
Abstract
Purpose
Immunoliposomes can be potentially used as carriers for drug delivery to specific cells. The aim of this paper was to exploit the overexpression of vascular cell adhesion molecule-1 (VCAM-1) on activated human endothelial cells (HEC) for targeting of anti-VCAM-1 coupled liposomes with the intent for further use as drug carriers.
Methods
TNF-α-activated HEC were exposed to liposomes, either plain or coupled with antibodies to VCAM-1 (L-VCAM-1) or to irrelevant IgG (L-IgG); nonactivated HEC subjected to the same conditions were used as control. For binding studies, the cells were incubated with fluorescently labeled liposomes at 4°C, and after 2 h, fluorescence intensity was assessed by flow cytometry; specificity of binding was determined by performing the experiments in the presence of excess anti-VCAM-1. Cellular internalization of liposomes was studied employing radioactively or fluorescently labelled liposomes; to detect the mechanisms of uptake, experiments were performed in the presence of agents that interfere in the endocytotic pathway. Transmigration of liposomes was monitored in a two-chamber culture model. The effect of L-VCAM-1 binding to HEC on intracellular calcium ([Ca2+]i) and distribution of actin was determined by fluorimetry and fluorescence microscopy.
Results
(1) L-VCAM-1 binds selectively and specifically to TNF-α activated HEC. (2) Approximately 50% of L-VCAM-1 is taken up by receptor-mediated endocytosis via clathrin-coated vesicles. (3) Binding of L-VCAM-1 to HEC surface induces a rise in [Ca2+]i and reorganization of actin filaments. (4) A small percentage of liposomes migrates across HEC.
Conclusion
The data indicate that VCAM-1 may be an appropriate target for specific drug delivery to activated HEC using immunoliposomes.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
For a successful therapy of human diseases, ideally the administered drugs should reach the right target site at the right time, at a correct concentration and at a proper rate to generate maximal healing benefits and minimal side effects. An approach to modulate the distribution of a drug in a specific location is to design vehicles that are able to selectively deliver the drug to the affected site. To achieve this purpose, liposomes are an attractive candidates due to their flexibility in size, composition, and vesicular structure that can incorporate a large variety of hydrophilic and hydrophobic therapeutic agents. The use of liposomes as vehicles for delivery of drugs and genes targeted selectively to endothelial cells (EC) is a desirable system in the treatment of cardiovascular diseases.
The vascular endothelium plays an important role in the pathogenesis of inflammation, vascular injury and repair, thrombosis, and atherosclerosis. Under the effect of plasma insults, endothelial cells undergo functional–structural modifications leading to an activated state, which at molecular level is translated in the expression of cell surface adhesion molecules such as E/P-selectin, ICAM-1, VCAM-1 (1). Induction or increased expression of specific cell adhesion molecules offers opportunities for specific drug delivery to the diseased EC in a specific vascular segment. Bloemen et al. (2) introduced the concept of targeting adhesion molecules with immunoliposomes, and further studies reported specific targeting of ICAM-1 or E-selectin-directed immunoliposomes (3–6).
Another potential molecular target is vascular cell adhesion molecule-1 (VCAM-1), an immunoglobulin-like transmembrane glycoprotein that is overexpressed in vivo by activated EC covering the developing atheromatous plaque. VCAM-1 plays a major role in monocyte adhesion and migration through EC (7). It is also recognized by leukocytes α4β1 (VLA-4) (8) and α4β7 integrins (9), thereby mediating their firm adhesion to endothelium and extravasation into inflammatory sites (10).
To use the surface-exposed VCAM-1 as a therapeutic approach, one has to understand the mechanisms involved in the interaction of activated EC with immunoliposomes targeted to VCAM-1 and their fate after binding to the cell surface. Because there are contradictory data concerning the internalization of VCAM-1 by EC (11,12), we decided to investigate the binding and the fate of specific immunoliposomes targeted to VCAM-1 expressed in the activated EC surface. It was recently reported that phosphatidylserine (PS)-containing liposomes targeted to VCAM-1 (for controlled thrombogenesis) bind efficiently to IL-1α-treated human umbilical vein endothelial cells (13). However, there are no reports on the fate of the VCAM-1-targeted immunoliposomes after binding to the surface of EC.
Taking advantage of the fact that VCAM-1 can be induced on the surface of TNF-α-activated EC in culture, in this study we examined the specificity of binding to activated EC surface of liposomes targeted to VCAM-1, as well as their internalization and transmigration into subendothelial space. Intracellular events that take place upon the binding of immunoliposomes to VCAM-1 were also investigated. We provide evidence that liposomes coupled with anti-VCAM-1 bound selectively and specifically to activated EC, and that some of them are taken up by clathrin-coated vesicles; under the conditions used, a small percentage of liposomes were transferred to the subendothelial space. Binding of immunoliposomes to EC surface induces an increase in intracellular calcium concentration and a redistribution of actin filaments.
The results extend and strengthen the concept that immunoliposomes directed to molecules expressed on activated EC surface may be used for the selective delivery of drugs as an efficient therapeutic tool in cardiovascular diseases.
Materials and Methods
Reagents
Reagents were obtained from the following sources: egg phosphatidyl choline (EPC), cholesterol (Chol), N-glutaryl-phosphatydilethanolamine (N-glutaryl-PE), and 1,2-dipalmitoyl-sn-glycero-3-phosphoetanolamine-N-(7-nitro-2-1, 3-benzoxadiazol-4-yl (NBD-PE) from Avanti Polar Lipids (Alabaster, AL, USA), 3H-Chol (1 mCi/mL) from the Institute of Physics and Nuclear Engineering (Magurele, Romania), N-(6-tetramethylrhodaminethiocarbamoyl)-1,2-dihexadecanoyl-sn-glycero-3-phosphoethanol-amine, triethylammonium salt (TRITC-DHPE) from Molecular Probes (Eugene, OR, USA), mouse antihuman VCAM-1 and irrelevant mouse IgG1 from R&D Systems (Abingdon, UK), Dulbecco's modified Eagle's medium (DMEM), fetal calf serum (FCS) from Gibco BRL (Gaithersburg, MD, USA), cell culture plates from Corning (New York, NY, USA), black 96-well plates from Nalge NUNC International (Rochester, NY, USA), transmigration chambers from Costar Europe Ltd. (Badhoevedorp, The Netherlands), antimouse IgG coupled with HRP, 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDC), kit for cholesterol assay from Dialab (Viena, Austria), bicinchoninic acid protein assay kit, antimycin A, cytochalasin D, filipin as well as all other chemicals were from Sigma (St. Louis, MO, USA).
Liposome Preparation
Unilamellar liposomes were prepared by extruding multilamellar vesicles through polycarbonate membranes, as previously described (14). Briefly, a mixture of phospholipids in chloroform was dried in a rotary evaporator under reduced pressure. The lipids in chloroform, EPC/Chol/N-glut-PE, were combined at a ratio of 65:30:5 mol%, and hydrated with buffer to reach the final lipid concentration of 10 μmol/mL. After vortexing, the lipid mixture was incubated overnight in a shaker. The resulting multilamellar vesicles were extruded ten times through a 100-nm polycarbonate membrane and ten times more through a 50-nm polycarbonate membrane using a Mini-Extruder (Avanti Polar Lipids). To prepare radiolabeled liposomes, 3H-Chol was incorporated in the lipid film composition at 0.064 nmol (5 μCi)/μmol lipid. Fluorescent liposomes were obtained by adding 3 mol% NBD-PE to the initial lipid film composition. To label only the outer phospholipid monolayer, 1 mol% TRITC-DHPE was subsequently added as an ethanol solution to liposome preparation. The average diameter of the resulting liposomes was determined by negative staining electron microscopy using 1% phosphotungstic acid.
