
Abstract
Plasmas formed in aqueous solutions dissociate water into highly oxidative and reductive radicals which can induce chemical changes in compounds present in the bulk liquid. As a result, electrical discharge plasmas have acquired significant importance in drinking and wastewater treatment. Part II of this manuscript reviews the chemistry of electrical discharges in liquid water and the chemical effects of plasmas on the degradation of organic molecules. Due to a wide range of work done with plasmas in water, this review is limited to streamer-like electrical discharges directly in water excluding the discharges with gases bubbling through the plasma zone and the presence of additives. The goal was to summarize and present major findings on the fundamental mechanisms related to the production of radicals in the plasma as well as to describe chemical pathways for the degradation of different groups of molecules.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Electrical discharge plasma formed in liquid water is under intensive investigation for many possible applications in biomedical, environmental and chemical engineering as well as for general scientific issues in plasma chemistry and other engineering applications [1–7]. A large fraction of these applications is oriented towards drinking and wastewater treatment using plasmas and they are the focus of this review article [2, 4, 8–12].
The earliest studies of electrical discharges in the presence of water date perhaps as far back as Faraday’s work [13] in 1832. The early studies showed that electrical discharge could dissociate water into hydroxyl (\( {\text{OH}}^{ \cdot } \)) and hydrogen (\( {\text{H}}^{ \cdot } \)) radicals [14] and that higher energy discharge could lead to the dissociation of \( {\text{OH}}^{ \cdot } \) into oxygen atoms (O) and \( {\text{H}}^{ \cdot } \). Later studies demonstrated the production of hydrogen peroxide (H2O2) [15–18], molecular oxygen (O2) and hydrogen (H2) [19, 20], hydroperoxyl (\( {\text{HO}}_{2}^{ \cdot } \)), and other radicals [21–23]. In addition, depending upon the solution conductivity and the magnitude of the discharge energy, shock waves and UV light can also be formed [4, 24, 25].
The real interest in the applications of electrical discharges in water for drinking and wastewater treatment began after Clements, Sato and Davis in 1987 [26] published the first results on the chemical effects of high-voltage electrical discharges formed directly in water on the decolorization of a dye. The result revealed that molecules inside the plasma channel dissociate into highly oxidative radicals which can induce chemical changes in compounds present in the bulk liquid.
The primary chemical reactions inside the aqueous plasma result in the formation of excited species (e.g. \( {\text{OH}}^{ \cdot } \), \( {\text{H}}^{ \cdot } \), O, \( {\text{HO}}_{2}^{ \cdot } \)) which either recombine to form stable by-products such as H2O2 and H2 [19, 27], or they return to a lower energetic state and emit UV light [28, 29]. However, in the presence of solutes (organic and inorganic compounds), primary and secondary molecular, ionic, or radical species produced by the discharge can attack these molecules and cause their degradation. Alternatively, solutes can be degraded indirectly through pyrolysis or photolysis.
Although several review articles on the electrical discharges in water already exist [1, 3, 30], Part II of the present one focuses exclusively on the chemistry of electrical discharges in liquid water and the chemical effects of plasmas on the degradation of organic molecules. Part I of this review article deals with plasma physic and streamer propagation mechanisms. Due to a wide range of work done with plasmas in water, this review is limited only to electrical discharges directly in water of any reactor geometry excluding discharges above the water surface, discharges with bubbling gases through the plasma zone and electrolysis. For the same reason only non-catalytic reactions are included (i.e. bulk liquid catalysis) even though the high-voltage electrode material can act as a catalyst. Unless stated otherwise, studies this manuscript reports were conducted with what is generally accepted as a non-catalytic electrode material (e.g. nickel chromium, stainless steel and tungsten, as indicated by linear production of hydrogen peroxide in the bulk liquid [31]).
The goal was to summarize and present major findings on the fundamental mechanisms related to the production of radicals in plasma as well as to describe the chemical pathways for the degradation of different groups of molecules. The authors realize this is a difficult task as plasma in water have been used to degrade numerous compounds including polychlorinated biphenyls [32], pentachlorophenol [33], trichloroethylene [34], BTEX [35], pesticides [12, 36–38], organic dyes [39–41], herbicides [36–38], phenols [18, 28, 42–49], substituted phenols [47, 50], chemical warfare agents [51], anthraquinone [26], dimethylsulfoxide [52], acetophenone [53], perchlorethylene [54], pharmaceuticals [12, 55, 56] and other organic contaminants. Nevertheless, the comparison of the pathways through which these molecules get degraded in the plasma might assist in building a better picture of the chemical processes taking place around and inside the plasma channels.
Chemistry of Electrical Discharges in Water
Primary Chemical Reactions in Plasma
Electron Collisions with Water Molecules
The dissociation of water molecules takes place inside the plasma channel by inelastic collisions with high-energy electrons. Depending on the electron energy, these collisions initiate various processes including ionization, vibrational/rotational excitation and electron attachment. Table 1 lists all the possible mechanisms of energy transfer between an electron and a neutral water molecule as a function of the reaction energy threshold. According to the literature, pulsed electrical discharges in water have average electron energies between 0.5 and 2 eV [57–59]. However, the actual energy of electrons in plasma is a distribution over a wide range of energies (EEDF) thus a small percentage of the electrons in the high tail of the distribution function must have energies greater than the average value. Because plasmas in water are weakly ionized, this energy must at least be high enough to cause the first ionization of the neutral water molecules (R17, Table 1). Estimates using electron density measurements at the streamer tip indicate that less than 1 % of plasma is ionized [60].
Examination of the energy thresholds for the reactions listed in Table 1 indicate that between 0.5 and 2 eV, vibrational and rotational excitation of water molecules are the most important mechanisms through which an electron can lose energy. Indeed, the cross sectional data for electron collisions with water molecules [61] reveal that these two modes of excitation predominate at electron energies of 1 eV. After the electron excitation, water relaxes into a lower energetic state through any of these three reactions [62–66]:
UV photolysis measurements indicate that the first process occurs with the highest probability (90 %) [67].
It can be noted that the by-products of reactions (1–3) are identical to the ones formed by electron collision with ground-state water molecules in reactions R8, R13 and R15 respectively, from the Table 1. However, the comparison of the energy thresholds between reactions (1) and R8 suggests that the former one is more plausible than the direct electron impact dissociation of water into \( {\text{H}}^{ \cdot } \) and \( {\text{OH}}^{ \cdot } \) (R8). The same is true for reactions (2) and (3) which are more likely to occur than reactions R13 (E treshold ~ 23.5 eV) and R15 (E treshold ~ 13.7 eV).
Direct ionization by electron impact includes the ionization of a neutral molecule which was not previously excited. For the ionization of water molecules, in addition to reactions R17–R21, Dolan [68] proposes the following set of reactions:
However, according to the same author, reactions R20, R21, (4) and (5) require a significant amount of energy (E threshold ≫ 19 eV) therefore they have very low probabilities. Itikawa and Mason [61] report the ionization cross sections for the reactions (4) and (5) with the addition of:
The information on the minimum electron energy in [61] confirms that reactions R20, R21 and (4–6) take place with very low probabilities.
Radiation chemistry measurements [69] have shown that when radiation interacts with water, direct ionization as well as the excitation dissociation are the main processes for active radicals formation. Mass spectroscopy studies confirmed that the main ions are H2O+ (~77 %) and OH+ (~18 %) with minor positive ions such as H+ and O+. Considering much lower electron energy in plasma (electron energies during water irradiation are on the order of MeV), it is safe to assume that liquid-phase electrical discharges yield very low concentrations of H2O+ and probably almost no OH+. Taking into account it requires the lowest energy threshold, the ionization of water molecules inside the plasma takes place via reaction R17 (Table 1). According to radiation chemistry studies [69], generated H2O+ radical ions quickly collide with neutral water molecules to yield \( {\text{OH}}^{ \cdot } \):
At electron energies of around 4 eV, the dissociative attachment is the main mechanism of energy transfer between an electron and a water molecule (R22, R23 and R24 in Table 1). After being formed in plasma, OH−, O− and H− undergo the following ion–molecule reactions:
Dissociative attachment most likely plays a minor role in the chemistry of electrical discharges in water.
Thermal Decomposition of Water
Apart from the water dissociation by electron collisions, water molecules can also be decomposed thermally. Thermal dissociation of water begins at temperatures greater than 2,000 K and at 5,000 K the molecule is completely dissociated into O and \( {\text{H}}^{ \cdot } \) [57]:
Taking into account that the gas plasma temperature could be much higher than 2,000 K [70], the primary chemical reactions in plasma are initiated by both, thermal (reaction 11) and electron impact excitation (R2–R7 in Table 1) and dissociation (R8–R24 in Table 1) of the water molecules. Recent studies suggest that plasmas formed using very short (ns) pulses initiate low-temperature non-thermal plasmas [71] whereas plasmas formed using longer, μs pulses are more thermal in nature [70]. The high temperature splitting of water molecules into \( {\text{H}}^{ \cdot } \) and \( {\text{OH}}^{ \cdot } \) is the primary reaction taking place during ultrasonic water treatment [72].
Experimental Evidence for the Primary Chemical Reactions in Plasma
Generation of radicals by the pulsed electrical discharge in water has been proven by optical emission spectroscopy (OES) [18, 22]. It has been shown that for a variety of discharge voltages [73] and solution conductivities [18, 73, 74], the main radical species in plasma are \( {\text{OH}}^{ \cdot } \) (294 and 308 nm), \( {\text{H}}^{ \cdot } \) (656 nm Hα and 486 Hβ), and O (777 and 845 nm) (Fig. 1). \( {\text{OH}}^{ \cdot } \) and \( {\text{H}}^{ \cdot } \) are produced directly by the electron impact dissociation of water. If the plasma temperature and/or electron energy is high enough, the complete dissociation of water molecules will also yield O. Otherwise, the results of a mathematical model describing chemical processes occurring during electrical discharge in water [75] predict that the formation of O proceeds via the following secondary reaction:
Both O and \( {\text{OH}}^{ \cdot } \) are oxidative whereas \( {\text{H}}^{ \cdot } \) is a reductive species.

Optical emission spectrum of an electrical discharge in water. Reproduced from [100]
Despite a variety of spectroscopic techniques available for the quantification of oxidative (\( {\text{OH}}^{ \cdot } \)) radicals in water (e.g. laser induced fluorescence (LIF) and electron spin resonance (ESR)), only the chemical probe method resulted in quantitative data. Indeed, a quantitative determination of \( {\text{OH}}^{ \cdot } \) concentration from spectroscopic data is extremely difficult since it requires a reliable simulation of the plasma physics inside the plasma channel. Hoeben et al. [48] attempted to quantify the concentration of \( {\text{OH}}^{ \cdot } \) produced during a pulsed electrical discharge in water using electron spin resonance and fluorescence spectroscopy. The ESR spin trap they used was 5,5-dimethyl-1-pyrroline N-oxide (DMPO) but because of the degradation of the DMPO-OH adduct by the plasma, they were not able to detect its signal. Furthermore, the high background signal in the water compared to the ones of fluorescent standards disabled them to quantify the radicals using the fluorescence spectroscopy.
