
Abstract
The design of new austenitic alloys based on the Fe–Ni–Cr system requires knowledge of their oxygen permeability. Data are available for pure Fe and Ni but not for Fe–Ni alloys. Wagner’s model for internal oxidation is used to evaluate the oxygen permeability in Fe–Ni alloys. Internal oxidation of Fe–Ni–Cr alloys carried out at 1150 and 1100 °C in Rhines packs and H2/H2O mixtures is described. Internal oxidation produces zones of FeCr2O4 and Cr2O3 precipitates, according to parabolic kinetics. Permeabilities are deduced taking into account interfacial diffusion contributions. Oxygen permeability decreases with nickel addition in a non-ideal way, and oxygen permeability in nickel-rich alloys is independent of the studied environments. However, the oxygen permeability in iron at the highest temperature studied is increased in H2/H2O.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Stainless steels and superalloys are based on the Fe–Ni–Cr system, and rely on formation of a chromia scale to achieve good temperature corrosion resistance. However, if the alloy chromium content is too low, chromium oxides are formed beneath the alloy surface and the material is internally oxidized. Wagner [1] derived Eq. (1) to calculate the minimum chromium content required to prevent internal oxidation, forming an external scale instead:
Here ν is internal oxide stoichiometric coefficient, V all and V Ox the molar volumes of alloy and oxide CrO ν , \(\tilde{D}_{\text{Cr}}\) the chromium interdiffusion coefficient, \(N_{\text{O}}^{{({\text{s}})}} D_{\text{O}}\) the alloy oxygen permeability and g the critical volume fraction of oxide required for transition from internal to external oxidation. If g reaches the critical value, internal oxides block the inward flux of oxygen and promote the growth of existing oxides instead of the nucleation of new particles and stop the progression of the oxidation front. A value g = 0.3 was found by Rapp [2] for Ag–In alloys, and this value is commonly used for other materials. However, recent results showed that the value of g could be affected by the presence of steam in the environment [3].
Clearly, the oxygen permeability value is required in designing new alloys, which may be needed as power generation technologies are improved. Such values have been measured for pure Fe and Ni by internal oxidation of binary alloys Fe–Al [4], Fe–Si [5], Fe–Cr [6, 7], Ni–Cr [8, 9], Ni–Al [9, 10], etc. Unfortunately, no data are available for Fe–Ni alloys, which form the basis of many commercial heat resisting alloys. A further complication arises from the presence of secondary oxidants, e.g. water vapour in the service environment. It has been suggested that water vapour accelerates internal alloy oxidation at high temperature [7, 11, 12], but the effect of water vapour on alloy oxygen permeability is unknown.
The purpose of this work was to determine the variation of oxygen permeability with composition of binary Fe–Ni alloys, and examine the effect thereon of the presence of water vapour.
Oxygen Permeability Measured by Internal Oxidation
According to Wagner [1], the internal oxidation kinetics are controlled by diffusion, and the depth of the internal oxidation zone (IOZ) is given by
where \(X_{{({\text{i}})}}\) is the IOZ depth, D O the oxygen diffusion coefficient, \(\gamma\) a kinetic parameter and \(k_{\text{p}}^{{({\text{i}})}}\) the internal oxidation parabolic constant. The ratio of oxygen and solute concentrations determines the relative diffusion contributions of these species:
with \(N_{\text{O}}^{{({\text{s}})}}\) the oxygen mole fraction at the sample surface and \(N_{\text{Cr}}^{(0)}\) is the original alloy chromium mole fraction. The secondary functions G and F are defined as
with \(hc = \gamma \varphi^{1/2}\) and \(\varphi = D_{\text{O}} /\tilde{D}_{\text{Cr}}\).