Preparation of Antibody Coupled Liposomes
An amide bound to N-glut-PE as membrane anchor was used for coupling of antibody to the liposome surface (15) because, compared to other techniques, this method (under neutral conditions) offers a good coupling efficiency (4). The free amino groups of proteins are linked to the carboxylic carrier in the presence of water-soluble carbodiimide; it is not required to modify the protein before coupling. For protein linkage, 2 mg EDC were added to 1 μmol liposome in buffer at pH 7.4 and incubated for 6 h at room temperature. Excess EDC was removed by passing through a Sephadex G-50 column. Next, anti-VCAM-1 or an irrelevant IgG were added at a molar ratio of 1:1,000 antibody/phospholipids and incubated overnight at room temperature. Unbound antibodies were removed using a Sepharose 4B column; loss of lipids (due to filtration) was quantitated by cholesterol enzymatic kit. The amount of antibody coupled at the surface of liposomes was quantified as previously described (16). Protein concentration was determined using a bicinchoninic acid protein assay kit and IgG as standard (17). Coupling efficiency was expressed as μg antibodies/μmol phospholipid and the number of antibody molecules/liposome.
Endothelial Cell Culture
A line of human endothelial cell (HEC), EA.hy926 (a kind gift from Dr. Cora-Jean Edgell, Department of Pathology, University of North Carolina, Chapel Hill, NC, USA) was cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal calf serum, 100 U penicillin, 100 μg streptomycin, 50 μg neomycin/mL, 100 μM hypoxanthine, and 16 μM thymidine, at 37°C in a 5% CO2 incubator as previously described (18). The cells express the typical characteristics of EC; they grow in culture as a monolayer of closely apposed polygonal cells and express von Willebrand factor.
Assessment of VCAM-1 Expression on Activated HEC as a Function of Time
The kinetics of VCAM-1 expression on the surface of HEC induced by cell activation with 1 ng/mL TNF-α for different time intervals was assessed by enzyme-linked immunosorbent assay (ELISA), as previously described (19). Briefly, confluent cultured cells were rinsed twice with washing buffer [phosphate buffer saline (PBS) and 1% bovine serum albumin (BSA)] and lightly fixed in 1% paraformaldehide at room temperature. After 15 min, the fixative was replaced with the washing buffer and the cells were successively incubated (1 h) in blocking buffer (PBS and 3% BSA), and then (1 h) with mouse monoclonal antibody to human VCAM-1 (5 μg/mL). Mouse IgG isotype, an irrelevant antibody, was used as control. After thorough rinsing with washing buffer, the cells were incubated with horseradish peroxidase conjugated antimouse IgG (1:1,000) for 1 h at room temperature and washed again with the same buffer. Substrate for the peroxidase reaction (2 mg/mL O-phenilenediamine, 0.03 H2O2 in 0.1 M citrate–phosphate buffer, pH 4) was added and after 30 min the reaction was stopped by the addition of 2 M sulfuric acid; the absorbance at 492 nm was read on a ELISA plate reader (Multiscan).
Evaluation of the Binding of Liposomes Coupled with Anti-VCAM-1 to the EC Surface
To quantify the liposome binding to the HEC surface, the activated or quiescent cells were detached from the culture dish using EDTA (0.02%) in HEPES-buffered saline for 15 min. After washing with DMEM, the resuspended cells (2.5 × 105) were incubated for 2 h at 4°C, with three fluorescently labeled liposome formulations: (1) specific immunoliposomes, namely liposomes coupled with anti-VCAM-1 (L-VCAM-1); (2) unspecific immunoliposomes, liposomes coupled with irrelevant IgG (L-IgG); and (3) plain liposomes (L). The total volume of incubation solution was 100 μl containing (in mM) the following: NaCl (115), KCl (5), NaHCO3 (10), HEPES (25), MgCl2 (0.5), CaCl2 (1), glucose (5.6), and BSA 1 mg/mL; the concentration used was 1 μmol lipid/106 cells. To remove unbound liposomes, the cell/liposome suspensions were washed three times with cold PBS containing 1 mM calcium chloride and 0.5 mM magnesium chloride (PBS-C), and then analyzed by flow cytometry (Coulter Epics XL-MCL/System II). Liposome binding, monitored by the presence of NBD-PE into the liposome bilayer (λex = 488 nm, λem=520 nm) was expressed as fluorescence intensity (X-axis) plotted on a logarithmic scale vs. cell number (counts of events) (Y-axis). From these graphs, the mean fluorescence intensity (fluorescence per cell, in arbitrary units; MFI) was calculated. To assess the specificity of L-VCAM-1 binding, the cells were preincubated with an excess concentration of anti-VCAM-1 (10 μg/mL) before exposure to immunoliposomes.
Internalization of Liposomes Coupled with Anti-VCAM-1 by HEC
Kinetic of Liposome Uptake
To record the kinetic of internalization, HEC were incubated with 3H-cholesterol labeled plain liposomes, specific and unspecific immunoliposomes at a concentration of 1 μmol liposomes/106 cells, for 1, 2, 4, and 24 h at 37°C. At indicated time target intervals, the cells were washed three times with cold PBS-C and incubated (5 min) with 40 mM citric acid, 120 mM NaCl, pH 3, a treatment that has been shown to remove all liposomes bound to the cell surface without disrupting the cell membrane (5,20). Then the cells were solubilized in 0.2 N NaOH (overnight at 4°C), and radioactivity was measured with a liquid scintillation counter (Beckman Counter LS 100C). The obtained values were expressed as nmol lipids/106 cells, using a standard curve constructed from 3H-cholesterol labeled liposomes with known phospholipid concentrations.
To visualize the internalization of specific immunoliposomes by activated HEC, the cells were incubated with fluorescently labeled liposomes with TRITC-DHPE for 1 h at 37°C. Internalization of liposomes was examined by epifluorescence using an inverted microscope Nikon Eclipse TE 300, with an excitation filter set in the range 510–560 nm, which allows observation of fluorescence emission at wavelengths above 575 nm.
Assay for the Internalization of Liposomes
Liposomes, fluorescently labeled with TRITC-DHPE, were successively incubated with activated HEC for 2 h at 4°C and then for 1 h at 37°C. Cells grown on black 96-well plates were pretreated for 30 min at 37°C with several known endocytosis inhibitors: antimycin A (1 μg/mL), which restricts the metabolic activity of the cells and inhibits both receptor-mediated and unspecific endocytosis (21); cytochalasin D (10 μg/mL), which disrupts microfilament network by inhibiting actin polymerization and thus blocking phagocytosis and pinocytosis (22); and filipin (5 μg/mL), which binds specifically cholesterol (23) and disintegrates the caveolae thus inhibiting the caveolae mediated transport (24). To test whether filipin affects the liposomes structure, the following experimental conditions were used: after pretreatment of cells with filipin, further incubation with liposomes was performed in the culture medium in the presence or absence of filipin. As the results of these experiments were similar, we considered that liposomes are not affected by filipin.
In other experiments, cells were exposed to hypertonic concentrations of sucrose, which blocks the clathrin-coated vesicles by preventing the assembly of adaptor proteins to clathrin (25) or to the culture medium depleted of potassium, a condition in which the clathrin is removed from the plasma membrane, impeding the clathrin-mediated endocytosis (26,27). In the latter case, the cells were washed once with potassium-free buffer (PFB) containing the following (in mM): NaCl (140), HEPES (20), CaCl2 (1), MgCl2 (1), and 1 mg/mL d-glucose, followed by a wash with hypotonic buffer (PFB/water, 1:1) and three washes with PFB. Control experiments consisted of exposing cells to PFB containing 10 mM KCl. Subsequent to these pretreatments, L-VCAM-1, L-IgG, and plain L were added and their cellular uptake was determined.