Joshi et al. [17] determined the rate of formation of \( {\text{OH}}^{ \cdot } \) using a pseudo-steady-state approximation. The approach involved scavenging of \( {\text{OH}}^{ \cdot } \) with carbonate ions in the presence of a probe (phenol). The reaction rate constant was calculated as a function of the applied voltage and it was on the order of 10−10 M s−1. In one of the latest studies, the quantification of \( {\text{OH}}^{ \cdot } \) produced by the high-voltage electrical discharge in water was performed using chemical probe measurements [27]. The authors used two chemical probes, dimethyl sulfoxide (DMSO) and disodium salt of terephthalic acid (NaTA). The results demonstrated that the rate of \( {\text{OH}}^{ \cdot } \) production for both probes follows zero-order kinetics even though the \( {\text{OH}}^{ \cdot } \) production rate was greater for DMSO. Moreover, the difference in the \( {\text{OH}}^{ \cdot } \) production rate between the two probes got significantly larger as the input power was increased. The authors explained the result with the low solubility of NaTA in water and thus insufficiently high probe concentration required to scavenge all the radicals. At 45 kV applied voltage, pulse energy of 1 J/pulse and the pulse repetition frequency of 60 Hz, they found the production rate of \( {\text{OH}}^{ \cdot } \) to be 6.7 × 10−8 M s−1 which is two orders of magnitude higher than the value measured by Joshi and coauthors [17]. The results of Sahni and Locke’s study [27] also experimentally confirmed that \( {\text{OH}}^{ \cdot } \) is the precursor to the H2O2 formation.
In a similar study, Gupta [76] attempted to quantify \( {\text{OH}}^{ \cdot } \) using two chemical probes: 4 chlorobenzoic acid (p-CBA) and disodium salt of terephthalic acid (DNTA). The results with p-CBA indicated a linear relation between the number of pulses (operating time) and the \( {\text{OH}}^{ \cdot } \) yield. However, limited solubility of p-CBA in water and the restricted pH operating range discouraged the authors to continue the measurements with this probe. Instead, they used DNTA. Experimental results with the second probe revealed that at the solution conductivity of 400 μScm−1, pulse energy of 4.68 J/pulse and the repetition frequency of 10 Hz, the production rate of \( {\text{OH}}^{ \cdot } \) is 1.5 × 10−9 M s−1.
Table 2 compiles and compares rates of production of molecular and radical species in plasma measured by different authors. The production rates of \( {\text{OH}}^{ \cdot } \), H2, O2 and H2O2 strongly depend on operating conditions and the type of the electrical discharge reactor. For a more detailed literature overview of the rates of H2O2 production in liquid-phase electrical discharges, readers are referred to Locke and Shih [77].
To quantify reductive species, Sahni and Locke [78] again used the chemical probe approach. The probes of choice included tetranitromethane (TNM) and tetrazolium chloride (NBT). TNM was chosen because it reacts with a high reaction rate with both \( {\text{H}}^{ \cdot } \) and \( {\text{O}}_{2}^{ \cdot - } \) whereas NBT reacts only with \( {\text{O}}_{2}^{ \cdot - } \). According to the authors, \( {\text{H}}^{ \cdot } \) and \( {\text{O}}_{2}^{ \cdot - } \) are the reductive species most likely to be produced by the discharge. In the presence of molecular oxygen, \( {\text{H}}^{ \cdot } \) quickly reacts to form \( {\text{HO}}_{2}^{ \cdot } \):
which is in a pH dependent equilibrium with \( {\text{O}}_{2}^{ \cdot - } \):
As it was the case with the probes for the oxidative radicals, the results of Sahni and Locke’s study show that different probes yield significantly different production rates of the radical species. Using qualitative measurements with NBT, the presence of \( {\text{O}}_{2}^{ \cdot - } \) was also confirmed by Gupta [76].
Aqueous Electrons
In radiation chemistry, the irradiation of water with high energy electrons results in the formation of aqueous (solvated) electrons \( {\text{e}}_{\text{aq}}^{ - } \) [79]:
Aqueous electrons are strong reducing agents which can react with numerous organic and inorganic compounds. The existence of \( {\text{e}}_{\text{aq}}^{ - } \) during an electrical discharge in water would imply that some of the free high-energy plasma electrons escape the plasma into the surrounding water and become solvated. Although there is no strong experimental evidence to support this hypothesis, many studies propose the involvement of \( {\text{e}}_{\text{aq}}^{ - } \) in chemical reactions of underwater discharges [22, 60, 80]. However, very few attempted to prove their existence. To detect \( {\text{e}}_{\text{aq}}^{ - } \), Gupta [76] used the well-known reaction between aqueous electrons and N2O to yield \( {\text{OH}}^{ \cdot } \):
By measuring the concentration of \( {\text{OH}}^{ \cdot } \) in N2O-saturated and unsaturated aqueous solutions, he found that the N2O-saturated solution yields 1.5 times more \( {\text{OH}}^{ \cdot } \). The result was attributed to the additional production of \( {\text{OH}}^{ \cdot } \) via reaction (16) and thus the presence of \( {\text{e}}_{\text{aq}}^{ - } \). This result was contrary to the one reported by Joshi et al. [17] who found that adding N2O does not play a significant role on the chemistry of electrical discharge in water.
As an indirect measure of \( {\text{e}}_{\text{aq}}^{ - } \), Thagard et al. [74] used the reaction of reduction of silver ions into silver metal. The experiment did not include any quantitative measurements and relied only on the observations of the metallic silver visible to the naked eye. Results showed that the reduction of silver ions takes place in the negative polarity discharges. Experiments conducted at different pH values suggest that \( {\text{H}}^{ \cdot } \) is not responsible for the reduction. Interestingly, the reduction of silver ions was not observed in the polarity discharge.
Clearly there is a need for a reliable method targeted at proving the existence of these species. While radiation chemistry uses spectroscopy (measurement of the electron’s absorption spectrum) [81], this technique is not suitable for characterizing electrical discharges in water due to very high plasma temperatures.
Secondary Chemical Reactions in Plasma
Due to their very short lifetime, the diffusion of the primary radicals from the plasma (\( {\text{OH}}^{ \cdot } \), \( {\text{H}}^{ \cdot } \) and O) into the surrounding water seems implausible. As the channel cools down or as the radicals diffuse towards the plasma-liquid boundary, many of them recombine to form other radical species and eventually stable molecular byproducts. Roots and Okada [82] calculated the lifetimes and diffusional distances of \( {\text{OH}}^{ \cdot } \), \( {\text{H}}^{ \cdot } \) and \( {\text{e}}_{\text{aq}}^{ - } \) using the reactivity of these radicals with a mammalian cell DNA. The authors found that the irradiation of mammalian cells with X-rays dissociate water surrounding the nucleus into radicals and electrons which then react with the DNA causing its degradation. Diffusional distance X was calculated by measuring the lifetime of these species in the DNA and in the presence of various alcohols as radical scavengers [82]. The following equation was used for the calculation:
where t (s) is the average life time and D (m2/s) is the diffusion coefficient of the radical species.
According to their results, the lifetime (~3.7 × 10−9 s) and thus the diffusion distance of \( {\text{OH}}^{ \cdot } \) is very short (~6 × 10−9 m). Assuming that radical species are produced around the center of the plasma channel, this would imply that even in the smallest experimentally measured plasma channel (r = 10 μm) [83], \( {\text{OH}}^{ \cdot } \) could not escape into the bulk liquid. The lifetime of \( {\text{H}}^{ \cdot } \) (~1.2 × 10−6 s) is approximately three orders of magnitude higher than that of \( {\text{OH}}^{ \cdot } \). Though this lifetime increases the \( {\text{H}}^{ \cdot } \) diffusional distance, the calculated value (~0.2 × 10−6 s) is still smaller than the radius of the smallest experimentally measured plasma channel. Finally, the average diffusion distance value of \( {\text{e}}_{\text{aq}}^{ - } \) (~1 × 10−6 s) implies that these are the only species that might reach the plasma-liquid boundary and thus be involved in the bulk liquid reactions. However, if any of the species (\( {\text{OH}}^{ \cdot } \), \( {\text{H}}^{ \cdot } \) or e −aq ) is formed very close to the plasma-liquid boundary, they could escape the plasma channel and diffuse no further than 6–20 nm. This number is an order of magnitude lower than the one reported for ultrasonic reactions where it has been estimated that the liquid reaction zone extends ~200 nm from the bubble surface [84].
As shown in Table 3 which compiles a large number of reactions in dissociated water vapor [75, 85, 86], the recombination reactions of \( {\text{H}}^{ \cdot } \), O and \( {\text{OH}}^{ \cdot } \) result in the formation of H2, O2 and H2O2. Indeed, experimental measurements confirm that liquid-phase electrical discharges in water result in the formation of three stable byproducts: hydrogen peroxide, hydrogen and oxygen. The production rates of hydrogen peroxide as a function of the applied voltage and solution conductivity have been measured by various groups [17, 19, 21, 28, 29] and some of the results are shown in Table 2. In almost all of the studies, the concentration of hydrogen peroxide was determined in a reaction with titanium sulfate [87]. A review of the different methods for the production of H2O2 including electrical discharges in water is given by Locke and Shih [77].
For a wide range of solution conductivity values and discharge powers, the production rates of molecular hydrogen, oxygen and hydrogen peroxide follow the stoichiometry described by reaction (18) [19].
The values of the hydrogen and oxygen production rates are shown in Table 2.
To explain the chemical pathways for the production of molecular and radical species in plasma, several mathematical models have been developed [17, 42, 51, 88–90]. In addition to a large number of reactions from radiation chemistry and combustion, the majority of these models used reaction (18) and its reaction rate constant as an adjustable parameter. One of the first models used to predict experimentally measured concentrations of molecular species in plasma without using the global reaction (18) took into account the pulsed nature of the plasma, however, it only included thermal dissociation reactions and neglected electron collisions [75]. According to the model, the most important reactions in the electrical discharge channel that result in the production of hydrogen, oxygen and hydrogen peroxide are the following:
where M is a third body. Results also accurately predicted the stoichiometry of reaction (18) and experimentally measured concentration of \( {\text{OH}}^{ \cdot } \) in the plasma [27].
Other Physical Processes in Plasma
The generation of active species in plasma is preceded by the formation and propagation of the plasma channel. Measurements have shown that as the plasma channel expands against the surrounding liquid, it forms shock waves, induces cavitation and emits UV light [1, 29]. While studies on chemical effects of the shock wave-induced cavitation and UV light are scarce, the biological effects of shockwaves on the soft animal tissue are currently under full investigation [91, 92]. Establishing the effects cavitation and UV light have on the degradation of molecules in the bulk phase is extremely important as ultraviolet radiation is one of the principal forms of energy which is dissipated from an electrical discharge in water [93].
Results obtained using emission spectroscopy showed that an electrical discharge in water is an intense radiation source ranging from far ultraviolet to near infrared spectral range [16, 18, 22, 26, 93, 94]. The emission of the UV light from plasma is a result of the collisions between electrons and neutral molecules. These collisions generate exited species which upon relaxation to lower energetic states emit UV light. The intensity of UV light produced by a pulsed corona liquid discharge in the spectral region 190–280 nm is known to increase with the solution conductivity (especially between 100 and 500 μScm−1) and the applied voltage [28, 95].
