
The kinetics and mechanism of hydrogenation of p-nitrobenzoic acid ethyl ester and transesterification of the resulting Anestezin (benzocaine) by diethylaminoethanol on palladium-containing polymers were studied. The kinetic data obtained here suggest a mechanism for transesterification in the presence of acid and alkali. Free functional groups of the weakly basic and strongly basic anion resins are shown to be involved in transesterification. An improved method for preparing novocaine in mild conditions (t = 45°C, hydrogen pressure 1 atm.) is proposed; this is more efficient than the industrial process.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
The synthesis of novocaine (I) from p-nitrobenzoic acid ethyl ester (II) is a multistage process. Large quantities of side and intermediate products are formed. This decreases the yield of I and increases the material costs associated with its manufacture.
With the aim of decreasing the number of intermediate stages and increasing the yield, we have studied the kinetics and mechanism of synthesis of novocaine by combining the hydrogenation of I and the transesterification of the in situ generated Anestezin (benzocaine) (III) by diethylaminoethanol (IV). Previous studies [1–3] demonstrated that the quantity and rate of formation of hydrogenation and transesterification products depend on the process conditions and catalyst.
The anion resins AB-17-8-Pd, AH-1-Pd, and AH-108-é-Pd, containing active palladium centers as nanoparticles tightly bound in a three-dimensional polymer matrix, were used as catalysts. Kinetic studies showed that in the conditions selected, hydrogenation of II and transesterification of III occur in the kinetic range and are first order with respect to catalyst and hydrogen. The relationship between changes in the effective rate constant of transesterification of III and the quantity of catalyst is shown in Fig. 1. Rate constants were calculated form kinetic data in terms of decreases in the concentration of II and increases in the concentration of III in the reaction mix, as determined by GLC. Figure 1 shows that although the first order relationship with catalyst applied to all three resins, AB-17-8-Pd was the most sensitive to increases in catalyst weight (curve 1). In this relationship, catalysts were in the following order: AB-17-8-Pd > AH-1-Pd > AH-108-é-Pd, i.e., in the same order as their basicities. We note that with the initial palladium concentration (4% w/w), most of the functional groups of the anionic resin remain free [4] and that these groups are active centers for the transesterification reaction. Thus, the structure of the catalysts used, i.e., their functional groups, can influence both the hydrogenation stage and the transesterification stage.

Relationship between rate constant of the reaction forming I (transesterification of III) and the quantity of catalyst: 1) AB-17-8-Pd; 2) AH-1-Pd; 3) AH-108-é-Pd. See Table 1 for conditions.
The relationship between the rate constant of formation of I and the initial concentration of II (Fig. 2 a) provides evidence that the hydrogenation reaction of II to III is null order, and this is consistent with previous data [1–3]. Thus, in conditions of simultaneous hydrogenation and transesterification, hydrogenation of the nitro compound occurs with a high yield of the amine and is subject to the same pattern as when the reactions are run separately. Losses of I associated with this stage of the reaction are therefore completely avoided. The relationships between changes in the concentration of IV and reaction time were different (Fig. 2 b). Thus, while catalysts AB-17-8-Pd and AH-1-Pd (curves 1 and 2) showed first-order reactions, the order of the reaction on AH-108-é-Pd (curve 3 ) was close to null, though there was a minor increase in the rate of the reaction with increases in the concentration of IV. Thus, IV can be used as solvent for AB-17-8-Pd and AH-1-Pd. Increases in the concentration of IV displace the equilibrium towards formation of the end product (Table 1). It should be noted that the use of one of the transesterification reaction components as solvent is not a drawback, as IV is easily regenerated with quantitative yields and reutilized in the reaction. However, this point is not addressed here.

Relationship between rate constant of formation of I and initial concentrations of: a) I; b) IV; 1) AB-17-8-Pd; 2) AH-1-Pd; 3) AH-108-é-Pd. See Table 1 for conditions; 100 mg of catalyst.
With the aim of improving the method of preparing I, the effects of adding acids and bases on the rate constant of transesterification of III were studied, as this reaction is known to be accelerated by acids or bases. Thus, addition of hydrochloric acid to the reaction mix (Fig. 3 a) increased the reaction rate to the greatest extent on AH-108-é-Pd (curve 3). At a hydrochloric acid concentration of 0.12 M, the reaction rate on AH-108-é-Pd became comparable to the rate on AH-1-Pd (curve 2), but remained less than that on AB-17-8-Pd (curve 1). Some difference in the patterns for catalysts AB-17-8-Pd and AH-1-Pd support their involvement not only in the hydrogenation of II, but also in the transesterification of III.