To trail only the internalization of liposomes, the fluorescence of noninternalized liposomes was quenched by incubation of HEC with 0.25 mg/mL Trypan blue in HEPES-buffered saline solution (HBSS), as previously described (28,29). The fluorescence corresponding to internalized liposomes was determined with a spectrofluorimeter plate reader TECAN equipped with an excitation filter at 530 nm and an emission filter at 580 nm. Internalization (percent) in the presence of inhibitors was calculated considering as 100% the fluorescence measured in untreated cells.
Evaluation of Liposome Transmigration Through HEC
The transport of liposomes across HEC was determined by using transmigration chambers (two-chamber culture model). Confluent endothelial cells grown on 0.4-μm-diameter filters, either quiescent or after TNF-α activation, were incubated with radioactively labeled specific or unspecific immunoliposomes or with plain liposomes. At different time intervals, radioactivity was measured in aliquots collected from the lower chamber and the liposome transmigration was expressed as a percentage of the initial radioactivity. To evaluate the distinct role of the cells in liposome transmigration, experiments were performed under the same conditions except that cell-free filters were employed. Values of the liposome transport across the cells were calculated as percentage of the values attained in the absence of cells (taken as 100%).
Assessment of Intracellular Changes Induced by L-VCAM-1 Binding to Activated HEC
Measurements of Intracellular Calcium Concentration
Intracellular free calcium ([Ca2+]i) concentration was determined using Fura 2 AM, as previously described (30). Briefly, TNF-α-activated HEC were detached with EDTA (0.02%) in HBSS, pH 7.4, containing (in mmol/l) NaCl 145, KCl 5, MgCl2 1, glucose 5, HEPES 10, and then washed with DMEM containing 5% FCS. After centrifugation, the cell pellet was resuspended in HBSS in the absence of Ca2+ but containing 3 μM Fura 2 AM, at final cell concentration of ∼500,000 cells/mL, and incubated (1 h) in a shaking water bath, at 37°C, in dark. After loading, Fura 2 AM was removed by centrifugation and HEC were resuspended in Ca2+-free HBSS buffer. [Ca2+]i concentration was measured by using a dual spectrofluorophotometer RF Shimadzu 5001 PC, connected to a computer for data acquisition. Continuous rapid alternating excitation was applied from monochromators set at 340 and 380 nm, while the emission was monitored at 510 nm.
To examine the effect of liposome binding to HEC on Ca2+ released from intracellular stores, the fluorescence signals were determined before and after liposomes (plain, unspecific, or specific immunoliposomes) were added to the cell suspension (for 5 min) in the absence of free extracellular Ca2+.
To determine whether incubation of cells (2 h) with three types of liposomes induced changes in basal [Ca2+]i concentration, the liposome-exposed cells were detached and loaded with Fura 2 AM and the release of calcium from intracellular stores was evaluated after cell stimulation with 25 μM histamine in the absence of free extracellular Ca2+. To assess Ca2+ entry into the cells, the increase in [Ca2+]i upon addition of 2.5 mM Ca2+ to the culture medium of histamine-stimulated cells was measured.
[Ca2+]i concentration was calculated from the ratio (R) of the fluorescence intensities at two different excitation wavelengths, according to the equation of Grynkiewicz et al. (30): [Ca2+]i (nM) = Kd × [(R − Rmin)/(Rmax − R)] × Sf/Sb, where R is the ratio of the fluorescence intensity of the sample excited at two different wavelengths (380/340 nm) and measured at 510 nm. Calibration measurements were performed at the end of each experiment and provided Rmin, Rmax, and Sf/Sb values for the calculation of [Ca2+]i. Rmax represents the fluorescence ratio of calcium-bound dye and was obtained after the addition of 12 mM CaCl2 (saturated solution) and cell permeabilization with 0.1% Triton X-100. Rmin is the minimum fluorescence ratio of Ca2+-free or unbound dye and was measured after treatment with 10 mM EGTA, the calcium ion chelator. Sf/Sb is the ratio of fluorescence values for calcium-free and calcium-saturated (12 mM) solution, respectively. Kd is the dissociation constant of calcium-bound Fura 2 AM and was considered to be 224 nM.
Distribution of Actin Filaments
The disposition of actin filaments was evaluated on confluent HEC grown on coverslips and incubated with specific and unspecific immunoliposomes. After 20 min at 37°C, the cells fixed in 3% paraformaldehide (PFA) were permeabilized with 0.5% Triton X-100, rinsed with PBS, and then incubated with 0.1 μg/mL Phalloidine-TRITC in PBS for 20 min at room temperature. Slides mounted in glycerol/PBS were viewed with a Nikon microscope with an excitation filter set in the range 510–560 nm, which allows observation of fluorescence emission at 590 nm wavelength.
Statistics
Results are presented as means ± SEM. Statistical analysis was performed by one-way analysis of variance (ANOVA) and differences were considered significant when p < 0.05.
Results
Characterization of Liposomes
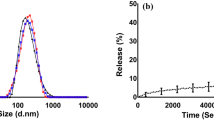
As determined by negative staining electron microscopy, the mean diameter of liposomes obtained after successive extrusion through 100 and 50 nm polycarbonate membranes was ∼80 nm (Fig. 1). Under all conditions employed, the resulting vesicles were unilamellar and of comparatively homogenous dimensions. For specific (L-VCAM-1) and unspecific (L-IgG) immunoliposomes, the calculated amount of antibody coupled to the liposome surface was ∼25 μg/μmol phospholipids corresponding to approximately 8 antibodies/liposome. No changes in size or structure of liposomes were induced by labeling with either 3H-cholesterol, NBD-PE, or TRITC-DHPE.

Negative staining electron microscopy of liposomes stained with phosphotungstic acid (1%). Note that the liposomes obtained appear as unilamellar vesicles of relative homogenous size. Bar, 100 nm.
Expression of VCAM-1 on Activated HEC
The experiments designed to test the expression of VCAM-1 on the surface of activated HEC were performed at 48 h postconfluency and after cell activation for different time intervals with 1 ng/mL TNF-α. As shown in Fig. 2, VCAM-1 expression on activated HEC increased as a function of time, attaining a maximal level after 24 h of cell activation, a time when a 4.5-fold increase above the control level (nonactivated cells) was determined. At 48 h after cell activation, VCAM-1 expression returned to basal level, having a value similar to that found in nonactivated cells (Fig. 2). Based on these data, all further experiments were performed on HEC after 24 h activation with TNF-α.

Induction of VCAM-1 expression on the surface of human endothelial cells as a function of time of TNF-α activation. Note that the expression of VCAM-1 is maximal after 24 h of activation. The data are representative of four experiments. The values represent the mean number of absorbance units ± SE for one experiment performed in triplicate.
Liposome Binding to the Surface of HEC
To evaluate liposome binding to the surface of HEC, activated and nonactivated cells were incubated with fluorescently labeled specific and unspecific immunoliposomes or plain liposomes, after which fluorescence intensity for each experimental condition was assessed by flow cytometry. Figure 3A illustrates the binding of L-VCAM-1, L-IgG, and L to nonactivated HEC, and reveals that the binding is closely comparable for all liposome types. Flow cytometry determinations showed that activated HEC bound with high efficiency to L-VCAM-1 compared to the binding of unspecific immunoliposomes (L-IgG) or plain (L) liposomes (Fig. 3B). Mean intensity values (determined from flow cytometry studies) are shown in Fig. 3C and indicate significant increase in L-VCAM-1 binding to activated HEC only. The specificity of L-VCAM-1 binding to activated endothelial cells was assessed by performing the experiments in the presence of an excess concentration of anti-VCAM-1 (10 μg/mL). In this case, the level of binding was reduced to that found in cells incubated with plain liposomes (not shown).