Shockwaves also induce chemical effects in bulk liquid. Using glass and plastic water filled cuvettes and placing them in water very closed to the high-voltage anode surface, Gupta [76] found that the \( {\text{OH}}^{ \cdot } \) yield inside the cuvettes increases with the number of pulses even though the solution inside the cuvette is not in a direct contact with the streamer channels. The effect was attributed to the shockwave-induced dissociation of water and the production of \( {\text{OH}}^{ \cdot } \) and H2O2 in the bulk liquid.
Chemistry of Electrical Discharges in Aqueous Solutions: Effect of Operating Parameters on the Production of Radical and Molecular Species
Input Power
An increase in the applied power should result in the faster decomposition of water molecules and consequently higher concentration of radical species in plasma and molecular species in the bulk liquid. Experiments have confirmed that densities of \( {\text{OH}}^{ \cdot } \) [22, 27], O [96], \( {\text{HO}}_{2}^{ \cdot } \) and \( {\text{O}}_{2}^{ \cdot - } \) [78], H2O2 [17, 19, 28, 97–99] and H2 and O2 [19] increase with an increase in the applied voltage. Lukes [28] also discovered that the rate of hydrogen peroxide formation increases linearly with the increase in mean power input regardless of the energy per pulse, frequency and pulse duration.
Solution Conductivity
An increase in solution conductivity has a strong effect on the appearance and physico-chemical characteristics of the plasma [100]. Specifically, when conductivity is increased, plasma channels become shorter, wider and brighter due to the strong emission of UV light [1]. Additionally, for the constant applied voltage, both electron and current density increase which results in the higher energy deposition into plasma. All these factors, in addition to the shorter duration of the plasma pulse, have an effect on plasma chemistry and the production of active species in plasma.
The effect of the solution conductivity on the production of molecular and radical species has been studied by several groups. Sun et al. [22] demonstrated that the intensity of \( {\text{OH}}^{ \cdot } \) in the conductivity range of 1–150 μScm−1 reaches a maximum between 10 and 80 μScm−1. To examine the role of the solution conductivity on the production of \( {\text{OH}}^{ \cdot } \), Sahni and Locke [27] conducted experiments with two different chemical probes, NaTA and DMSO. At low concentrations of both probes and the applied voltage of 45 kV, their results indicate that between 100 and 840 μScm−1 \( {\text{OH}}^{ \cdot } \) concentration decreases. Contrarily, experiments with high concentrations of DMSO show that the initial conductivity of the solution does not affect the \( {\text{OH}}^{ \cdot } \) concentration (production rate). To explain the different results, the authors hypothesized that probe concentration has an effect on the physical location where the reaction between \( {\text{OH}}^{ \cdot } \) and the probe take place. At low probe concentrations, DMSO reacts with \( {\text{OH}}^{ \cdot } \) close to the plasma-liquid boundary and as a result, changes in the bulk solution conductivity have a strong effect on the \( {\text{OH}}^{ \cdot } \) concentration. However, at high probe concentrations, the probe molecules diffuse into the plasma channel and completely scavenge \( {\text{OH}}^{ \cdot } \). Because chemical reactions in plasma are assumed to be independent of the solution conductivity, so is the concentration of \( {\text{OH}}^{ \cdot } \).
Shih and Locke [100] found that at 45 kV applied voltage the concentration of \( {\text{OH}}^{ \cdot } \) and H2O2 decreases with an increase in solution conductivity. They also observed that as the solution conductivity is increased, the intensities of both O and \( {\text{H}}^{ \cdot } \) exhibit a maximum at 150 μScm−1 after which they slowly start to decrease. Further increase in the solution conductivity above 500 μScm−1 decreases the intensities of all the radical species. The results were explained by the physical and chemical changes occurring in the plasma when solution conductivity is increased.
Sahni and Locke also studied the effect of the solution conductivity on the production of reductive species [78]. Using TNM as the probe, the authors found that at 45 kV and the initial solution pH of 4 and 7 the concentration of reductive species in the conductivity range 150–500 μScm−1 decreases.
Between 100 and 500 μScm−1 the concentration of hydrogen peroxide in the bulk liquid decreases with an increase in the solution conductivity [28, 95, 101]. The effect is attributed to the increased intensity of the UV light and direct photolysis of H2O2:
The production rates of H2 and O2 also decrease with increasing conductivity [90, 100].
Thagard et al. [74] found that for the positive polarity discharge, an increase in the solution conductivity (from 50 to 2,000 μScm−1) strongly effects the concentration of hydrogen peroxide in the bulk liquid. At low pH (= 3.5) an increase in the solution conductivity increases the concentration of H2O2 in the bulk liquid due to the higher power deposited into plasma. At high pH (= 10.5), the increase in conductivity lowers the H2O2 concentration and the effect was attributed to the scavenging of \( {\text{OH}}^{ \cdot } \) (precursors to H2O2) with hydroxide ions. In the negative polarity discharge, regardless of the pH value, an increase in the solution conductivity lowers the production of H2O2. To explain the result, the authors proposed the decomposition of H2O2 at the plasma-liquid boundary by the aqueous electrons.
Due to a large number of studies with contradictory results, more research is needed to quantify oxidative and reductive radicals in plasma. The knowledge of the concentrations of these species might assist in assessing the oxidative power of electrical discharges compared to other advanced oxidation technologies.
pH
The solution pH changes with solution conductivity therefore the effects of the two parameters are difficult to differentiate. Nevertheless, experimental evidence on the effects of pH on the production of molecular and radical species exists. OES studies indicate that the \( {\text{OH}}^{ \cdot } \) emission intensity is pH-dependent and increases when neutral and alkaline conditions are used [22]. The same was discovered for \( {\text{HO}}_{2}^{ \cdot } \) and \( {\text{O}}_{2}^{ \cdot - } \) [78] and explained by the \( {\text{HO}}_{2}^{ \cdot } \)/\( {\text{O}}_{2}^{ \cdot - } \) pH dependent equilibrium and the increased concentration of \( {\text{O}}_{2}^{ \cdot - } \) at the plasma-liquid boundary at higher pH values. Lukes [28] on the other hand found that between pH = 3.2 and pH = 10.6 the formation of H2O2 is independent of pH.
The effect of the solution pH on the production of H2O2 and the emission intensity of \( {\text{OH}}^{ \cdot } \), \( {\text{H}}^{ \cdot } \) and O was also studied by Thagard et al. [74]. The results of that study reveal that at low solution conductivities (<200 μScm−1) solution pH has a minimal effect on the intensity of plasma radicals and the production rate of H2O2. Contrarily, at very high salt concentrations (conductivities >500 μScm−1), the production rate of H2O2 begins to strongly depend on the pH. The results of this study are presented in “Solution Conductivity” section.
Polarity
Though physical and electrical characteristic of negative and positive polarity plasma channels are quite different [22], there are only a few published studies that report the effects of electrode polarity on plasma chemistry. Lukes [28] and Thagard et al. [74] both report that the production rate of H2O2 in the negative polarity is always lower than that of the positive polarity. Thagard’s study hypothesized this is due to the differences in the concentration of radicals inside plasma as the relative intensities of \( {\text{OH}}^{ \cdot } \), \( {\text{H}}^{ \cdot } \) and O are higher for the positive than for the negative polarity discharge [22, 96]. Intensity of UV light produced by the electrical discharge is also much higher for the positive than for the negative polarity discharge [28].
Chemistry of Electrical Discharges in Aqueous Solutions Containing Organic Molecules
Oxidation
Hydroxyl Radical
Of all the radical species that exist in plasma, \( {\text{OH}}^{ \cdot } \) is probably the most important one. It is one of the strongest oxidizers among the oxygen-based oxidizers with a standard oxidation potential (\( E_{{{\text{OH}}^{ \cdot } /{\text{H}}_{2} {\text{O}}}}^{0} \)) of 2.85 V [102]. In strongly alkaline solution \( {\text{OH}}^{ \cdot } \) is rapidly converted to its conjugate base \( {\text{O}}^{ \cdot - } \) [103]:
\( {\text{O}}^{ \cdot - } \) reacts much more slowly than \( {\text{OH}}^{ \cdot } \) with inorganic anions whereas in reaction with organic molecules \( {\text{OH}}^{ \cdot } \) behaves as an electrophile and \( {\text{O}}^{ \cdot - } \) is a nucleophile. As a result, \( {\text{OH}}^{ \cdot } \) easily undergoes electrophilic addition (reactions 26 and 27) but \( {\text{O}}^{ \cdot - } \) does not. Both radical forms abstract hydrogen from C–H bonds (reactions 28 and 29), and this can result in the formation of different products when the pH is raised to the value where \( {\text{O}}^{ \cdot - } \) , rather than \( {\text{OH}}^{ \cdot } \), is the reactant [48, 104].
Addition reactions usually take place with aromatic and unsaturated aliphatic compounds. Attack of the \( {\text{OH}}^{ \cdot } \) on an aromatic compound followed by reactions with molecular oxygen yields a variety of intermediate radicals which finally decompose to unsaturated aliphatic hydrocarbons with polyfunctional groups (e.g. carboxyl, aldehyde, etc.). The reaction between \( {\text{OH}}^{ \cdot } \) and an alkene proceeds in a similar manner; the initial \( {\text{OH}}^{ \cdot } \) attack on the alkene yields a radical which further reacts with oxygen to give α-hydroxyalkyl radicals, aldehydes/ketones and oxygen. Additionally, a competitive reaction between the α-hydroxyalkyl radical and oxygen can result in the production of an aldehyde or ketone and a hydroperoxyl radical.
\( {\text{OH}}^{ \cdot } \) (and \( {\text{O}}^{ \cdot - } \)) can also abstract a hydrogen atom and this can occur either with saturated hydrocarbons or alcohols. The reaction results in the formation of water (or hydroxide ions) and an organic radical \( {\text{R}}^{ \cdot } \).
Oxygen Atom
According to the table of standard oxidation potentials, O has an oxidation potential of \( {\text{E}}_{{{\text{O/H}}_{2} {\text{O}}}}^{0} \) = 2.42 V [102] which is slightly higher than that of ozone (\( {\text{E}}_{{{\text{O}}_{3} /{\text{O}}_{2} }}^{0} \)=2.07 V). In alkaline solutions, it reacts with hydroxide ions to give \( {\text{HO}}_{2}^{ - } \) and \( {\text{OH}}^{ \cdot } \) [103]:
Because the mechanism of the O-induced degradation of organic molecules is not well understood, it is assumed to be similar to that of \( {\text{OH}}^{ \cdot } \).
