Handle10.1016/j.pcad.2004.04.006
M. Kamiwatari Y. Nagata H. Kikuchi A. Yoshimura T. Sumizawa N. Shudo R. Sakoda K. Seto S. Akiyama (1989) ArticleTitleCorrelation between reversing of multidrug resistance and inhibiting of [3H]azidopine photolabeling of P-glycoprotein by newly synthesized dihydropyridine analogues in a human cell line Cancer Res. 49 3190–3195 Occurrence Handle2566378 Occurrence Handle1:CAS:528:DyaL1MXkslSnsb4%3D
T. Tsuruo H. Iida S. Tsukagoshi Y. Sakurai (1981) ArticleTitleOvercoming of vincristine resistance in P388 leukemia in vivo and in vitro through enhanced cytotoxicity of vincristine and vinblastine by verapamil Cancer Res. 41 1967–1972 Occurrence Handle7214365 Occurrence Handle1:CAS:528:DyaL3MXitVaks7s%3D
T. Tsuruo H. Iida S. Tsukagoshi Y. Sakurai (1983) ArticleTitlePotentiation of vincristine and adriamycin effects in human hemopoietic tumor cell lines by calcium antagonists and calmodulin inhibitors Cancer Res. 43 2267–2272 Occurrence Handle6831450 Occurrence Handle1:CAS:528:DyaL3sXktVShsbg%3D
H. Tanabe S. Tasaka H. Ohmori N. Gomi Y. Sasaki T. Machida M. Iino A. Kiue S. Naito M. Kuwano (1998) ArticleTitleNewly synthesized dihydropyridine derivatives as modulators of P-glycoprotein-mediated multidrug resistance Bioorg. Med. Chem. 6 2219–2227 Occurrence Handle9881113 Occurrence Handle1:CAS:528:DyaK1MXhvFyl
U. Vanhoefer M. R. Muller R. A. Hilger B. Lindtner U. Klaassen N. Schleucher Y. M. Rustum S. Seeber A. Harstrick (1999) ArticleTitleReversal of MDR1-associated resistance to topotecan by PAK-200S, a new dihydropyridine analogue, in human cancer cell lines Br. J. Cancer 81 1304–1310 Occurrence Handle10604726 Occurrence Handle10.1038/sj.bjc.6694384 Occurrence Handle1:CAS:528:DC%2BD3cXls1KltQ%3D%3D
U. Vanhoefer S. Cao H. Minderman K. Toth R. J. Scheper M. L. Slovak Y. M. Rustum (1996) ArticleTitlePAK-104P, a pyridine analogue, reverses paclitaxel and doxorubicin resistance in cell lines and nude mice bearing xenografts that overexpress the multidrug resistance protein Clin. Cancer Res. 2 369–377 Occurrence Handle9816180 Occurrence Handle1:CAS:528:DyaK28XhvVaqs7o%3D
T. Sumizawa Z. S. Chen Y. Chuman K. Seto T. Furukawa M. Haraguchi A. Tani N. Shudo S. I. Akiyama (1997) ArticleTitleReversal of multidrug resistance-associated protein-mediated drug resistance by the pyridine analog PAK-104P Mol. Pharmacol. 51 399–405 Occurrence Handle9058594 Occurrence Handle1:CAS:528:DyaK2sXhvFektLs%3D
K. Takara T. Sakaeda Y. Tanigawara K. Nishiguchi N. Ohmoto M. Horinouchi F. Komada N. Ohnishi T. Yokoyama K. Okumura (2002) ArticleTitleEffects of 12 Ca2+ antagonists on multidrug resistance, MDR1-mediated transport and MDR1 mRNA expression Eur. J. Pharm. Sci. 16 159–165 Occurrence Handle12128170 Occurrence Handle1:CAS:528:DC%2BD38XlsVKrtLw%3D
X. F. Zhou L. Zhang E. Tseng E. A. Scott-Ramsay J. J. Schentag R. A. Coburn M. E. Morris (2005) ArticleTitleNew 4-aryl-1,4-dihydropyridines and 4-arylpyridines as P-glycoprotein inhibitors Drug Metab. Dispos. 33 321–328 Occurrence Handle15585608 Occurrence Handle10.1124/dmd.104.003558 Occurrence Handle1:CAS:528:DC%2BD2MXntVamsb4%3D
M. Lehnert K. Mross J. Schueller B. Thuerlimann N. Kroeger H. Kupper (1998) ArticleTitlePhase II trial of dexverapamil and epirubicin in patients with non-responsive metastatic breast cancer Br. J. Cancer 77 1155–1163 Occurrence Handle9569055 Occurrence Handle1:CAS:528:DyaK1cXisVKhs7Y%3D