Flow cytometry charts showing the binding of plain liposomes (L), unspecific immunoliposomes (L-IgG) and specific immunoliposomes (L-VCAM-1) to nonactivated (A) or TNF-α activated (B) human endothelial cells (HEC). Data are from a single experiment and are representative of three such experiments. (C) Values of mean fluorescence intensity (fluorescence per cell in arbitrary units; MFI) as determined by flow cytometry experiments and plotted from a single experiment using triplicate probes (p < 0.05). Note that the binding of L-VCAM-1 to the surface of activated HEC is significantly higher than the binding to nonactivated cells. The binding of L-IgG and plain L is lower than that of L-VCAM-1 to either activated or nonactivated cells.
Uptake of Liposomes by HEC
Kinetic of Liposome Uptake
The uptake of liposomes by endothelial cells was tracked via liquid scintillation counting after incubation of activated or nonactivated HEC at different time intervals with 3H-colesterol labeled specific and unspecific immunoliposomes or plain liposomes. Cellular uptake was monitored at different time intervals and expressed as nmol/106 cells.
It was found that at all time points studied, activated HEC take up L-VCAM-1 at a constantly higher rate than L-IgG or plain L. The uptake increased as a function of time and attained the highest value at 24 h (Fig. 4A).

Internalization of 3H-cholesterol labeled specific (L-VCAM-1) or unspecific (L-IgG) immunoliposomes and plain liposomes (L) by activated (A) or nonactivated (B) human endothelial cells (HEC) as a function of time. Note that in the case of the latter, all three liposome types are taken up at similar levels; in contradistinction, the activated cells internalize at a significantly higher degree the specific immunoliposomes. Data represent the means of three independent experiments ± SE. Insert A: fluorescence micrographs depicting the internalization of fluorescently labeled L-VCAM-1 by activated HEC.
The insert to Fig. 4A shows the internalization of fluorescently labeled immunoliposomes by activated HEC after extracellular fluorescence is quenched. The immunoliposomes appeared within the cytoplasm as small fluorescent dots particularly concentrated in perinuclear area. In the case of nonactivated HEC, internalization of L-VCAM-1, L-IgG, and plain L showed a comparable level (Fig. 4B), and after 24 h of incubation of cells with liposomes, the internalization reached values of ∼3% of the initial liposome concentration; this value was comparable to that obtained for activated cells incubated with unspecific immunoliposomes or plain liposomes. In contrast, the calculated uptake of specific immunoliposomes by activated HEC reached a double value (∼6%).
Mechanisms Involved in the Internalization Pathway of Liposomes
The experiments described above indicated that HEC internalize labeled liposomes. To assess the mechanisms involved in the uptake of liposomes, endothelial cells were exposed to various inhibitors (known to affect a specific endocytotic pathway) prior to incubation with fluorescently labeled liposomes. As shown in Fig. 5, antimycin A and cytochalasin D reduced the uptake of L-IgG and L by approximately 60 and 80%, respectively, whereas the internalization of L-VCAM-1 was decreased by 30 ± 7 and 11 ± 3%. Filipin treatment led to a higher inhibition of internalization in L-IgG and plain L (43 ± 5 and 45 ± 4%, respectively) compared to that of L-VCAM-1 (25 ± 6%). Exposure of the cells to hypertonic concentration of sucrose or incubation in potassium-depleted culture medium induced an inhibition of internalization that was significantly higher for L-VCAM-1 (50 ± 6% in the presence of sucrose and 46 ± 7% in the case of potassium-depleted medium) than for L-IgG and L (∼5% under both incubation conditions).

The effect of various inhibitors that interfere with the endocytosis of DHPE-TRITC labeled specific (L-VCAM-1), unspecific (L-IgG) immunoliposomes and plain liposomes (L) by human endothelial cells (HEC). Note that antimycin A, cytochalasin D, and filipin treatment of HEC lead to a high percentage of inhibition in internalization of L-IgG and plain L, whereas potassium depletion from the medium and use of hypertonic concentration of sucrose mainly inhibit the uptake of L-VCAM-1. Data represent means ± SE (n = 3).
These data indicate that HEC took up L-IgG and plain liposomes via nonspecific endocytosis and that caveolae play a role in this process. In contrast, nonspecific endocytosis had a minor role in the internalization of specific immunoliposomes, and uptake of L-VCAM-1 by endothelial cells occurred mainly by a receptor-mediated mechanism, a process in which clathrin-coated vesicles played a major role.
Liposome Transmigration into the Subendothelial Space
For transport studies, HEC cultured in a double chamber system were incubated with radioactively labeled specific and unspecific liposomes; transport across the cells was assessed by counting the radioactivity amassed in the lower chamber at 1, 2, and 24 h. Similar experiments were carried out using cell-free filters, and values obtained for the liposome transport across HEC were calculated as percentage of the values attained in the absence of cells (taken as 100%). As presented in Fig. 6, a time-dependent increase in the liposome transmigration, particularly in the case of L-VCAM-1 (expressed as percentage from the initial radioactivity), was detected. A significant difference was found in the transport of specific liposomes across HEC as compared to unspecific IgG-L or plain L. The calculated liposome transmigration was ∼3% for L-VCAM-1 and ∼1.3% for L-IgG and plain L of the maximum transmigration value obtained in the experiments that were performed under the same conditions except that the filters were without cells.

Transmigration across activated HEC monolayer of 3H-cholesterol labeled specific (L-VCAM-1) or unspecific (L-IgG) immunoliposomes and plain liposomes (L). The data show that the percentage of L-VCAM-1 transmigration across the cells is higher than for L-IgG or plain L. Data are means of at least three experiments ± SE (p < 0.05).
Intracellular Events Induced by L-VCAM-1 Binding to Activated HEC
The findings that immunoliposomes against VCAM-1 specifically recognized molecules on the surface of activated HEC, which they bound, were internalized and transported across the cell at a higher rate than nontargeted liposomes put forward the hypothesis that certain intracellular events may occur that enable the cells to respond to the challenge. We investigated the effect of immunoliposome binding to the specific target on the endothelial cell's surface upon calcium release (from intracellular stores) and distribution of actin filaments.
Intracellular Calcium
As determined by measurements of calcium-bound Fura-2 AM, the incubation of activated HEC for 5 min with liposomes resulted to an increase in cytosolic free calcium, the increase being significantly higher in the case of specific immunoliposomes directed toward VCAM-1 than when incubation is performed with unspecific immunoliposomes or with plain liposomes (Fig. 7A). Data revealed that upon binding of L-VCAM-1 to HEC, both the peak and the plateau levels of [Ca2+]i were significantly increased in comparison with L-IgG or plain L binding to cells. The obtained data (peak and basal levels) were used to calculate the differential increase in [Ca2+]i release in HEC under the experimental conditions used (Fig. 7B). Data indicated that as a result of L-VCAM-1 binding, the release of [Ca2+]i attained a value that was about fivefold higher than the figures obtained for L-IgG and plain L (p < 0.05).

(A) Intracellular calcium concentration ([Ca2+]i) in human endothelial cells (HEC) exposed to L-VCAM-1, L-IgG, and plain L. Confluent cells were harvested, washed, and exposed for 1 h to calcium free medium in the presence of Fura 2 AM, then incubated with liposomes; changes in [Ca2+]i were subsequently recorded for 5 min. In comparison with L-IgG and L, the cell incubation with L-VCAM-1 induces a significant augmentation of [Ca2+]i. (B) The differential increase in [Ca2+]i release in HEC, calculated from the data obtained in (A) as the difference (Δ) between the values obtained upon liposomes addition and the basal values (p < 0.05 vs. L-IgG, L). The graph represents the means of three independent experiments.