Hydroperoxyl Radical and Superoxide Radical Anion
\( {\text{HO}}_{2}^{ \cdot } \) is an oxidative species produced in a reaction between molecular oxygen and \( {\text{H}}^{ \cdot } \) (reaction 13). Its oxidation potential (\( {\text{E}}_{{{\text{HO}}_{2}^{ \cdot } /{\text{H}}_{2} {\text{O}}}}^{0} \)) is 1.44 V [105] which makes it the weakest of all the oxidative species produced in plasma. This radical exists in a pH dependent equilibrium with \( {\text{O}}_{2}^{ \cdot - } \) (reaction 14) whose reduction potential (\( {\text{E}}_{{{\text{O}}_{2}^{ \cdot - } /{\text{H}}_{2} {\text{O}}}}^{0} \)) is −0.33 V [102]. Both \( {\text{HO}}_{2}^{ \cdot } \) and \( {\text{O}}_{2}^{ \cdot - } \) react with various inorganic and organic substances in aqueous solutions. However, because of the pH equilibrium, the concentration of only one species is dominant in the certain pH range. Simple calculations show that at pH = 3.5 only 5 % of \( {\text{HO}}_{2}^{ \cdot } \) exist as \( {\text{O}}_{2}^{ \cdot - } \) whereas at pH = 6, almost 94 % of \( {\text{HO}}_{2}^{ \cdot } \) is \( {\text{O}}_{2}^{ \cdot - } \). Thus, chemical reactions involving \( {\text{HO}}_{2}^{ \cdot } \) will be important only at low pH values. Both \( {\text{HO}}_{2}^{ \cdot } \) and \( {\text{O}}_{2}^{ \cdot - } \) react by hydrogen abstraction [105]. However, there is no direct experimental evidence that organic molecules are degraded by either of these two radicals during the electrical discharge.
Hydrogen Peroxide
The standard oxidation potential of H2O2 (\( {\text{E}}_{{{\text{H}}_{2} {\text{O}}_{2} /{\text{H}}_{2} {\text{O}}}}^{0} \)) is around 1.77 V [102] which is much lower than the one for \( {\text{OH}}^{ \cdot } \), O and even O3. Similarly, its reduction potential (\( {\text{E}}^{0}_{{{\text{O}}_{2} /{\text{H}}_{2} {\text{O}}_{2} }} \)) = −0.70 V [102]) is also very low compared to the ones of other reductive species produced in plasma. Considering that most organic compounds have standard oxidation potential of around 0.5 V, during an electrical discharge, H2O2 could react with organic species directly. Indirectly, it can be involved in the additional production of \( {\text{OH}}^{ \cdot } \) in the bulk liquid via photolysis (reaction (24)) and metal-based catalytic reactions (e.g. Fenton’s reagent). At very high pH values, H2O2 is in equilibrium with its conjugated base \( {\text{HO}}_{2}^{ - } \):
which can also be involved in reactions with radicals. However, the typical pH values of aqueous solutions treated by an electrical discharge are never high enough to shift the equilibrium of reaction (32) to the right, so the concentration of \( {\text{HO}}_{2}^{ - } \) usually remains very low. When present in sufficient quantities, H2O2 reacts with various radical species and acts as a radical scavenger [103]:
Reactions (33–36) are especially important when organic compounds getting degraded are present in low concentrations (<mM range).
Reduction
Hydrogen Radical
\( {\text{H}}^{ \cdot } \) is formed directly by the electron collision with water molecules. This species is a powerful reducing agent with a reduction potential (\( {\text{E}}_{{{\text{H}}_{2} {\text{O}}/{\text{H}}^{ \cdot } }}^{0} \)) of -2.30 V [102]. In the absence of organic molecules in the bulk liquid, \( {\text{H}}^{ \cdot } \) quickly recombine to form stable byproducts (H2) and/or other radicals (e.g. \( {\text{HO}}_{2}^{ \cdot } \)) (see Table 3). In the alkaline region, \( {\text{H}}^{ \cdot } \) react with hydroxide ions to give \( {\text{e}}_{\text{aq}}^{ - } \):
In the presence of organic and inorganic compounds, \( {\text{H}}^{ \cdot } \) react according to any of the following reactions [106]:
-
(a)
Electron transfer:
$$ {\text{H}}^{ \cdot } + {\text{M}}^{n + } \to {\text{H}}^{ + } + {\text{M}}^{(n - 1) + } $$(38) -
(b)
Atom transfer:
$$ {\text{H}}^{ \cdot } + {\text{X}}_{2} \to {\text{HX}} + {\text{X}}^{ \cdot } $$(39) -
(c)
Addition:
$$ {\text{H}}^{ \cdot } + {\text{X}} - {\text{CH}} = {\text{CH}} - {\text{Y}} \to {\text{X}} - {\text{CH}}_{2} \mathop C\limits^{ \cdot } {\text{H}} - {\text{Y}} $$(40) -
(d)
Abstraction:
$$ {\text{H}}^{ \cdot } + {\text{C}}_{6} {\text{H}}_{5} -{\text{Y}} \to \mathop {\text{C}_{6}}\limits^{ \cdot }{\text{H}_{6}} - {\text{Y}} $$(41)
where X–CH = CH–Y is an unsaturated, C6H5–Y an aromatic and x–CH2–Y a saturated compound.
For the hydrogen abstraction from an aliphatic compound or an addition to an aromatic ring, the reactions of \( {\text{H}}^{ \cdot } \) usually follow the same pathway as those of \( {\text{OH}}^{ \cdot } \). However, the reaction rate constants for the abstraction are usually higher for \( {\text{OH}}^{ \cdot } \) than for \( {\text{H}}^{ \cdot } \), and the selectivity of \( {\text{OH}}^{ \cdot } \) is lower.
Aqueous Electrons
Though both \( {\text{H}}^{ \cdot } \) and \( {\text{e}}_{\text{aq}}^{ - } \) are reductive in nature, the reactions of \( {\text{H}}^{ \cdot } \) follow those of \( {\text{e}}_{\text{aq}}^{ - } \) only to a limited extent because the latter are pure electron transfer reactions whereas \( {\text{H}}^{ \cdot } \) can react both by electron and atom transfer. The reductive potential of \( {\text{e}}_{\text{aq}}^{ - } - \left( {{\text{E}}_{{{\text{H}}_{2} {\text{O}}/{\text{e}}_{\text{aq}}^{ - } }}^{0} } \right) \) is –2.77 V which makes it a strong reducing agent [102]. It reacts with numerous organic compounds and the extent of reactivity is usually determined by the electron density of a specific functional group. Nevertheless, the bond dissociation of the organic molecule usually proceeds via a dissociative electron capture [107]:
where X is a halogen atom. Because of its high reactivity with most carbon-halogen bonds, \( {\text{e}}_{\text{aq}}^{ - } \) has the potential to be effective even when other organic species are present in much higher concentrations. In pure water, these species also react with \( {\text{OH}}^{ \cdot } \), H2O2 and O2 [103]:
To avoid scavenging of \( {\text{OH}}^{ \cdot } \) with \( {\text{e}}_{\text{aq}}^{ - } \), electrical discharge experiments should be carried out at lower pH where \( {\text{e}}_{\text{aq}}^{ - } \) are removed by reacting with hydronium ions. In these conditions, reaction (37) would be negligible because of the low concentration of hydroxide ions.
Pyrolysis
Pyrolysis is a thermochemical decomposition of organic compounds at elevated temperatures in the absence of oxygen. The reaction involves cleavage of chemical bonds to form smaller molecules and proceeds via a Rice radical mechanism. According to this mechanism [108, 109], the reaction is initiated via C–C bond cleavage of the given aromatic hydrocarbon to form two radicals:
or, if the starting molecule is a chained hydrocarbon, via C–H bond cleavage to give a hydrocarbon radical and \( {\text{H}}^{ \cdot } \):
The radicals subsequently propagate by β-scission and/or H abstraction and finally recombine to give stable molecular byproducts. The most common byproducts or thermal decomposition of hydrocarbons include short-chained hydrocarbons such as methane, ethane, ethylene and acetylene [110].
During an electrical discharge in an aqueous solution, pyrolytic reactions take place only inside the high-temperature plasma channel. In the absence of organic molecules, high temperatures dissociate water molecules into radicals (reaction 11). However, if solution contains organic compounds, these molecules, assuming they are volatile enough, can penetrate the plasma channel and also get degraded thermally. Because the decomposition of water inside the plasma yields oxygen, subsequent reactions of organic radicals formed by the dissociation of organic molecules do not strictly follow the Rice mechanism which implies the absence of oxygen and oxidation mechanisms. Instead, during the discharge, organic radicals are oxidized by any of the oxidative species produced by the plasma (e.g. \( {\text{OH}}^{ \cdot } \)) and result in the formation of CO2, organic and inorganic acids.
UV Photolysis
When a molecule is irradiated by UV light, it absorbs the radiation and gets promoted to an excited state. The most probable transition occurs from the ground to the singlet state [102]:
The molecule in the excited state (M*) has a very short lifetime (10−9 to 10−8 s) after which it returns to the ground state either by one or several mechanisms (internal conversion, fluorescence, phosphorescence, etc.):
The excited molecule can also decompose to give a new molecule:
In the presence of oxygen, the electron in the excited state can be transferred to the oxygen molecule in its ground state to form the superoxide radical anion [102]:
In addition to direct excitation, the organic molecule can also get photo-dissociated via a carbon-hydrogen bond homolysis followed by the reaction with oxygen and the formation of organic peroxyl radicals:
The most common use of UV radiation in water treatment is in disinfection and oxidation (e.g. UV/H2O2 process). For those purposes, UV–C radiation at 254 nm is normally used although other types of UV radiation have also been reported [102].
Electrical discharge in water is an intense radiation source ranging from far ultraviolet to near infrared spectral range. The fraction of radiation in the wavelength range between 75 and 200 nm is usually absorbed by the water molecules immediately surrounding the plasma channel [111]:
Thus only the UV light with wavelengths greater than 200 nm can penetrate the bulk solution and react with organic molecules. In the absence of organics, \( {\text{OH}}^{ \cdot } \) produced in reaction (55) is contributing to the formation of H2O2 in the bulk liquid [101].
Organic Molecules
Dyes
Synthetic dyes are relatively large molecules with molecular weights typically greater than 200 g mol−1 and a characteristic chromophore responsible for the color of the dye. They differ greatly in their chemical composition although the most investigated chemical classes of dyes include the azo, anthraquinone, sulfur, indigoid, triphenylmethyl (trityl), and phthalocyanine. Due to the ease of the method involved in their quantification, dyes are one of the most used model compounds for investigating the efficiency of liquid-phase electrical discharge reactors. As a result, there have been many publications on the degradation of different types of dyes [39, 112–116] and the major findings are summarized in Table 4.
All the studies on the degradation of dyes during an electrical discharge in water agree that \( {\text{OH}}^{ \cdot } \) are the major species responsible for decolorization and the extent to which a dye will be degraded depends on the reactivity between \( {\text{OH}}^{ \cdot } \) and the dye functional groups and substitutes [117]. Experimental results by Sugiarto et al. [39] show that in addition to \( {\text{OH}}^{ \cdot } \), UV photolysis is another dominant process involved in the degradation. In the case of diaphragm discharge, electrolysis also plays a role in the dye removal [117].