W. H. Wilson S. E. Bates A. Fojo G. Bryant Z. Zhan J. Regis R. E. Wittes E. S. Jaffe S. M. Steinberg J. Herdt et al. (1995) ArticleTitleControlled trial of dexverapamil, a modulator of multidrug resistance, in lymphomas refractory to EPOCH chemotherapy J. Clin. Oncol. 13 1995–2004 Occurrence Handle7636540 Occurrence Handle1:CAS:528:DyaK2MXntlKkurY%3D
X. F. Zhou X. Yang Q. Wang R. A. Coburn M. E. Morris (2005) ArticleTitleEffects of dihydropyridines and pyridines on multidrug resistance mediated by breast cancer resistance protein: in vitro and in vivo studies Drug Metab. Dispos. 33 1220–1228 Occurrence Handle15908473 Occurrence Handle10.1124/dmd.104.003558 Occurrence Handle1:CAS:528:DC%2BD2MXntVamsb4%3D
R. W. Robey Y. Honjo K. Morisaki T. A. Nadjem S. Runge M. Risbood M. S. Poruchynsky S. E. Bates (2003) ArticleTitleMutations at amino-acid 482 in the ABCG2 gene affect substrate and antagonist specificity Br. J. Cancer 89 1971–1978 Occurrence Handle14612912 Occurrence Handle10.1038/sj.bjc.6601370 Occurrence Handle1:CAS:528:DC%2BD3sXovVers7g%3D
A. Gupta Y. Zhang J. D. Unadkat Q. Mao (2004) ArticleTitleHIV protease inhibitors are inhibitors but not substrates of the human breast cancer resistance protein (BCRP/ABCG2) J. Pharmacol. Exp. Ther. 310 334–341 Occurrence Handle15007102 Occurrence Handle10.1124/jpet.104.065342 Occurrence Handle1:CAS:528:DC%2BD2cXlsVOjsbc%3D
R. W. Robey Y. Honjo A. Laarvan Particlede K. Miyake J. T. Regis T. Litman S. E. Bates (2001) ArticleTitleA functional assay for detection of the mitoxantrone resistance protein, MXR (ABCG2) Biochim. Biophys. Acta 1512 171–182 Occurrence Handle11406094 Occurrence Handle1:CAS:528:DC%2BD3MXkt12itbo%3D
R. R. Vethanayagam H. Wang A. Gupta Y. Zhang F. Lewis J. D. Unadkat Q. Mao (2005) ArticleTitleFunctional analysis of the human variants of breast cancer resistance protein: I206L, N590Y, and D620N Drug Metab. Dispos. 33 697–705 Occurrence Handle15743976 Occurrence Handle10.1124/dmd.105.003657 Occurrence Handle1:CAS:528:DC%2BD2MXkslelu7g%3D
G. C. Williams G. T. Knipp P. J. Sinko (2003) ArticleTitleThe effect of cell culture conditions on saquinavir transport through, and interactions with, MDCKII cells overexpressing hMDR1 J. Pharm. Sci. 92 1957–1967 Occurrence Handle14502536 Occurrence Handle10.1002/jps.10458 Occurrence Handle1:CAS:528:DC%2BD3sXnvFOmsrY%3D
S. Zhang X. Yang M. E. Morris (2004) ArticleTitleFlavonoids are inhibitors of breast cancer resistance protein (ABCG2)-mediated transport Mol. Pharmacol. 65 1208–1216 Occurrence Handle15102949 Occurrence Handle10.1124/mol.65.5.1208 Occurrence Handle1:CAS:528:DC%2BD2cXjsFKgtro%3D
S. P. Hammerle B. Rothen-Rutishauser S. D. Kramer M. Gunthert H. Wunderli-Allenspach (2000) ArticleTitleP-glycoprotein in cell cultures: a combined approach to study expression, localisation, and functionality in the confocal microscope Eur. J. Pharm. Sci. 12 69–77 Occurrence Handle11121735 Occurrence Handle1:CAS:528:DC%2BD3cXot1yiu7g%3D Occurrence Handle10.1016/S0928-0987(00)00142-1
T. Litman M. Brangi E. Hudson P. Fetsch A. Abati D. D. Ross K. Miyake J. H. Resau S. E. Bates (2000) ArticleTitleThe multidrug-resistant phenotype associated with overexpression of the new ABC half-transporter, MXR (ABCG2) J. Cell. Sci. 113 IssueIDPt 11 2011–2021 Occurrence Handle10806112 Occurrence Handle1:CAS:528:DC%2BD3cXksVKntrg%3D