Similar differences were found after the incubation of endothelial cells for 2 h with three types of liposomes: L-VCAM-1 induced an increase in cytosolic free calcium above the values obtained in the case of cell incubation with L-IgG or plain L. The mean value of basal cytosolic free calcium in L-VCAM-1-treated cells was ∼110 nM, whereas for cells exposed to L-IgG or plain L the figures were ∼83 nM in both cases (Fig. 8A).

(A) The effect of 2 h incubation of human endothelial cells (HEC) with different liposome types on the basal level of calcium, response to histamine, and calcium addition in the extracellular medium. Note that by comparison with HEC incubated with L-IgG or L, the incubation of cells with L-VCAM-1 induces a cellular increase in basal calcium (p < 0.05), a decreased response to histamine stimulation (p < 0.05), whereas the addition of calcium stimulates an augmentation of [Ca2+]i that is comparable for all three liposome types. (B) The differential increases in [Ca2+]i release in HEC treated with the three types of liposomes calculated from (A) as the difference (Δ) between the values obtained upon addition of histamine and the basal value. (C) The differential increase in calcium entry in HEC calculated from (A) as the difference (Δ) between the values obtained upon calcium addition and the basal values. The graphs represent the media of three independent experiments.
Application of histamine induced an elevation of [Ca2+]i under all three conditions used. The peak increase in [Ca2+]i in response to histamine was lower in HEC treated with L-VCAM-1 than in cells treated with L-IgG or plain L. The differential increase in [Ca2+]i release in HEC treated with three types of liposomes calculated (from the data presented in Fig. 8A) as the difference (Δ) between the values obtained upon addition of histamine and the basal value is presented in Fig. 8B; the Δ[Ca2+] in the case of L-VCAM-1 was found significantly lower than that for L-IgG and plain L (8.36 nM for L-VCAM-1 vs. 17.48 nM for L-IgG and 20.08 nM for L).
As shown in Fig. 8A subsequent addition of extracellular calcium (2.5 mM) to HEC incubated with L-VCAM-1, L-IgG, and plain L generated a great enhancement in fluorescence intensity. However, calculations of Ca2+ entry into the cells, considered as the difference between the values obtained upon Ca2+ addition and the basal values, showed that the differential increase in [Ca2+]i was similar under all experimental conditions used (Fig. 8C).
Distribution of Actin Filaments
Having established that as a result of L-VCAM-1 binding to the surface of HEC an increase in [Ca2+]i occurred, we investigated whether this event was associated with a rearrangement of cytoskeletal actin filaments. Monolayers of HEC were treated with 1 ng/mL TNF-α for 24 h to induce VCAM-1 expression, then incubated for 20 min with L-IgG or L-VCAM-1, fixed, permeabilized, and stained with TRITC-phalloidin. Figure 9 illustrates the F-actin distribution in TNF-α-activated HEC incubated with L-IgG (A) or L-VCAM-1 (B). It was observed that when the cells were incubated with L-IgG, the actin filaments were distributed along the cell plasmalemma as depicted by the fine fluorescence bordering the cell membrane (Fig. 9A); in the central region of the cells the filament bundles were almost absent. Exposure of HEC to L-VCAM-1 induced a redistribution of the actin network that in fluorescence microscopy appeared as intense fluorescent staining distributed throughout the cytoplasm and often aligned in parallel to the long axis of the cells (Fig. 9B).

Fluorescence micrographs depicting the distribution of F-actin in human endothelial cell monolayers exposed to L-IgG or L-VCAM-1. Note that rare actin filaments appear within the cytoplasm of cells incubated with L-IgG (A), whereas a prominent network of actin filaments distributed throughout the cytoplasm of monolayers exposed to L-VCAM-1 (B).
Discussion
The concept of targeted drug delivery providing an effective concentration of drugs to reach the diseased sites is an appealing strategy for therapy of human diseases. In this respect, the vascular endothelium is an accessible target due to its large surface area exposed to the circulating blood. There are studies in which immunoliposomes (liposomes coupled with antibodies directed against endothelial cell surface antigens) have been used for specific targeting of compounds to endothelium (2,31–36); in some instances, the mechanism of uptake was determined (6,37).
Vascular cell adhesion molecule-1 (VCAM-1) is a convenient target receptor because it plays a major role in the pathogenesis of diseases such as atherosclerosis (7), rheumatoid arthritis (38), and multiple sclerosis (39). VCAM-1 expression is selectively restricted to activated endothelium under different pathological conditions, such as early developing atheromatous plaque (40) or human malignant tumors (41). Other cells such as macrophages, dendritic cells, bone marrow stromal cells, neurons, fibroblasts, fibroblast-like synovial cells from inflamed joints, and smooth muscle cells express constitutively or after activation VCAM-1, but they do not interfere with binding of immunoliposomes directed to endothelial VCAM-1 because they are not accessible after intravenous administration. However, a weak expression of VCAM-1 was detected under normal conditions in few vessels of kidney, thymus, and thyroid gland (42); nevertheless, the low degree of expression cannot interfere largely with the targeting of L-VCAM-1 to the diseased vascular endothelium. Therefore, the liposomes directed against VCAM-1 could be useful as tools for selective targeting to the activated endothelium. There is one report illustrating that anti-VCAM-1 conjugated phosphatidylserine liposomes bind to IL-1α-treated human umbilical vein endothelial cells (13), but there are no data about the fate of VCAM-1-targeted immunoliposomes after binding to the EC surface. The goal of this study was to exploit the specific expression of VCAM-1 on the surface of activated HEC for selective targeting of liposomes coupled with anti-VCAM-1 (L-VCAM-1) and to study the fate of L-VCAM-1 after binding to the cell surface; the results could be further used for targeting drugs to activated endothelial cells covering diseased vascular segments.
Results showed that liposomes coupled with anti-VCAM-1 bind selectively to activated endothelial cells by a specific mechanism, as substantiated by the competitive binding assay.
In addition, we observed the uptake of L-VCAM-1 by HEC and found that the internalization of L-VCAM-1 (at various time intervals) by activated cells was approximately twofold higher than the values obtained with control liposomes (L-IgG or plain L). Interestingly, nonactivated HEC take up L-VCAM-1, L-IgG, and plain L at comparable amount, and the values were similar with those obtained for the uptake of L-IgG or plain L by activated cells.
Previous reports on the internalization of VCAM-1 by endothelial cells are contradictory (11,12). Our data suggest that the internalization of L-VCAM-1 by activated EC can be safely attributed to VCAM-1 internalization, because the cellular uptake of control liposomes is significantly lower. The results corroborate well with our previous reports showing that transferrin (Tf)-coupled liposomes are specifically taken up by EC via a receptor-mediated mechanism employing the pathway of Tf receptors (43); it is to be expected that L-VCAM-1, after specific binding to the EC surface, follows the same pathway as its receptor (VCAM-1).
There are some reports on the role of other cell adhesion molecules (CAM), such as ICAM-1 and E-selectin, in internalization after targeting with liposomes (5,6,37). Although it is difficult to compare these data because different CAM were used as targets, our results for internalization of liposomes targeted to VCAM-1 are in line with those of Mastrobatista E et al. (5) for liposomes targeted to ICAM-1: after 1 h of incubation at 37°C, we have found that 5.6 nmol of VCAM-1 targeted immunoliposomes are internalized by 106 activated HEC vs. 4.2 nmol of ICAM-1 targeted immunoliposomes internalized by 106 epithelial cells. However, Kessner et al. (6) reported a lower percentage of internalization for E-selectin targeted immunoliposomes by activated human umbilical vein endothelial cells. One can assume that upon targeting with immunoliposomes, ICAM-1 and VCAM-1 (both belonging to Ig superfamily CAM) have a prominent role in the internalization profiles, whereas the role of selectin is relatively low.