One common finding for all the studies in Table 4 is that the degradation of dyes proceeds faster at lower (pH ~ 3) compared to the higher pH (pH > 7) values regardless of the dye structure. Sugiarto et al. [39, 112] explains the phenomenon by scavenging of \( {\text{OH}}^{ \cdot } \) by carbonate ions, the byproducts of the dye degradation. According to the same authors, at pH > 7 \( {\text{OH}}^{ \cdot } \) are nonselective and react with carbonate ions thereby substantially reducing the efficiency of the oxidation process. Xiaoqiong et al. [118] offers another explanation which involves scavenging of \( {\text{OH}}^{ \cdot } \) by hydroxide ions (reaction 25). Scavenging of \( {\text{OH}}^{ \cdot } \) by hydroxide ions was also used to explain the removal of atrazine [38] and 2,2′,4,4′-tetrachlorobiphenyl (TCB) [32] at high pH values. At pH = 10 for example, the concentration of hydroxide ions could be several orders higher than that of the targeted compounds which would make the reaction rate between the \( {\text{OH}}^{ \cdot } \) and hydroxide ions higher. In the case of dyes, the acid–base equilibrium of the dye molecules also plays a significant role in the dye removal from water and could be used to explain different removal rates. The pH-dependent equilibrium was not considered by the studies presented in Table 4.
It must be mentioned that an aqueous solution containing organic molecules (dyes and other compounds as well) could also contain dissolved nitrogen (solubility of N2 at 20 °C is 13.8 mgL−1) and oxygen (solubility of O2 at 20 °C is 8.84 mgL−1) which could compete for the plasma radicals. In plasma, nitrogen dissociates and reacts with oxygen to give nitrite and nitrate ions whereas oxygen most likely yields ozone (oxygen is also produced by the dissociation of water and could be involved in the production of ozone). However, there are no studies reporting that ozone, nitrate and nitrite ions are formed from dissolved air in measurable quantities and it is generally assumed these species have a minute or no effect on the plasma and bulk liquid chemistry.
Contrary to pH, the results in [118] reveal that between 50 and 250 μScm−1 conductivity does not play a role in the degradation of Sunset yellow dye. The reason for that could be the fact that the concentration of \( {\text{OH}}^{ \cdot } \) responsible for the dye degradation does not change significantly in the conductivity range studied. Intensity of \( {\text{OH}}^{ \cdot } \) increases up to 200 μScm−1 and sharply decreases above that value [100]. Stara et al. [117] report that not only conductivity, but also the type of electrolyte strongly affect the dye removal during a DC diaphragm discharge in water.
Xiaoqiong et al. [118] reports that pulse repetition frequency is yet another factor affecting the degradation of dyes. Assuming each pulse produces the same concentration of \( {\text{OH}}^{ \cdot } \), an increase in the pulse repetition rate should increase the \( {\text{OH}}^{ \cdot } \) yield and thus the removal rate of the dye. However, in the capacitor-charging power supplies, an increase in the pulse repetition frequency increases the concentration of plasma radicals only up to a certain value; after reaching a frequency threshold, the discharging of the capacitors might become incomplete and result in lower radical concentrations.
Phenols
After dyes, phenol and its derivatives are the most studied model compounds for the assessment of electrical discharge reactors in pollutant removal. Due to their very low biodegradability, finding an effective oxidation technology for the removal of phenols is still an active area of research. Table 5 summarizes the majority of work that has been done on the degradation of phenol and its derivatives using electrical discharge in water. Studies presented in Table 5 as well as many others [18, 42, 80, 119–125] reveal that the degradation of phenol and its derivatives during an electrical discharge in water depends on numerous parameters including pH, solution conductivity, applied voltage, voltage polarity etc.
While Sharma et al. [45] reported that phenol degradation is independent of the solution pH, Lukes [28] observed that the phenol removal rate increases with an increase in solution pH and that under alkaline conditions the rate of oxidation of phenol is almost twice that under acidic conditions. The result was explained by the acid–base equilibrium of the phenol molecule and the existence of phenoxide, an alkaline form of phenol, which reacts very fast with \( {\text{OH}}^{ \cdot } \). The same effect was observed during the phenol removal from water using gas-phase electrical discharges [126] and other advanced oxidation technologies [127].
Mizeraczyk et al. [125] showed that between 1 and 600 μScm−1 phenol removal decreases with an increase in solution conductivity. The effect was attributed to the lower production of \( {\text{OH}}^{ \cdot } \), the main species responsible for the degradation of phenol, in the water of higher conductivity. That explanation is again supported by the results in [100] where it was shown that the concentration of \( {\text{OH}}^{ \cdot } \) decreases with an increase in solution conductivity. An identical effect was observed by Tothova et al. [128] during the degradation of nonylphenol. Lukes [28] reported that between 100 and 500 μScm−1 solution conductivity does not have an effect on the degradation of phenol. The decreased rate of \( {\text{OH}}^{ \cdot } \) production at higher solution conductivity should have lowered the removal rate of phenol, however, as Lukes suggested, direct photolysis of H2O2 into \( {\text{OH}}^{ \cdot } \) which increases with conductivity makes up for the “lost” \( {\text{OH}}^{ \cdot } \).
All the studies that analyzed degradation pathways of phenols and chlorophenols [28, 129] agree that \( {\text{OH}}^{ \cdot } \) attack on the molecule is the primary mechanism by which these compounds get degraded. The reaction usually proceeds by the electrophilic addition of the \( {\text{OH}}^{ \cdot } \) to the aromatic ring and the formation of hydroxylated byproducts. Final byproducts include organic and inorganic acids as well as carbonyl byproducts. Lukes [28] and Bian et al. [129] give a detailed description of the degradation pathways of both phenol and chlorophenol. Experiments in [28] also suggest the correlation between the degradation rates and structural parameters; higher removal rates of chlorophenols (CP) compared to that of phenol were explained by the electron donating character of chlorine group which increases the electron density on the aryl ring and renders the compound more susceptible towards the electrophilic characteristics of oxidizing agents. Measured removal rates in the order 3-CP > 2-CP > 4-CP were explained by the decreased aromatic electron density of individual chlorophenol isomers depending on the position of chlorine relatively to OH.
Other Compounds
Unlike for dyes and phenols, studies conducted with other compounds report that other species, apart from \( {\text{OH}}^{ \cdot } \) are involved in the degradation of these molecules. One hour degradation of 250 mg/L of acetophenone by a pulsed electrical discharge in water at 30 kV results in 45 % removal of this compound [130]. Results showed that acetophenone is converted into the following byproducts; toluene (C6H5CH3), 1-phenylethanol (C6H5CH(OH)CH3), and 2-acetylphenol (HOC6H4COCH3). The formation of toluene was explained by the pyrolysis and/photolysis of acetophenone:
2-acetylphenol is formed by the reaction between acetophenone and \( {\text{OH}}^{ \cdot } \):
The presence of 1-phenylethanol was attributed to attack of acetophenone carbonyl group by aqueous electrons:
This is one of the first studies that proposed the involvement of other species, apart from \( {\text{OH}}^{ \cdot } \), in the degradation of organic compounds by the pulsed electrical discharge in water.
While studying the degradation of polychlorinated biphenyls (PCB), Sahni et al. [32]. found that at 45 kV and KCl as electrolyte (conductivity is 100 μScm−1), 70 % of 68 ppm TCB can be removed in 1 h. Due to the low initial concentration of TCB, it was concluded that the molecule gets degraded by \( {\text{OH}}^{ \cdot } \) in the bulk of the solution (no diffusion into the plasma). When KCl was replaced by FeSO4, due to the Fenton’s reaction (reaction between ferrous ions and hydrogen peroxide), TCB concentration dropped to zero in about 30 min. Though they were not confirmed experimentally, results of the mathematical model developed in the study suggested that in the case of Fenton’s reaction TCB could also get degraded in reactions with reductive species produced by the plasma. Experiments with \( {\text{OH}}^{ \cdot } \) scavengers (mainly alcohols) confirmed that these are the species responsible for the degradation of atrazine [38], s-triazine [37], H agent simulant (2-chloroethyl phenyl sulfide (CEPS)), G agent simulant (diphenyl chlorophosphate (DPCP)), tricholoroetylene [131] and dimethyl sulfoxide (DMSO) [132]. The structures of these compounds are shown in Fig. 2.

Chemical structures of selected compounds degraded by liquid-phase electrical discharges
The effect of the solution initial pH on the removal efficiency was analyzed using compounds such as PCBs [32], atrazine [38] and s-triazine [37]. The role of pH on the degradation of TCB was assessed by performing experiments at three different pH values (3.5, 5.5 and 10.1). It was shown that at pH = 3.5 and 5.5 the TCB degradation is approximately the same whereas increasing the solution pH to 10 lowers the extent of degradation. The latter result was explained by the scavenging of \( {\text{OH}}^{ \cdot } \) by hydroxide ions (reaction 25).
Mededovic and Locke [38] investigated the degradation of herbicide atrazine at 45 kV and 130 μScm−1 and also at pH = 3.5, pH = 5.0, and pH = 10.0. Adjusting the solution pH to 3.5 resulted in the highest atrazine removal: 90 % in 1 h. Increasing the pH to 5.0 decreased the removal to 20 % and increasing it to pH = 10.0 resulted in only 10 % atrazine removal. The results were again explained by the scavenging of \( {\text{OH}}^{ \cdot } \) with hydroxide ions. The same reaction was used to explain the pH-dependent removal of PCBs [32].
The type of the electrolyte used to adjust solution conductivity strongly effects the pollutant removal. To analyze the effect of the chloride ions (form KCl) on the degradation of TCB, Sahni et al. [32] conducted experiments with KNO3 and K2SO4. Chloride ions are well-known \( {\text{OH}}^{ \cdot } \) scavengers (the reaction may be explained by the values of the standard oxidation potentials of the systems \( {\text{OH}}^{ \cdot } /{\text{H}}_{2} {\text{O}} \) and ClOH/Cl− in acid medium) and their presence might lower the extent of TCB degradation. However, experimental results showed similar degradation of TCB with sulfate and chloride salts while nitrate salts resulted in lower degradation. Similar degradation in sulfate and chloride experiments implies the chloride ion scavenging of the hydroxyl radicals is not a major factor in TCB degradation. The lower degradation observed in the nitrate experiments was explained by the scavenging of reductive species (\( {\text{H}}^{ \cdot } \) and \( {\text{O}}_{2}^{ \cdot - } \)) by the nitrate ions which indicated that other species, apart from \( {\text{OH}}^{ \cdot } \), could also be involved in the degradation.
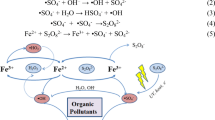
Replacement of halogen-based salts with metal ion salt such as ferrous sulfate usually results in much higher extent of compound degradation on a much shorter time scale [37, 38, 51]. This is due to the high concentration of \( {\text{OH}}^{ \cdot } \) produced by the metal-ion catalyzed decomposition of hydrogen peroxide. Highly reactive radicals can also be formed directly from an electrolyte such as sodium persulfate. UV light and/or heat produced by the plasma decompose persulfate ions and result in the formation of highly oxidative sulfate radical anions (\( {\text{SO}}_{4}^{ \cdot - } \)) which are capable of degrading organic molecules (M):
The standard oxidation potential of \( {\text{SO}}_{4}^{ \cdot - } \) (\( {\text{E}}_{{{\text{S}}_{2} {\text{O}}_{8}^{2 - } /{\text{SO}}_{4}^{ \cdot - } }}^{0} \)) is around 2.01 V, which is only slightly lower than that of \( {\text{OH}}^{ \cdot } \). Compared to potassium chloride, the use of sodium persulfate as an electrolyte significantly increases the removal rate of atrazine [38].