In most cases of internal oxidation without an external scale, \(\gamma \ll 1\) and Eq. (3) becomes
In addition, if during internal oxidation, \(N_{\text{Cr}}^{(0)} \tilde{D}_{\text{Cr}} \ge N_{\text{O}}^{(s)} D_{\text{O}}\), chromium enrichment occurs in the IOZ as a result of diffusion from the alloy interior. Wagner defined the enrichment factor, \(\alpha\), and evaluated it from his diffusion analysis
where \(N_{\text{Cr}}^{\text{Ox}}\) is the mole fraction of chromium as oxide in the IOZ. Finally, the internal oxidation parabolic constant is given by
Experimental Procedures
The Fe–Cr and Fe–Ni–Cr alloys used in this study were cast from high purity iron (99.98%), nickel (99.995%) and chromium (99.99%) in an arc melter using a non-consumable electrode and under flowing Ar–5%H2. Pure iron and 4 ratios N Fe/(N Fe + N Ni) were selected as base alloy compositions, and 3 chromium levels used, 2, 4 and 7.5 at.%. For high nickel alloys, 1 at.% of chromium was used because alloys containing 7.5 at.% of chromium formed an external chromium scale. After casting, alloys were annealed for 72 h under flowing hydrogen at 1100 °C to ensure homogeneity. Alloy compositions were checked by Energy Dispersive Spectroscopy (EDS) and were in agreement with nominal compositions presented in Table 1. Samples were then cut in slices 1.3 mm thick, polished to a 4000 grit finish using SiC paper grade, cleaned with ethanol in an ultrasonic bath and dried before exposure.
Internal oxidation experiments were all performed at the Fe/FeO dissociation pressure corresponding to an oxygen partial pressure of 2.4 × 10−13 and 4.7 × 10−14 atm at 1150 and 1100 °C, respectively. Anhydrous conditions were achieved using the Rhines Pack technique (RP) [13]. The samples were oxidized in vacuum-sealed quartz capsules, which contained a mixture of powdered iron and wüstite (molar ratio 2:3) to control the oxygen potential. The amount of powder provided oxygen in excess of the quantity of chromium being oxidized. To avoid interaction between the powder and the sample, the two were separated by a constriction in the capsule.
The Fe/FeO powder mixture was obtained by oxidizing an iron powder in Ar–18%H2O–4%H2 at 950 °C for 5 h. The co-existence of Fe and FeO was checked by X-Ray Diffraction (XRD) analysis after synthesis and the powder composition was determined by mass change. The powder was sieved to a particle size range of 100–850 μm. After reaction, powder mixtures were analysed by XRD to confirm that wüstite was still present.
Flowing H2/H2O gas mixtures were used to control the oxygen partial pressure at values equal to the Fe/FeO dissociation pressure. Gas mixtures were generated by passing mixtures of Ar + H2 through a thermostated water saturator set at a temperature to produce an excess of water vapour. The excess was then removed by passing the gas through a condenser set at a temperature corresponding to the desired water content. Gas compositions were Ar–14%H2O–19%H2 for reaction at 1150 °C, and Ar–14%H2O–20%H2 for reaction at 1100 °C.
After exposure, sample surfaces were analysed by grazing angle XRD. Reaction morphology was examined using metallography, and IOZ depths measured using image analysis software ImageJ. In addition, precipitates were analysed by EDS in a scanning electron microscope.
Results and Discussion
Internal Oxidation Kinetics and Internal Oxide Morphology
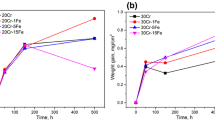
Kinetics measured at 1150 and 1100 °C in RP [14] and H2/H2O environments were all parabolic. Internal oxidation constants were determined using linear fits of data in X 2(i) = f(t) plots. The internal oxidation rates increase with N Fe/(N Fe + N Ni) (Fig. 1). In Fig. 2, internal oxidation constants measured in wet condition are plotted as a function of internal oxidation constants evaluated in dry condition.