Recently it was reported that in endothelial cells, ICAM-1 has the capacity to recycle, signifying that a single ICAM-1 molecule could participate in multiple rounds of binding and internalization of nanocarriers coupled to ICAM-1 (44). A good hypothesis to explain the large capacity for internalization of VCAM-1 targeted liposomes in HEC is to presume that VCAM-1 has the capacity to recycle; however, this assumption should be confirmed by further experiments. Nevertheless, the considerable capacity for internalization of VCAM-1 targeted liposomes opens new perspectives for the intracellular delivery of drugs or genes to the vascular endothelium, to correct altered cellular functions by a site-directed therapy.
To investigate the mechanisms involved in L-VCAM-1 internalization, experiments were performed under conditions known to inhibit a specific endocytotic pathway. Results suggest that clathrin-coated pits and vesicles mediate a major internalization pathway of VCAM-1-directed immunoliposomes, as the internalization process was inhibited (∼50%) by potassium depletion from the culture medium or by the exposure of the cells to hypertonic sucrose concentration. Phagocytosis and pinocytosis are not effective in the internalization of L-VCAM-1 (cytochalasin D induced ∼11% inhibition of internalization), whereas caveolae mediated transport represents ∼26% inhibition (as revealed by filipin experiments). At the concentrations used, antimycin A inhibits the internalization of L-VCAM-1 by ∼31%; higher concentrations could not be employed because they are toxic to the cells.
In contrast, unspecific immunoliposomes and plain liposomes are taken up by nonspecific endocytosis (antimycin A and cytochalasin D inhibit internalization by 60 and 80%, respectively) and by caveolae (filipin treatment inhibits endocytosis by 45%).
Recently, it was proposed that a novel endocytic pathway for ICAM-1 and PECAM-1 in endothelial cells, namely a CAM-mediated endocytosis, which is similar but distinct from “classical” macropinocytosis, is dependent on antigen clustering and requires Rho-kinase and PKC-mediated rearrangements of actin cytoskeleton (45). This type of endocytosis cannot be excluded for L-VCAM-1 internalization, because in HEC only ∼50% of liposomes are internalized by a receptor-mediated mechanism; however, further studies are needed to validate this theory.
A small percentage of VCAM-1 targeted liposomes migrate across the endothelial monolayer; under the experimental conditions used, data revealed that the transmigration of L-VCAM-1 is about 15-fold lower than its internalization. Nonetheless, the percentage of L-VCAM-1 transmigration is twofold higher when compared with that of L-IgG or plain L. Among the possible factors that can play a role in the transport of immunoliposomes are intracellular calcium concentration and actin filaments.
The functional implication of Ca2+ in the increased EC monolayer permeability and leukocyte diapedesis was previously reported (46). In addition, it was demonstrated that monoclonal antibodies directed against VCAM-1 increase [Ca2+]i and induce cytoskeleton rearrangements (47) in HUVEC. More recent studies have shown that VCAM-1 acts as a signaling receptor by interacting with membrane-actin cytoskeleton linkers, moesin and ezrin, promoting endothelial cell signaling and actin remodeling (48) and inducing Rac activation resulting in loss of cell-to-cell adhesion (49). Therefore, we searched whether the binding of L-VCAM-1 to the surface of HEC generates intracellular signals that determine an increase in [Ca2+]i concentration and cytoskeleton reorganization, which in turn may induce an increase in endothelium permeability.
The data revealed that after 5 min exposure of HEC to liposomes, the differential increase in [Ca2+]i (calculated as the difference between values obtained upon liposome addition and basal values) in the case of L-VCAM-1 was fivefold higher above the values obtained for L-IgG or plain L. Although significantly lower than in the case of L-VCAM-1, the control liposomes (L-IgG, plain L) also induced an increase in [Ca2+]i; this could be explained by the fact that some intracellular signaling are induced by nonspecific liposomes in EC.
Incubation of endothelial cells with L-VCAM-1 for 2 h induces a 1.2-fold increase in the basal level of [Ca2+]i, whereas no changes are recorded for L-IgG or L incubated under the same conditions. In response to histamine addition, the decrease in [Ca2+]i concentration is more pronounced when cells are incubated with L-VCAM-1 than with L-IgG or L, suggesting a larger depletion of the intracellular calcium stores in the case of specific immunoliposomes.
Having established that L-VCAM-1 binding to EC surface induces an increase in [Ca2+]i, we investigated whether this signaling event is associated with a redistribution of cytoskeleton actin filaments. We found that the actin filaments form a prominent network distributed throughout the cytoplasm of the cell monolayer exposed to L-VCAM-1, a feature that was absent when HEC were incubated with L-IgG. These data suggest that redistribution of actin is associated with specific immunoliposome internalization. However, internalization experiments performed in the presence of cytochalasin D indicate only a minor role of actin in L-VCAM-1 uptake. We assume that there are two different events: cytochalasin D, which inhibits actin polymerization, does not interfere with the endocytic pathway of immunoliposomes; conversely, the binding of L-VCAM-1 to the EC surface expressed VCAM-1 (that acts as a signaling receptor by interacting with membrane actin cytoskeleton linkers) induces a rearrangement of actin filaments and the ensuing increase in paracellular transport of specific immunoliposomes.
These results suggest that [Ca2+]i may play a role in the regulation of binding, internalization, or transendothelial migration of L-VCAM-1, being a part of a signal cascade that determines cytoskeleton reorganization and consequently, may induce opening of EC junctions and transmigration of liposomes.
Taken together, our results indicate that (1) liposomes targeted to VCAM-1 bind specifically to activated EC; (2) approximately 50% of the internalized L-VCAM-1 is taken up by clathrin-coated vesicles; (3) L-VCAM-1 binding to EC surface determines signaling events that may lead to opening of the junctions and transendothelial migration of L-VCAM-1.
The data suggest that VCAM-1 may be an appropriate molecular target for specific delivery of drugs to activated EC, using immunoliposomes.