In order to gain understanding of the chemical mechanism involved in the degradation of DMSO during an electrical discharge in water, Lee et al. [132] calculated the volume of the active region (V a ) where \( {\text{OH}}^{ \cdot } \) are concentrated around the plasma channel and the concentration of \( {\text{OH}}^{ \cdot } \) in that region (OHeff). Using the model proposed by Willberg et al. [133], they derived the following expression for calculating V a:
where
V T is the total solution volume and r is the pulse repetition rate. According to equation (62), V a can be calculated from the pseudo-first order rate constant of DMSO degradation (k DMSO). Calculations have shown that in the absence of methanol, V a is 0.21 μL (1/105 of the reaction volume (20 mL)). With the addition of methanol, the size of V a exponentially decreases. OH eff was defined as a time and volume-averaged concentration of \( {\text{OH}}^{ \cdot } \) in the active region during one discharge pulse and was expressed as:
where t d is the duration of the discharge (0.1 μs), k 1 is the reaction rate constant for the formation of hydrogen peroxide by \( {\text{OH}}^{ \cdot } \) recombination, and \( k_{{{\text{H}}_{2} {\text{O}}_{2} }} \) is experimentally determined zero order reaction rate constant for the production of H2O2.
In the absence of methanol, the concentration of \( {\text{OH}}^{ \cdot } \) was calculated to be 5.0 × 10−3 M which is significantly higher than the concentration calculated for other AOPs (10−13 ~ 10−11 M) [134, 135]. Contrary to the results with V a , the addition of methanol did not change the calculated concentration of OHeff in the active region volume.
Based on the performed calculations and the analyses of the experimental data, it was concluded that due to the high local concentration of \( {\text{OH}}^{ \cdot } \), their recombination reaction is significant and competes with the reaction between \( {\text{OH}}^{ \cdot } \) and substrates present in the bulk of the liquid. If the substrate concentration is low, \( {\text{OH}}^{ \cdot } \) will predominantly recombine to form H2O2 without being effectively used for the removal of organic contaminants. That hypothesis is supported by Sahni and Locke’s [27] results who propose that low concentrations of non-volatile substrates degrade primarily in the bulk liquid or at the plasma interface. If the substrate is volatile, it can penetrate the plasma and get degraded inside the plasma channel by pyrolysis and other radical-based mechanisms. This physical picture of the location of degradation of molecules is identical to the one in sonochemistry where it has been experimentally proven that inside cavitation bubbles molecules are pyrolyzed in addition to being degraded by radicals [136]. Since pyrolytic reactions result in the formation of low molecular weight hydrocarbons, CO and CO2, chromatographic analysis of gas-phase decomposition byproducts of volatile compounds formed during the discharge might yield insight into different mechanisms of degradation. Also, emission spectroscopy of aqueous solutions containing organic compounds could assist in understanding the diffusion of volatile (and non-volatile) molecules into the plasma.
Conclusions
Electric discharges in liquids, and the resulting streamers in particular, are being intensively investigated due to the applications ranging from the decomposition of volatile organic compounds and dyes in waste water, to the development of water purification systems. The emphasis of this review was on the chemical aspects of electrical discharges formed directly in water. Though these discharges can be created using various electrode geometries, the majority of the work is still conducted in a point-to-plane electrode geometry.
Basic mechanisms for the dissociation of water are also presented. Though water splitting can take place via electronic and/or thermal pathways, there is no direct experimental evidence supporting either of the mechanisms. Some studies suggest that electrical discharges with longer pulse duration result in the formation of non-thermal whereas those with longer pulses more thermal-like plasmas. Nevertheless, both mechanisms result in the formation of \( {\text{OH}}^{ \cdot } \), \( {\text{H}}^{ \cdot } \) and O. Due to their relatively short lifetimes, these species are unlikely to diffuse into the bulk liquid; as they move toward the plasma-liquid boundary they recombine to form other radicals and long-lived species. Chemical probe and analytical measurements have identified the presence of \( {\text{HO}}_{2}^{ \cdot } \), \( {\text{O}}_{2}^{ \cdot - } \), H2, O2, and H2O2. Though only for negative polarity discharges, qualitative measurements have also confirmed the presence of aqueous electrons. The production of primary (radicals) and secondary (stable molecules) species in plasma strongly depends on the input power, solution pH and conductivity and electrode polarity.
Electrical discharge in water results in the production of shockwaves, cavitation and the emission of UV light. Experimental evidence suggests that shockwaves contribute to the production of \( {\text{OH}}^{ \cdot } \) and H2O2 in the bulk liquid. On the other hand, the intensity of UV light emitted by the plasma increases with solution conductivity and results in the production of additional \( {\text{OH}}^{ \cdot } \) via direct photolysis of H2O2.
Electrical discharges directly in water without any gases bubbled through the plasma and in absence of additives have been studied for the degradation of a variety of compounds including aromatics, biphenyls, triazines, etc. Regardless of the type of the compound investigated, research indicates that the main species involved in the degradation of these molecules are \( {\text{OH}}^{ \cdot } \). One of the most common methods for proving the occurrence of the oxidative degradation is by measuring the extent of the degradation in the presence of an \( {\text{OH}}^{ \cdot } \) scavengers (e.g. alcohols). Though some studies suggest the involvement of reductive species and UV light in the degradation of organic molecules, there is no experimental evidence to support these hypotheses. The location of the degradation of organic compounds strongly depends on the compound concentration and volatility. Highly volatile compounds diffuse into the plasma channel where they are able to react with higher concentrations of (\( {\text{OH}}^{ \cdot } \)) radicals and get degraded faster. Less volatile compounds get degraded at the plasma-liquid boundary. However, if the compound is primarily degraded by reacting with \( {\text{OH}}^{ \cdot } \), it competes with the recombination of \( {\text{OH}}^{ \cdot } \) into H2O2 which also takes place at the boundary. As a rule, all the compounds get degraded faster at higher input powers, higher pulse repetition rates and in the presence of ferrous ions (Fenton’s reaction). Effects of other parameters on the removal rate strongly depend on the type of the molecule getting degraded.
One of the obstacles in achieving higher degradation efficiencies lies in the relatively small plasma volume compared to the volume of the treated solution and the loss of \( {\text{OH}}^{ \cdot } \) by recombination into H2O2 which oxidize molecules relatively slowly. The problem has been partially solved by using alternate electrode arrangements, bubbling oxygen through the plasma zone as well as by adding metal ions to decompose H2O2 into \( {\text{OH}}^{ \cdot } \). Until the ultimate reactor design and plasma optimization are achieved, coupling electrical discharge with another advanced oxidation technology remains the only option for degrading parent molecules into less toxic byproducts.
References
Locke BR, Sato M, Sunka P, Hoffmann MR, Chang JS (2006) Electrohydraulic discharge and nonthermal plasma for water treatment. Ind Eng Chem Res 45(3):882–905
Akiyama H (2000) Streamer discharges in liquids and their applications. IEEE Trans Dielectr Electr Insul 7(5):646–653
Malik MA, Ghaffar A, Malik SA (2001) Water purification by electrical discharges. Plasma Sources Sci Technol 10(1):82–91
Sunka P (2001) Pulse electrical discharges in water and their applications. Phys Plasmas 8(5):2587–2594
Bruggeman P, Leys C (2009) Non-thermal plasmas in and in contact with liquids. J Phys D Appl Phys 42(5):053001–053012
Brisset J-L, Moussa D, Doubla A, Hnatiuc E, Hnatiuc B, Kamgang Youbi G, Herry J-M, Naitali M, Bellon-Fontaine M-N (2008) Chemical reactivity of discharges and temporal post-discharges in plasma treatment of aqueous media: examples of gliding discharge treated solutions. Ind Eng Chem Res 47(16):5761–5781
Chang J-S (2001) Recent development of plasma pollution control technology: a critical review. Sci Technol Adv Mater 2(3–4):571–581
Schoenbach KH, Joshi RP, Stark RH, Dobbs FC, Beebe SJ (2000) Bacterial decontamination of liquids with pulsed electric fields. IEEE Trans Dielectr Electr Insul 7(5):637–645
Abou-Ghazala A, Katsuki S, Schoenbach KH, Dobbs FC, Moreira KR (2002) Bacterial decontamination of water by means of pulsed-corona discharges. IEEE Trans Plasma Sci 30(4):1449–1453
Bystritskii VM, Wood TK, Yankelevich Y, Chauhan S, Yee D, Wessel F (1997) Pulsed power for advanced waste water remediation. In: Pulsed power conference, 1997 11th IEEE international, 29 June–2 July 1997, pp 79–84
Lisitsyn IV, Nomiyama H, Katsuki S, Akiyama H (1999) Streamer discharge reactor for water treatment by pulsed power. Rev Sci Instrum 70(8):3457–3462
Gerrity D, Stanford BD, Trenholm RA, Snyder SA (2010) An evaluation of a pilot-scale nonthermal plasma advanced oxidation process for trace organic compound degradation. Water Res 44(2):493–504
Faraday M (1832) Experimental researches in electricity. Printed by R. Taylor, London
Rodebush WH, Wahl MW (1933) The reactions of the hydroxyl radical in the electrodeless discharge in water vapor. J Chem Phys 1:696–702
Davies RA, Hickling A (1952) 686. Glow-discharge electrolysis. Part I. The anodic formation of hydrogen peroxide in inert electrolytes. J Chem Soc 3595–3602. doi:10.1039/JR9520003595
Sato M, Ohgiyama T, Clements JS (1996) Formation of chemical species and their effects on microorganisms using a pulsed high-voltage discharge in water. IEEE Trans Ind Appl 32(1):106–112