Relationship between rate constant of formation of I and concentrations of: a) HCl; b) KOH; 1) AB-17-8-Pd; 2) AH-1-Pd; 3) AH-108-é-Pd. See Table 1 for conditions.
Studies of the effects of a base also showed this (Fig. 3 b). An increase in the KOH concentration had no effect on the reaction using AB-17-8-Pd and AH-1-Pd (curves 1 and 2). Potassium hydroxide had an even greater influence than hydrochloric acid on the transesterification reaction with AH-108-é-Pd (Fig. 3 a, curve 3). The null order with respect to KOH provides evidence that AB-17-8-Pd and AH-1-Pd had direct roles in the transesterification of III. The active centers here were free functional groups of weakly bound, strongly basic, and weakly basic anionic resins. This is evidenced by the first order kinetics of the transesterification reaction with respect to catalyst (Fig. 1, curves 1 and 2).
These data, along with theoretical considerations, allow the following reaction mechanisms to be described: 1) for transesterification catalyzed by acid:
-
1)
for transesterification catalyzed by acid:
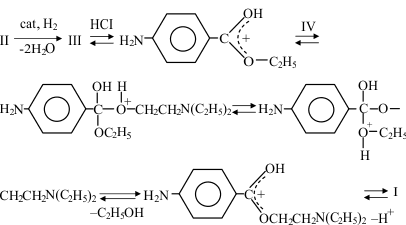
-
2)
for transesterification catalyzed by base:
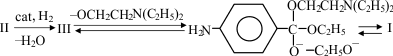