References
M. P. Bevilacqua J. S. Pober D. L. Mendrick R. S. Cotran M. A. Gimbrone SuffixJr (1987) ArticleTitleInducible endothelial functions in inflammation and coagulation Semin. Thromb. Hemost. 13 425–433 Occurrence Handle3122325
P. G. M. Bloemen P. A. J. Henricks L. Bloois Particlevan M. C. Tweel Particlevan den A. C. Bloem F. P. Nijkamp D. J. A. Crommelin G. Storm (1995) ArticleTitleAdhesion molecules: a new target for immunoliposome-mediated drug delivery FEBS Lett. 357 140–144 Occurrence Handle10.1016/0014-5793(94)01350-A Occurrence Handle7805880
M. Everts G. A. Koning R. J. Kok S. A. Asgeirsdottir D. Vestweber D. K. Meijer G. Storm G. Molema (2003) ArticleTitleIn vitro cellular handling and in vivo targeting of E-selectin-directed immunoconjugates and immunoliposomes used for drug delivery to inflamed endothelium Pharm. Res. 20 64–72 Occurrence Handle10.1023/A:1022298725165 Occurrence Handle12608538
G. Bendas A. Krause R. Schmidt J. Vogel U. Rothe (1998) ArticleTitleSelectins as new targets for immunoliposome-mediated drug delivery. A potential way of anti-inflammatory therapy Pharm. Acta Helv. 73 19–26 Occurrence Handle10.1016/S0031-6865(97)00043-5 Occurrence Handle9708035
E. Mastrobattista G. Storm L. Bloois Particlevan R. Reszka P. G. M. Bloemen D. J. A. Crommelin P. A. J. Henricks (1999) ArticleTitleCellular uptake of liposomes targeted to intercellular adhesion molecule-1 (ICAM-1) on bronchial epithelial cells Biochim. Biophys. Acta 1419 353–363 Occurrence Handle10407086
S. Kessner A. Krause U. Rothe G. Bendas (2001) ArticleTitleInvestigation of the cellular uptake of E-selectin-targeted immunoliposomes by activated human endothelial cells Biochim. Biophys. Acta 1514 177–190 Occurrence Handle11557019
M. I. Cybulsky K. Iiyama H. Li S. Zhu M. Chen M. Iiyama V. Davis J. C. Gutierez-Ramos P. W. Connely D. S. Milstone (2001) ArticleTitleA major role for VCAM-1, but nor ICAM-1, in early atherosclerosis J. Clin. Invest. 107 1255–1262 Occurrence Handle11375415
B. M. Chan M. J. Elices E. Murphy M. E. Hemler (1992) ArticleTitleAdhesion to vascular cell adhesion molecule 1 and fibronectin. Comparison of alpha 4 beta 1 (VLA-4) and alpfa4 beta 7 on human T cell line JY J. Biol. Chem. 267 8366–8370 Occurrence Handle1373725
C. Ruegg A. A. Postigo E. E. Sikorski E. C. Butcher R. Pytela D. J. Erle (1992) ArticleTitleRole of integrin alpha 4 beta 7/alpha 4 beta P in lymphocyte adherence to fibronectin and VCAM-1 and in homotypic cell clustering J. Cell Biol. 117 179–189 Occurrence Handle10.1083/jcb.117.1.179 Occurrence Handle1372909
B. A. Imhof D. Dunon (1995) ArticleTitleLeukocyte migration and adhesion Adv. Immunol. 58 345–416 Occurrence Handle7537936
T. W. Kuijpers M. Raleigh T. Kavanagh H. Janssen J. Calafat D. Roos J. M. Harlan (1994) ArticleTitleCytokine-activated endothelial cells internalize E-selectin into a lysosomal compartment of vesiculotubular shape J. Immunol. 152 5060–5069 Occurrence Handle7513727
I. Ricard M. D. Payet G. Dupuis (1998) ArticleTitleVCAM-1 is internalized by a clathrin-related pathway in human endothelial cells but its alpha 4 beta 1 integrin counter-receptor remains associated with the plasma membrane in human T lymphocytes Eur. J. Immunol. 28 1708–1718 Occurrence Handle10.1002/(SICI)1521-4141(199805)28:05<1708::AID-IMMU1708>3.0.CO;2-Y Occurrence Handle9603478
G. N. C. Chiu M. Bally L. D. Mayer (2003) ArticleTitleTargeting of antibody conjugated, phosphatidylserine-containing liposomes to vascular cell adhesion molecule 1 for controlled thrombogenesis Biochim. Biophys. Acta 1613 115–121 Occurrence Handle12832092
M. Paternostre M. Ollivan J. Bolard (1996) Description of four selected techniques of liposome preparation R. Prasad (Eds) Manual on Membrane Lipids Springer-Verlag Berlin 218–226
I. Ezpeleta J. M. Irache S. Stainmesse C. Chabenat J. Gueguen A. M. Orecchioni (1996) ArticleTitlePreparation of lectin–vicilin nanoparticle conjugates using the carbodiimide coupling technique Int. J. Pharm. 142 227–233 Occurrence Handle10.1016/0378-5173(96)04668-6
D. Wessel U. I. Flugge (1984) ArticleTitleA method for the quantitative recovery of protein in dilute solution in the presence of detergents and lipids Anal. Biochem. 138 141–143 Occurrence Handle10.1016/0003-2697(84)90782-6 Occurrence Handle6731838
P. K. Smith R. I. Krohn G. T. Hermansen A. K. Malia F. H. Gartner M. D. Provenzano E. K. Fujimoto N. M. Goeke B. J. Olson D. C. Klenk (1985) ArticleTitleMeasurement of protein using bicinchoninic acid Anal. Biochem. 150 76–85 Occurrence Handle10.1016/0003-2697(85)90442-7 Occurrence Handle3843705
C. J. S. Edgell C. C. McDonald J. B. Graham (1983) ArticleTitlePermanent cell line expressing human actor VIII-related antigen established by hybridization Proc. Natl. Acad. Sci. 80 3734–3737 Occurrence Handle6407019
S. Hashemi D. C. Smith C. A. Izaguirre (1987) ArticleTitleAnti-endothelial cell antibodies: detection and characterization using a cellular enzyme-linked immunosorbent assay J. Lab. Clin. Med. 109 434–440 Occurrence Handle3546564
K. K. Matthay T. D. Heath C. C. Badger I. D. Bernstein D. Papahadjopoulos (1986) ArticleTitleAntibody-directed liposomes: comparison of various ligands for association, endocytosis and drug delivery Cancer Res. 46 4904–4910 Occurrence Handle3756852
K. D. Lee S. Nir D. Papahadjopoulos (1993) ArticleTitleQuantitative analysis of liposome-cell interactions in vitro: rate constants of binding and endocytosis with suspension and adherent J774 cells and human monocytes Biochemistry 32 889–899 Occurrence Handle10.1021/bi00054a021 Occurrence Handle8422393
J. L. Salisbury G. A. Keller (1983) ArticleTitleStructural investigations on the role of microfilaments in ligand translocation Methods Enzymol. 98 368–375 Occurrence Handle6142407
J. Bolard (1986) ArticleTitleHow do the polyene macrolide antibiotics affect the cellular membrane properties? Biochim. Biophys. Acta 864 257–304 Occurrence Handle3539192
J. E. Schnitzer P. Oh E. Pinney J. Allard (1994) ArticleTitleFilipin-sensitive caveolae-mediated transport in endothelium: reduced transcytosis, scavenger endocytosis and capillary permeability of select macromolecules J. Cell Biol. 127 1217–1232 Occurrence Handle10.1083/jcb.127.5.1217 Occurrence Handle7525606
J. E. Heuser R. G. Anderson (1989) ArticleTitleHypertonic media inhibit receptor-mediated endocytosis by blocking clathrin-coated pit formation J. Cell Biol. 108 389–400 Occurrence Handle10.1083/jcb.108.2.389 Occurrence Handle2563728
J. M. Larkin M. S. Brown J. L. Goldstein R. G. Anderson (1983) ArticleTitleDepletion of intracellular potassium arrests coated pit formation and receptor-mediated endocytosis in fibroblasts Cell 33 273–285 Occurrence Handle10.1016/0092-8674(83)90356-2 Occurrence Handle6147196
J. Rejman V. Oberle I. S. Zuhorn D. Hoekstra (2004) ArticleTitleSize-dependent internalization of particles via the pathways of clathrin- and caveolae-mediated endocytosis Biochem. J. 377 159–169 Occurrence Handle10.1042/BJ20031253 Occurrence Handle14505488