Joshi AA, Locke BR, Arce P, Finney WC (1995) Formation of hydroxyl radicals, hydrogen peroxide and aqueous electrons by pulsed streamer corona discharge in aqueous solution. J Hazard Mater 41(1):3–30
Sunka P, Babicky V, Clupek M, Lukes P, Simek M, Schmidt J, Cernak M (1999) Generation of chemically active species by electrical discharges in water. Plasma Sour Sci Technol 8(2):258–265
Kirkpatrick MJ, Locke BR (2005) Hydrogen, oxygen, and hydrogen peroxide formation in aqueous phase pulsed corona electrical discharge. Ind Eng Chem Res 44(12):4243–4248
Tezuka M (1993) Anodic hydrogen evolution in contact glow-discharge electrolysis of sulfuric acid. Denki Kagaku 61:794–795
Sun B, Sato M, Harano A, Clements JS (1998) Non-uniform pulse discharge-induced radical production in distilled water. J Electrostat 43(2):115–126
Sun B, Sato M, Sid Clements J (1997) Optical study of active species produced by a pulsed streamer corona discharge in water. J Electrostat 39(3):189–202
Hickling A (1971) Electrochemical processes in glow discharge at the gas-solution interface. In: Bockris JOM, Conway BE (eds) Modern aspects of electrochemistry. Plenium Press, New York
Hubbard CD, Rv Eldik (1997) Chemistry under extreme or non-classical conditions. John Wiley and Spektrum, New York
Willberg DM, Lang PS, Hochemer RH, Kratel AW, Hoffman MR (1996) Electrohydraulic destruction of hazardous wastes. ChemTech 26(4):52–57
Clements JS, Sato M, Davis RH (1987) Preliminary Investigation of Prebreakdown Phenomena and Chemical Reactions Using a Pulsed High-Voltage Discharge in Water. IEEE Trans Ind Appl IA-23(2):224–235
Sahni M, Locke BR (2006) Quantification of hydroxyl radicals produced in aqueous phase pulsed electrical discharge reactors. Ind Eng Chem Res 45(17):5819–5825
Lukes P (2001) Water treatment by pulsed streamer corona discharge. PhD dissertation, Institute of Chemical Technology, Prague, Czech Republic
Anpilov AM, Barkhudarov EM, Bark YB, Zadiraka YV, Christofi M, Kozlov YN, Kossyi IA, Kop’ev VA, Silakov VP, Taktakishvili MI, Temchin SM (2001) Electric discharge in water as a source of UV radiation, ozone and hydrogen peroxide. J Phys D Appl Phys 34(6):993–999
Cheng H-H, Chen S-S, Wu Y-C, Ho D-L (2007) Non-thermal plasma technology for degradation of organic compounds in wastewater. J Environ Eng Manage 17(6):427–433
Mededovic S, Locke BR (2006) Platinum catalysed decomposition of hydrogen peroxide in aqueous-phase pulsed corona electrical discharge. Appl Catal B-Environ 67(3–4):149–159
Sahni M, Finney WC, Locke BR (2005) Degradation of aqueous phase polychlorinated biphenyls (PCB) using pulsed corona discharges. J Adv Oxid Technol 8(1):105–111
Brisset JL (1998) Removal of pentachlorophenol from water by AC corona discharge treatment in air. J Trace Microprobe Tech 16(3):363–370
Even-Ezraa I, Mizrahia A, Gerrityb D, Snyderb S, Salvesonc A, Lahavd O (2009) Application of a novel plasma-based advanced oxidation process for efficient and cost-effective destruction of refractory organics in tertiary effluents and contaminated groundwater. Desalin Water Treat 11:236–244
Manolache S, Shamamian V, Denes F (2004) Dense medium plasma-enhanced decontamination of water of aromatic compounds. J Environ Eng 130(1):17–25
Koh IO, Wohlers J, Thiemann W, Rotard W (2009) Application of an air ionization device using an atmospheric pressure corona discharge process for water purification. Water Air Soil Poll 196(1–4):101–113
Mededovic S, Finney WC, Locke BR (2007) Aqueous-phase mineralization of s-triazine using pulsed electrical discharge. IJPEST 1(1):82–90
Mededovic S, Locke BR (2007) Side-chain degradation of atrazine by pulsed electrical discharge in water. Ind Eng Chem Res 46(9):2702–2709
Sugiarto AT, Ito S, Ohshima T, Sato M, Skalny JD (2003) Oxidative decoloration of dyes by pulsed discharge plasma in water. J Electrostat 58(1–2):135–145
Mededovic S, Takashima K (2008) Decolorization of indigo carmine dye by spark discharge in water. IJPEST 2(1):56–66
Yano T, Shimomura N, Uchiyama I, Fukawa F, Teranishi K, Akiyama H (2009) Decolorization of indigo carmine solution using nanosecond pulsed power. IEEE Trans Dielectr Electr Insul 16(4):1081–1087
Grymonpre DR, Finney WC, Locke BR (1999) Aqueous-phase pulsed streamer corona reactor using suspended activated carbon particles for phenol oxidation: model-data comparison. Chem Eng Sci 54(15–16):3095–3105
Lukes P, Clupek M, Sunka P, Peterka F, Sano T, Negishi N, Matsuzawa S, Takeuchi K (2005) Degradation of phenol by underwater pulsed corona discharge in combination with TiO2 photocatalysis. Res Chem Intermediat 31(4–6):285–294
Yang XL, Bai MD, Han F (2009) Treatment of phenol wastewater using hydroxyl radical produced by micro-gap discharge plasma technique. Water Environ Res 81(4):450–455
Sharma AK, Locke BR, Arce P, Finney WC (1993) A preliminary study of pulsed streamer corona discharge for the degradation of phenol in aqueous solutions. Hazard Waste Hazard 10(2):209–219
Sun B, Sato M, Clements JS (1999) Oxidative processes occurring when pulsed high voltage discharges degrade phenol in aqueous solution. Environ Sci Technol 34(3):509–513
Hoeben WFLM, van Velduizen EM, Rutgers WR, Kroesen GMW (1999) Gas phase corona discharges for oxidation of phenol in an aqueous solution. J Phys D Appl Phys 32(24):L133–L137
Hoeben WFLM (2000) Pulsed corona-induced degradation of organic materials in water. PhD dissertation, Eindhoven, Netherlands
Tezuka M, Iwasaki M (1998) Plasma induced degradation of chlorophenols in an aqueous solution. Thin Solid Films 316(1–2):123–127
Hao XL, Zhou MH, Lei LC (2007) Non-thermal plasma-induced photocatalytic degradation of 4-chlorophenol in water. J Hazard Mater 141(3):475–482
Sahni M, Locke BR (2006) Degradation of chemical warfare agent simulants using gas–liquid pulsed streamer discharges. J Hazard Mater 137(2):1025–1034
Yoon J, Lee C, Lee Y (2006) Oxidative degradation of dimethylsulfoxide by locally concentrated hydroxyl radicals in streamer corona discharge process. Chemosphere 65(7):1163–1170
Wen Y-Z, Jiang X-Z (2001) Pulsed corona discharge-induced reactions of acetophenone in water. Plasma Chem Plasma Process 21(3):345–354
Yee DC, Chauhan S, Yankelevich E, Bystritskii V, Wood TK (1998) Degradation of perchloroethylene and dichlorophenol by pulsed-electric discharge and bioremediation. Biotechnol Bioeng 59(4):438–444
Krause H, Schweiger B, Prinz E, Kim J, Steinfeld U (2011) Degradation of persistent pharmaceuticals in aqueous solutions by a positive dielectric barrier discharge treatment. J Electrostat 69(4):333–338
Magureanu M, Piroi D, Mandache NB, David V, Medvedovici A, Bradu C, Parvulescu VI (2011) Degradation of antibiotics in water by non-thermal plasma treatment. Water Res 45(11):3407–3416
Fridman AA (2008) Plasma chemistry. Cambridge University Press, Cambridge
Bruggeman P, Schram DC (2010) On OH production in water containing atmospheric pressure plasmas. Plasma Sources Sci Technol 19(4):045025–045034
Givotov VK, Fridman AA, Krotov MF, Krasheninnikov EG, Patrushev BI, Rusanov VD, Sholin GV (1981) Plasmochemical methods of hydrogen production. Int J Hydrogen Energy 6(5):441–449
Gupta SB, Bluhm H (2008) The potential of pulsed underwater streamer discharges as a disinfection technique. IEEE Trans Plasma Sci 36(4):1621–1632
Itikawa Y, Mason N (2005) Cross sections for electron collisions with water molecules. J Phys Chem Ref Data 34(1):1–22
Yousfi M, Benabdessadok MD (1996) Boltzmann equation analysis of electron-molecule collision cross sections in water vapor and ammonia. J Appl Phys 80(12):6619–6630
Zheng WJ, Jewitt D, Kaiser RI (2006) Formation of hydrogen, oxygen, and hydrogen peroxide in electron-irradiated crystalline water ice. Astrophys J 639(1):534–548
Slanger TG, Black G (1982) Photodissociative channels at 1216 A for H2O, NH3, and CH4. J Chem Phys 77(5):2432–2437
Black G, Porter G (1962) Vacuum ultra-violet flash photolysis of water vapour. Proc R Soc Lond A 266(1325):185–197
Nishimura T, Itikawa Y (1995) Electron-impact vibrational excitation of water molecules. J Phys B-At Mol Opt 28(10):1995–2003
Black G, Porter G (1962) Vacuum ultra-violet flash photolysis of water vapour. Proc R Soc Lond A 266(1325):185–197
Dolan TJ (1993) Electron and ion collisions with water vapour. J Phys D Appl Phys 26(1):4–8
Mozumder A (1999) Fundamentals of radiation chemistry. Academic Press, San Diego
Namihira T, Sakai S, Yamaguchi T, Yamamoto K, Yamada C, Kiyan T, Sakugawa T, Katsuki S, Akiyama H (2007) Electron temperature and electron density of underwater pulsed discharge plasma produced by solid-state pulsed-power generator. IEEE Trans Plasma Sci 35(3):614–618
Staack D, Fridman A, Gutsol A, Gogotsi Y, Friedman G (2008) Nanoscale corona discharge in liquids, enabling nanosecond optical emission spectroscopy. Angew Chem Int Ed 47(42):8020–8024
Mason T, Lorimer J (2002) Applied sonochemistry: the uses of power ultrasound in chemistry and processing. Wiley-VCH, Darmstadt
Kai-Yuan S, Locke BR (2011) Optical and electrical diagnostics of the effects of conductivity on liquid phase electrical discharge. IEEE Trans Plasma Sci 39(3):883–892
Thagard SM, Takashima K, Mizuno A (2009) Chemistry of the positive and negative electrical discharges formed in liquid water and above a gas-liquid surface. Plasma Chem Plasma Process 29(6):455–473
Mededovic S, Locke BR (2007) Primary chemical reactions in pulsed electrical discharge channels in water. J Phys D Appl Phys 40(24):7734–7746
Gupta SB (2007) Investigation of a physical disinfection process based on pulsed underwater corona discharges. PhD dissertation, Karlshrue, Germany
Locke BR, Shih K-Y (2011) Review of the methods to form hydrogen peroxide in electrical discharge plasma with liquid water. Plasma Sour Sci Technol 20(3):034006–034021
Sahni M, Locke BR (2006) Quantification of reductive species produced by high voltage electrical discharges in water. Plasma Process Polym 3(4–5):342–354
Yasui K, Tuziuti T, Iida Y, Mitome H (2003) Theoretical study of the ambient-pressure dependence of sonochemical reactions. J Chem Phys 119(1):346–356
Sun B, Sato M, Clements JS (1999) Use of a pulsed high-voltage discharge for removal of organic compounds in aqueous solution. J Phys D Appl Phys 32(15):1908–1915