In both cases, IV functions as a nucleophile. Transesterification is an equilibrium reaction. To displace the equilibrium to the right, a large excess of alcohol or removal of one of the reaction products from the reaction mix is required. This latter approach is not applicable in our case, as the reaction occurs in conditions of hydrogenation. However, the first method is more appropriate and does not require special conditions. The resulting ethanol is easily evaporated.
Transesterification in the presence of acid involves in situ protonation of the compound III which is formed. H+ binds to an oxygen atom in the ester carboxyl group. Protonation of the amino group of III is unlikely, as the electron-acceptor ester group is known [8] to produce strong reductions in the basicity of the amino group. Compound IV, operating as a nucleophile, then binds to the positively charged carbon atom and the proton moves to the oxygen of the departing group, on cleavage of which the newly formed ester loses the proton and becomes the product of the transesterification reaction.
In the presence of bases, molecule III is attacked by the stronger nucleophile (potassium salt IV), which, displacing the leaving group (the ethylate ion), forms the reaction product.
As shown by the experimental data (Fig. 3 a, b; Table 1), the rate of the compound III transesterification reaction and the yield of I are quite high. Transesterification catalyzed by base is nonetheless preferred, as in this case the number of intermediate products is much smaller and, most importantly, reactions on catalysts AB-17-8-Pd and AH-1-Pd (Fig. 3 b, curves 1 and 2) do not require use of high concentrations of base. Metallopolymers, as noted above, contain free functional groups, which also catalyze transesterification. The data in Table 1 show that the relationship between the yield of I (GLC) and the KOH concentration has a peak, evidently because of hydrolysis of I. Increases in the concentration of the nucleophile IV result in uniform increases in the yield of I.
Thus, these experimental kinetic data and theoretical considerations suggest that the one-step catalytic synthesis of I from II by combining the hydrogenation of II to III and its transesterification has potential as a method for making novocaine.
Experimental Section
Preparation of palladium-containing anionic resins involves several stages.
Conversion of Anionic Resin to the OH Form
Anionic resin AB-17-8 (10 g) was placed in a 100-ml conical flask and 20 ml of 1 N hydrochloric acid was added; the suspension was held for 3 h. The anionic resin was then collected by filtration and washed with distilled water until the filtrate no longer gave an acidic reaction. Washed anionic resin was then transferred to a 100-ml flask and 60 ml of 1 N sodium hydroxide was added; the suspension was periodically stirred and held for 3 h. The anionic resin was then washed with 50 ml of acetone, 50 ml of diethyl ether or alcohol, and dried in air.
This method was used to convert anionic resins AH-1 and AH-108-é to the OH form. Where necessary, anionic resins of different sizes were prepared by grinding in a mortar and passage through a sieve.
Preparation of Palladium-Containing Anionic Resin
Palladium-containing anionic resins were prepared by dissolving 0.1208 g of potassium tetrachloropalladate in 10 ml of water. This solution was poured into a 50-ml flask preloaded with 1 g of anionic resin AB-17-8 in the OH form and 1 ml of water. The contents of the flask were mixed on a magnetic stirrer for 1 – 2 h at 25°C. Catalyst was then collected by filtration, washed with 100 ml of water and 50 ml of acetone, and dried in air. The palladium content in the catalyst was 3.99 ± 0.01% by weight. Catalyst AH-1-Pd and samples with different metal contents were prepared by the same method.
Activation of Catalyst
A 10-g portion of catalyst was placed in a thermostatted glass reactor fitted with a stirrer. Ethanol (50 ml) and sodium borohydride (0.5 g) were added at 45°C. The reactor was ventilated with hydrogen and, with intense mixing, catalyst was activated by passing hydrogen for 60 min. Catalyst was then collected by filtration and washed with water and 50 ml of acetone. Ready catalyst is stored beneath a layer of acetone.
Assay of Palladium in Catalyst
Palladium contents in catalyst were measured in terms of decreases in the concentration of [PdCl4]2- ions during attachment to the carrier; [PdCl4]2- concentrations were estimated in the mother solution using a Specord-UV instrument in quartz cuvettes with a path length of 1 cm, at 280 nm.
Chromatographic Analysis
Hydrogenation products were analyzed using a model 3700 serial chromatograph (Russia) with a flame ionization detector. A glass chromatography column was used, of size 200 × 3 mm, filled with Lukopren G-1000 (5%) in Chromaton. The carrier gas was helium. The evaporator temperature was 523 K, the column temperature was 473 K, the carrier gas flow rate was 1.6 liters/h, sample volume was 0.1 – 0.5 μl, and analysis run time was 40 – 80 min. The internal standard was tridecane. Calibration coefficients were determined for analysis of artificial mixtures.
The content of each component in the mixture (%) was measured in terms of the internal standard and normalized using the calibration coefficients.
Thin Layer Chromatography
Hydrogenation and transesterification reaction products were analyzed qualitatively by TLC on Silufol plates run in acetone:toluene:ammonia. Detection was with UV light.
Method for Hydrogenation and Transesterification
A glass reactor filled with a thermostatting jacket and a magnetic stirrer for mixing was loaded, in a flow of hydrogen with a portion of catalyst (100 – 500 mg) beneath a layer of solvent (10 – 50 mg/kg); the catalyst was then activated with hydrogen for 20 – 30 min. Portions of substrates II and IV (0.1 – 0.7 and 0.04 – 0.18 M respectively) were then added to the reactor in a flow of hydrogen. Reaction mixtures were stirred constantly throughout the experiment at a rate of 900 – 1100 rpm at a hydrogen pressure of 98 – 103 kPa. Alkali was added at the end of the reaction. Novocaine base, an oily liquid, was then extracted with chloroform. The resulting base was then converted to the hydrochloride salt by treatment with the calculated quantity of ethanolic hydrogen chloride. The reaction rate was measured volumetrically in terms of the absorption of hydrogen and by analyzing samples of reaction mix by GLC.
Effective rate constants for the catalytic hydrogenation and transesterification reactions were calculated from GLC data using standard equations for low hydrogen pressures and allowing for the partial pressure of solvent vapor.
These experiments used purified p-nitrobenzoic acid ethyl ester recrystallized from alcohol, reagent grade, and diethylaminoethanol, reagent grade.
The purity of the novocaine synthesized here, determined by TLC and GLC, was at least 98.0%. The yield of I was 72 – 97%.
References
M. G. Abdullaev, Khim.-Farm. Zh., 35(1), 42 – 45 (2001).
M. V. Klyuev and M. G. Abdullaev, Izv. Vuzov. Khimiya Khim. Tekhnol., 42(5), 3 – 13 (1999).
M. V. Klyuev, M. G. Abdullaev, and Z. Sh. Abdullaeva, Khim.-Farm. Zh., 44(8), 31 – 37 (2010).
V. D. Kopylova, T. V. Pogodina, and M. V. Klyuev, Zh. Fiz. Khim., 64(3), 724 – 728 (1990).
Author information
Authors and Affiliations
Corresponding author
Additional information
Translated from Khimiko-Farmatsevticheskii Zhurnal, Vol. 48, No. 5, pp. 43 – 46, May, 2014.
Rights and permissions
About this article

Cite this article
Abdullaev, M.G., Abdullaeva, Z.S., Klyuev, M.V. et al. Kinetics and Mechanism of Synthesis of Novocaine in the Presence of Palladium-Containing Polymers. Pharm Chem J 48, 343–346 (2014). https://doi.org/10.1007/s11094-014-1107-6
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11094-014-1107-6