J. Hed G. Hallden S. G. Johansson P. Larsson (1987) ArticleTitleThe use of fluorescence quenching in flow cytofluorometry to measure the attachment and ingestion phases in phagocytosis in peripheral blood without prior cell separation J. Immunol. Methods 101 119–125 Occurrence Handle10.1016/0022-1759(87)90224-9 Occurrence Handle3112235
E. S. Amersfoort ParticleVan J. A. Strijp ParticleVan (1994) ArticleTitleEvaluation of a flow cytometric fluorescence quenching assay of phagocytosis of sensitized sheep erythrocytes by polymorphonuclear leukocytes Cytometry 17 294–301 Occurrence Handle7875036
G. Grynkiewicz M. Poenie R. Y. Tsien (1985) ArticleTitleA new generation of Ca2+ indicators with greatly improved fluorescence properties J. Biol. Chem. 260 3440–3450 Occurrence Handle3838314
O. V. Trubetskaya V. S. Trubetskoy S. P. Domogatsky A. V. Rudin N. V. Popov S. M. Danilov M. N. Nikolayeva A. L. Klibanov V. P. Torchilin (1988) ArticleTitleMonoclonal antibody to human endothelial cell surface internalization and liposome delivery in cell culture FEBS Lett. 228 131–134 Occurrence Handle10.1016/0014-5793(88)80601-X Occurrence Handle3342871
B. J. Hughes S. Kennel R. Lee L. Huang (1989) ArticleTitleMonoclonal antibody targeting of liposomes to mouse lung in vivo Cancer Res. 49 6214–6220 Occurrence Handle2478282
K. Maruyama E. Holmberg S. J. Kennel A. Klibanov V. P. Torchilin L. Huang (1990) ArticleTitleCharacterization of in vivo immunoliposome targeting to pulmonary endothelium J. Pharm. Sci. 79 978–984 Occurrence Handle2292774
K. Maruyama T. Takizawa T. Yuda S. J. Kennel L. Huang M. Iwatsuru (1995) ArticleTitleTargetability of novel immunoliposomes modified with amphipathic poly(ethylene glycol)s conjugated at their distal terminals to monoclonal antibodies Biochim. Biophys. Acta 1234 74–80 Occurrence Handle7880861
V. P. Torchilin (1994) ArticleTitleImmunoliposomes and PEGylated immunoliposomes: possible use for targeted delivery of imaging agents Immunomethods 4 244–258 Occurrence Handle10.1006/immu.1994.1027 Occurrence Handle7820455
P. Benzinger G. Martiny-Baron P. Reusch G. Siemeister J. T. Kley D. Marme C. Unger U. Massing (2000) ArticleTitleTargeting of endothelial KDR receptors with 3G2 immunoliposomes in vitro Biochim. Biophys. Acta 1466 71–78 Occurrence Handle10825432
D. D. Spragg D. R. Alford R. Greferath C. E. Larsen K. D. Lee G. C. Gurtner M. I. Cybulsky P. F. Tosi C. Nicolau (1997) ArticleTitleImmunotargeting of liposomes to activated vascular endothelial cells: a strategy for site-selective delivery in the cardiovascular system Proc. Natl. Acad. Sci. 94 8795–8800 Occurrence Handle10.1073/pnas.94.16.8795 Occurrence Handle9238057
R. A. Carter I. P. Wicks (2001) ArticleTitleVascular cell adhesion molecule 1 (CD106): a multifaceted regulator of joint inflammation Arthritis Rheum. 44 985–994 Occurrence Handle10.1002/1529-0131(200105)44:5<985::AID-ANR176>3.0.CO;2-P Occurrence Handle11352261
P. Vajkoczy M. Laschinger B. Engelhardt (2001) ArticleTitleAlpha4-integrin-VCAM-1 binding mediates G protein-independent capture of encephalitogenic T cell blasts to CNS white matter microvessels J. Clin. Invest. 108 557–565 Occurrence Handle10.1172/JCI200112440 Occurrence Handle11518729
K. Iiyama L. Hajra M. Iiyama H. Li M. DiChiara B. D. Medoff M. I. Cybulsky (1999) ArticleTitlePatterns of vascular cell adhesion molecule-1 and intercellular adhesion molecule-1 expression in rabbit and mouse atherosclerotic lesions and at sites predisposed to lesion formation Circ. Res. 85 199–207 Occurrence Handle10417402
S. Guccione K. C. Li M. D. Bednarski (2004) ArticleTitleMolecular imaging and therapy directed at the neovasculature in pathologies. How imaging can be incorporated into vascular-targeted delivery systems to generate active therapeutic agents Eng. Med. Biol. Mag. 23 50–56 Occurrence Handle10.1109/MEMB.2004.1360408
I. Kuzu R. Bicknell C. D. Fletcher K. C. Gatter (1993) ArticleTitleExpression of adhesion molecules on the endothelium of normal tissue vessels and vascular tumors Lab. Invest. 69 322–328 Occurrence Handle7690867
M. Voinea E. Dragomir I. Manduteanu M. Simionescu (2002) ArticleTitleBinding and uptake of transferrin-bound liposomes targeted to transferrin receptors of endothelial cells Vascul. Pharmacol. 39 13–20 Occurrence Handle10.1016/S1537-1891(02)00165-9 Occurrence Handle12616986
S. Muro C. Gajewski M. Koval V. R. Muzykantov (2005) ArticleTitleICAM-1 recycling in endothelial cells: a novel pathway for sustained intracellular delivery and prolonged effects of drugs Blood 105 650–658 Occurrence Handle10.1182/blood-2004-05-1714 Occurrence Handle15367437
S. Muro R. Wiewrodt A. Thomas L. Koniaris S. M. Albelda V. R. Muzykantov M. Koval (2003) ArticleTitleA novel endocytic pathway induced by clustering endothelial ICAM-1 or PECAM-1 J. Cell Sci. 116 1599–1609 Occurrence Handle10.1242/jcs.00367 Occurrence Handle12640043
A. J. Huang J. E. Manning T. M. Bandak M. C. Ratau K. R. Hanser S. C. Silverstein (1993) ArticleTitleEndothelial cell cytosolic free calcium regulates neutrophil migration across monolayers of endothelial cells J. Cell Biol. 120 1371–1380 Occurrence Handle10.1083/jcb.120.6.1371 Occurrence Handle8449983
P. Lorenzon E. Vecile E. Nardon E. Ferrero J. M. Harlan F. Tedesco A. Dobrina (1998) ArticleTitleEndothelial cell E- and P-selectin and vascular cell adhesion molecule-1 function as signaling receptors J. Cell Biol. 142 1381–1391 Occurrence Handle10.1083/jcb.142.5.1381 Occurrence Handle9732297
O. Barreiro M. Yanez-Mo J. M. Serrador M. C. Montoya M. Vicente-Manzanares R. Tejedor H. Furthmayr F. Sanchez-Madrid (2002) ArticleTitleDynamic interaction of VCAM-1 and ICAM-1 with moesin and ezrin in a novel endothelial docking structure for adherent leukocytes J. Cell Biol. 157 1233–1245 Occurrence Handle10.1083/jcb.200112126 Occurrence Handle12082081
S. Wetering Particlevan N. Berk Particlevan den J. D. Buul Particlevan F. P. Mul I. Lommerse R. Mous J. P. Klooster Particleten J. J. Zwaginga P. L. Hordijk (2003) ArticleTitleVCAM-1-mediated Rac signaling controls endothelial cell-cell contacts and leukocyte transmigration Am. J. Physiol., Cell Physiol. 285 C343–C352
Acknowledgments
The authors are indebted to G. Mesca, D. Rogoz, and F. Georgescu for the excellent technical assistance. We are grateful to Dr. Cora-Jean S. Edgell (Department of Pathology, University of North Carolina, Chapel Hill, NC, USA) for kindly providing the HEC line. This work was supported by a grant obtained from the Romanian Ministry of Education and Research, National Program VIASAN (Grant nr. 031).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Voinea, M., Manduteanu, I., Dragomir, E. et al. Immunoliposomes Directed Toward VCAM-1 Interact Specifically with Activated Endothelial Cells—A Potential Tool for Specific Drug Delivery. Pharm Res 22, 1906–1917 (2005). https://doi.org/10.1007/s11095-005-7247-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11095-005-7247-3