Hart EJ, Boag JW (1962) Absorption spectrum of the hydrated electron in water and in aqueous solutions. J Am Chem Soc 84(21):4090–4095
Roots R, Okada S (1975) Estimation of life times and diffusion distances of radicals involved in X-ray-induced DNA strand breaks or killing of mammalian cells. Radiat Res 64(2):306–320
Klimkin VF, Ponomarenko AG (1979) Interferometric study of pulsed breakdown in a liquid. Sov Phys-Tech Phys 24(1):1067–1071
Adewuyi YG (2001) Sonochemistry: environmental science and engineering applications. Ind Eng Chem Res 40(22):4681–4715
NIST Chemical Kinetics Database. Standard Reference Database 17, Version 7.0 (Web Version), Release 1.6.5 Data Version 2012.02. http://kinetics.nist.gov/kinetics/index.jsp
Liu DX, Bruggeman P, Iza F, Rong MZ, Kong MG (2010) Global model of low-temperature atmospheric-pressure He + H2O plasmas. Plasma Sour Sci Technol 19(2):025018–025040
Eisenberg G (1943) Colorimetric determination of hydrogen peroxide. Ind Eng Chem Anal Ed 15(5):327–328
Grymonpré DR, Sharma AK, Finney WC, Locke BR (2001) The role of Fenton’s reaction in aqueous phase pulsed streamer corona reactors. Chem Eng J 82(1–3):189–207
Grymonpre DR, Finney WC, Clark RJ, Locke BR (2004) Hybrid gas-liquid electrical discharge reactors for organic compound degradation. Ind Eng Chem Res 43(9):1975–1989
Kirkpatrick M (2004) Plasma-catalyst interactions in treatment of gas phase contaminants and electrical discharge in water. PhD dissertation, Tallahassee, Florida
Sunka P, Benes J, Lukes P, Zadinova M, Hoffer P, Pouckova P (2009) Biological effects of tandem shock waves on soft animal tissues- preliminary “in vivo” experiments. In: ICOPS 2009 IEEE international conference on plasma science, 1–5 June 2009, pp 1–1
Sunka P, Babicky V, Clupek M, Benes J, Pouckova P (2004) Localized damage of tissues induced by focused shock waves. IEEE Trans Plasma Sci 32(4):1609–1613
Robinson JW, Ham M, Balaster AN (1973) Ultraviolet radiation from electrical discharges in water. J Appl Phys 44(1):72–75
Bolton JR (1999) Ultraviolet applications handbook. Bolton Photosciences Inc., Ayr
Lukes P, Clupek M, Babicky V, Sunka P (2008) Ultraviolet radiation from the pulsed corona discharge in water. Plasma Sour Sci Technol 17(2):024012–024023
Sun B, Zhang L, Sato M (2008) Characteristics of atomic oxygen produced by a pulsed streamer corona discharge. Int J Environ Waste Manage 2(11):447–457
Gupta SB, Bluhm H (2007) Pulsed underwater corona discharges as a source of strong oxidants: OH and H2O2. Water Sci Technol 55(12):7–12
De Baerdemaeker F, Simek M, Leys C (2007) Efficiency of hydrogen peroxide production by ac capillary discharge in water solution. J Phys D Appl Phys 40(9):2801–2809
Yang SD, Zhang LH, Cui FG, Ma J (2009) Production of hydrogen peroxide by pulsed high voltage discharge in water. In: 2009 3rd international conference on bioinformatics and biomedical engineering, vol 1–11, pp 5346–5349
Shih KY, Locke BR (2011) Optical and electrical diagnostics of the effects of conductivity on liquid phase electrical discharge. IEEE Trans Plasma Sci 39(3):883–892
Goryachev VL, Rutberg FG, Ufimtsev AA (1998) Photolytic properties of a pulsed discharge in water. Tech Phys Lett +24(2):122–123
Tarr MA (2003) Chemical degradation methods for wastes and pollutants: environmental and industrial applications. M. Dekker, New York
Notre Dame Radiation Database. http://kinetics.nist.gov/solution/
Dorfman LM, Adams GE (1973) Reactivity of the hydroxyl radical in aqueous solutions. National Standard Reference Data Series 46 (NSRDS-NBS46). U. S. Department of Commerce
Bielski BHJ, Cabelli DE, Arudi RL, Ross AB (1985) Reactivity of HO2/O2- radicals in aqueous solution. J Phys Chem Ref Data 14(4):1041–1100
Neta P (1972) Reactions of hydrogen atoms in aqueous solutions. Chem Rev 72(5):533–543
Anbar M, Hart EJ (1964) The reactivity of aromatic compounds toward hydrated electrons. J Am Chem Soc 86(24):5633–5637
Rice FO, Herzfeld KF (1934) The thermal decomposition of organic compounds from the standpoint of free radicals. VI. The mechanism of some chain reactions. J Am Chem Soc 56(2):284–289
Rice FO (1931) The thermal decomposition of organic compunds from the standpoint of free radicals. I. Saturated hydrocarbons. J Am Chem Soc 53(5):1959–1972
Mededovic Thagard S, Prieto G, Takashima K, Mizuno A (2012) Identification of gas phase byproducts formed during electrical discharges in liquid fuels”. IEEE Trans Plasma Sci 40:2016–2111
Jakob L, Hashem TM, Bürki S, Guindy NM, Braun AM (1993) Vacuum-ultraviolet (VUV) photolysis of water: oxidative degradation of 4-chlorophenol. J Photoch Photobio A 75(2):97–103
Tri Sugiarto A, Ohshima T, Sato M (2002) Advanced oxidation processes using pulsed streamer corona discharge in water. Thin Solid Films 407(1–2):174–178
Vujevic D, Koprivanac N, Bozic AL, Locke BR (2004) The removal of direct orange 39 by pulsed corona discharge from model wastewater. Environ Technol 25(7):791–800
Bozic AL, Koprivanac N, Sunka P, Clupek M, Babicky V (2004) Organic synthetic dye degradation by modified pinhole discharge. Czech J Phys 54:C958–C963
Maehara T, Miyamoto I, Kurokawa K, Hashimoto Y, Iwamae A, Kuramoto M, Yamashita H, Mukasa S, Toyota H, Nomura S, Kawashima A (2008) Degradation of methylene blue by RF plasma in water. Plasma Chem Plasma Process 28(4):467–482
Stara Z, Krcma F, Nejezchleb M, Skalny JD (2008) Influence of solution composition and chemical structure of dye on removal of organic dye by DC diaphragm discharge in water solutions. J Adv Oxid Technol 11(1):155–162
Stara Z, Krcma F, Nejezchleb M, Skalny JD (2009) Organic dye decomposition by DC diaphragm discharge in water: effect of solution properties on dye removal. Desalination 239(1–3):283–294
Xiaoqiong W, Ming W, Zhenfeng D, Guishi L (2012) Decoloration of azo dye sunset yellow by a coaxial insulated-rod-to-cylinder underwater streamer discharge system. Plasma Sci Technol 14(4):293
Kunitomo S, Bing S (2001) Removal of phenol in water by pulsed high voltage discharge. PPPS-2001: pulsed power plasma science 2001, vols I and Ii, Digest of Technical Papers 1138–1141
Kunitomo S, Ohbo T, Sun B (2003) The effects of using various types of pulsed discharge reactors for phenol removal in waste water. J Adv Oxid Technol 6(1):70–74
Lukes P, Clupek M, Sunka P, Babicky V, Janda V (2002) Effect of ceramic composition on pulse discharge induced processes in water using ceramic-coated wire to cylinder electrode system. Czech J Phys 52:800–806
Lukes P, Clupek M, Babicky V, Sunka P, Winterova G, Janda V (2003) Non-thermal plasma induced decomposition of 2-chlorophenol in water. Acta Phys Slovaca 53(6):423–428
Dang TH, Denat A, Lesaint O, Teissedre G (2008) Degradation of organic molecules by streamer discharges in water: coupled electrical and chemical measurements. Plasma Sour Sci Technol 17(2):024013–024021
Sugiarto AT, Sato M, Ohshima T, Skalny JD (2002) Characteristics of ring-to-cylinder type electrode system on pulsed discharge in water. J Adv Oxid Technol 5(2):211–216
Mizeraczyk J, Dors M, Metel E (2006) Phenol degradation in water by pulsed streamer discharge and Fenton reaction. Prog Green Oxid /Reduct Technol 162–166
Lukes P, Locke BR (2005) Plasmachemical oxidation processes in a hybrid gas-liquid electrical discharge reactor. J Phys D Appl Phys 38(22):4074–4081
Esplugas S, Giménez J, Contreras S, Pascual E, Rodríguez M (2002) Comparison of different advanced oxidation processes for phenol degradation. Water Res 36(4):1034–1042
Tothova I, Lukes P, Clupek M, Babicky V, Janda V (2009) Removal of nonylphenol by pulsed corona discharge in water. In: 19th international symposium on plasma chemistry, Bochum, July 26–31, 2009
Bian WJ, Song XH, Liu DQ, Zhang J, Chen XH (2011) The intermediate products in the degradation of 4-chlorophenol by pulsed high voltage discharge in water. J Hazard Mater 192(3):1330–1339
Wen YZ, Jiang XZ (2001) Pulsed corona discharge-induced reactions of acetophenone in water. Plasma Chem Plasma Process 21(3):345–354
Sahni M (2002) Degradation of trichloroethylene using a pulsed corona reactor: experiments and simulation. Florida State University, Tallahassee
Lee C, Lee Y, Yoon J (2006) Oxidative degradation of dimethylsulfoxide by locally concentrated hydroxyl radicals in streamer corona discharge process. Chemosphere 65(7):1163–1170
Willberg DM, Lang PS, Hochemer RH, Kratel A, Hoffmann MR (1996) Degradation of 4-chlorophenol, 3,4-dichloroaniline, and 2,4,6-trinitrotoluene in an electrohydraulic discharge reactor. Environ Sci Technol 30(8):2526–2534
Elovitz MS, von Gunten U, Kaiser HP (2000) Hydroxyl radical/ozone ratios during ozonation processes. II. The effect of temperature, pH, alkalinity, and DOM properties. Ozone Sci Eng 22(2):123–150
Lee Y, Lee C, Yoon J (2003) High temperature dependence of 2,4-dichlorophenoxyacetic acid degradation by Fe(3+)/H2O2 system. Chemosphere 51(9):963–971
Suslick KS, Didenko Y, Fang MM, Hyeon T, Kolbeck KJ, McNamara WB, Mdleleni MM, Wong M (1999) Acoustic cavitation and its chemical consequences. Philos Trans R Soc Lond Ser A-Math Phys Eng Sci 357(1751):335–353
Koprivanac N, Kusic H, Vujevic D, Peternel I, Locke BR (2005) Influence of iron on degradation of organic dyes in corona. J Hazard Mater 117(2–3):113–119
Grymonpré DR, Sharma AK, Finney WC, Locke BR (2001) The role of Fenton’s reaction in aqueous phase pulsed streamer corona reactors. Chem Eng J 82(1–3):189–207
Sun B, Sato M, Clements JS (2000) Oxidative processes occurring when pulsed high voltage discharges degrade phenol in aqueous solution. Environ Sci Technol 34(3):509–513
Sugiarto AT, Sato M (2001) Pulsed plasma processing of organic compounds in aqueous solution. Thin Solid Films 386(2):295–299
Acknowledgments
One of the authors (S.M.T.) would like to acknowledge the support by the National Science Foundation (CBET: BRIGE 1125592).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Joshi, R.P., Thagard, S.M. Streamer-Like Electrical Discharges in Water: Part II. Environmental Applications. Plasma Chem Plasma Process 33, 17–49 (2013). https://doi.org/10.1007/s11090-013-9436-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11090-013-9436-x