H. Minderman A. Suvannasankha K. L. O'Loughlin G. L. Scheffer R. J. Scheper R. W. Robey M. R. Baer (2002) ArticleTitleFlow cytometric analysis of breast cancer resistance protein expression and function Cytometry 48 59–65 Occurrence Handle12116365 Occurrence Handle10.1002/cyto.10111 Occurrence Handle1:CAS:528:DC%2BD38Xlt1Cgtro%3D
P. M. Gerk L. Hanson M. C. Neville P. J. McNamara (2002) ArticleTitleSodium dependence of nitrofurantoin active transport across mammary epithelia and effects of dipyridamole, nucleosides, and nucleobases Pharm. Res. 19 299–305 Occurrence Handle11934237 Occurrence Handle10.1023/A:1014495018640 Occurrence Handle1:CAS:528:DC%2BD38XisFGgu7s%3D
G. Merino, J. W. Jonker, E. Wagenaar, A. E. van Herwaarden, and A. H. Schinkel. The breast cancer resistance protein (BCRP/ABCG2) affects pharmacokinetics, hepatobiliary excretion and milk secretion of the antibiotic nitrofurantoin. Mol. Pharmacol. (2005).
C. Borchers R. Boer K. Klemm V. Figala T. Denzinger W. R. Ulrich S. Haas W. Ise V. Gekeler M. Przybylski (2002) ArticleTitleCharacterization of the dexniguldipine binding site in the multidrug resistance-related transport protein P-glycoprotein by photoaffinity labeling and mass spectrometry Mol. Pharmacol. 61 1366–1376 Occurrence Handle12021398 Occurrence Handle10.1124/mol.61.6.1366 Occurrence Handle1:CAS:528:DC%2BD38XksVWrtL4%3D
C. Pascaud M. Garrigos S. Orlowski (1998) ArticleTitleMultidrug resistance transporter P-glycoprotein has distinct but interacting binding sites for cytotoxic drugs and reversing agents Biochem. J. 333 IssueIDPt 2 351–358 Occurrence Handle9657975 Occurrence Handle1:CAS:528:DyaK1cXltVyks78%3D
P. Pavek G. Merino E. Wagenaar E. Bolscher M. Novotna J. W. Jonker A. H. Schinkel (2005) ArticleTitleHuman breast cancer resistance protein: interactions with steroid drugs, hormones, the dietary carcinogen 2-amino-1-methyl-6-phenylimidazo(4,5-b)pyridine, and transport of cimetidine J. Pharmacol. Exp. Ther. 312 144–152 Occurrence Handle15365089 Occurrence Handle1:CAS:528:DC%2BD2MXhtVWmt7c%3D
T. Lenz A. Wilson (2003) ArticleTitleClinical pharmacokinetics of antiplatelet agents used in the secondary prevention of stroke Clin. Pharmacokinet. 42 909–920 Occurrence Handle12885264 Occurrence Handle1:CAS:528:DC%2BD3sXntlKhsr8%3D
B. Terhaag F. Donath G. Petit ParticleLe K. Feller (1986) ArticleTitleThe absolute and relative bioavailability of dipyridamole from different preparations and the in vitro–in vivo comparison Int. J. Clin. Pharmacol. Ther. Toxicol. 24 298–302 Occurrence Handle3733279 Occurrence Handle1:CAS:528:DyaL28XkvVGjtb0%3D
Acknowledgments
The authors acknowledge financial support from NIH grant HD044404 (to QM and JDU) and from the Department of Pharmaceutics, University of Washington. We thank Dr. Robert W. Robey and Dr. Susan E. Bates (National Cancer Institute, Bethesda, MD, USA) for providing the pcDNA-482R plasmid, FTC, and the HEK cell lines. We also thank Dr. Alberto Lazarowski (Clinical Chemistry Laboratory, Instituto de Investigaciones Neurológicas “Raul Carrea,” Buenos Aires, Argentina) for his useful comments in the initial stage of this study.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Zhang, Y., Gupta, A., Wang, H. et al. BCRP Transports Dipyridamole and is Inhibited by Calcium Channel Blockers. Pharm Res 22, 2023–2034 (2005). https://doi.org/10.1007/s11095-005-8384-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11095-005-8384-4