\(k_{\text{p}}^{{({\text{i}})}}\) in wet (filled symbols) and dry (opened symbols) environments at 1150 °C (a) and 1100 °C (b)

\(k_{\text{p}}^{{({\text{i}})}}\) in dry and wet environments at 1150 °C (a) and 1100 °C (b)
The \(k_{\text{p}}^{{({\text{i}})}}\) values are similar in dry and wet atmosphere for all compositions at the two temperatures.
The IOZ was composed of discrete oxide particles, and no preferential oxidation of grain boundaries was observed. Particles were spherical and randomly distributed in the IOZ with no preferential growth orientation. In addition, the size of the internal oxides was increasing with increased chromium and nickel content of the alloy (Fig. 3). No significant difference was observed in the internal oxide morphology after reaction in dry and wet environments.

Internal oxide structure after 8 h of reaction at 1150 °C in H2/H2O mixture. (a) N Fe/(N Fe + N Ni) = 0.8 and N Cr = 0.02, (b) N Fe/(N Fe + N Ni) = 0.8 and N Cr = 0.075. (c) N Fe/(N Fe + N Ni) = 0.4 and N Cr = 0.02
Oxygen Permeability Calculation
Calculation of \(N_{\text{O}}^{{({\text{s}})}} D_{\text{O}}\) values from the dependence of \(k_{\text{p}}^{{({\text{i}})}}\) on \(N_{\text{Cr}}^{(0)}\) using Eq. (8) requires knowledge of the internal oxide stoichiometry and enrichment factor. The IOZ is made up of subzones containing FeCr2O4 and Cr2O3 (Fig. 4). For this reason, an effective stoichiometric coefficient was evaluated from the following equation [14]
where \(X_{{({\text{t}})}}\) is the depth of the boundary between the spinel and the chromium oxide subzones, and \(X_{{({\text{i}})}}\) the IOZ depth, as shown in Fig. 4. Effective stoichiometric coefficients are reported in Table 2 and the measurement error was estimated to be 5% of the measured value. As results show, the effective stoichiometric coefficient is sensitive to the ratio N Fe/(N Fe + N Ni); however, the temperature and the environment have no significant effect on the stoichiometry value.

Internal oxidation zone of alloy CC exposed 16 h in H2/H2O at 1150 °C (SEM–BSE)
The enrichment factor was calculated with Eq. (7). The variable \(hc\) was evaluated from the value of \(k_{\text{p}}^{{({\text{i}})}}\) and the interdiffusion coefficient of chromium published by Duh and Dayananda [15] (Table 3).
In order to check the validity of Wagner’s model, the enrichment factor was also measured from internal oxide volume fraction measurement using image analysis [14], and the results obtained were in agreement with the enrichment factors calculated by the diffusional approach. Proceeding on this basis, the enrichment factors calculated using Eq. (7) were used and are reported in Table 1. The errors in the enrichment calculation arise from error in the internal oxidation constant determination. In nickel-rich alloy, the IOZ depth is measured with larger uncertainty due to the smaller number of particles, as shown in Fig. 3. In addition, the error evaluated for one alloy was the same in all experimental conditions. The errors are reported in Table 1 as relative errors. Like the effective stoichiometric coefficient, the enrichment factor is determined mainly by the alloy composition
In Fig. 5, the product \(\alpha \nu_{\text{eff}} k_{\text{p}}^{{({\text{i}})}}\) is plotted versus the inverse of the chromium content. Values of \(\alpha\) and \(\nu_{\text{eff}}\) are taken from Tables 1 and 2, respectively. The product \(\alpha \nu_{\text{eff}} k_{\text{p}}^{{({\text{i}})}}\) varies linearly with the inverse of the chromium content of the alloy, as predicted by Eq. (8), but extrapolations do not pass through the origin. This disagreement with Wagner’s model was attributed to fast oxygen diffusion at the incoherent matrix/oxide interfaces as reported in previous studies [7, 9]. The contribution of fast oxygen diffusion at matrix/oxide interfaces to the oxygen permeability is proportional to the internal oxide volume fraction which is proportional to the chromium content of the alloy [1, 9]. To consider enhanced oxygen diffusion at interfaces, an effective oxygen permeability was defined by the following equation [7]
where \(N_{\text{O}}^{{({\text{s}})}} D_{\text{O}}^{\text{eff}}\) is the effective permeability, \(N_{\text{O}}^{{({\text{s}})}} D_{\text{O}}^{\text{matrix}}\) the oxygen permeability of the matrix and A a constant which characterizes oxygen diffusion at oxide/matrix interface. Combining Eqs. (8) and (11), the relation between the internal oxidation constant and oxygen permeability becomes

Internal oxidation constant as a function of inverse of the chromium concentration in wet (filled symbols) and dry (open symbols) atmosphere at 1150 °C (a) and 1100 °C (b)
In Fig. 6, the oxygen permeability of the matrix and the constant A are plotted with the alloy composition in dry and wet atmospheres at 1150 and 1100 °C. In addition, permeability values measured by Guo et al. [16] in nickel at the Ni/NiO equilibrium were extrapolated to the Fe/FeO dissociation pressure using Sievert’s law to adjust the oxygen solubility, and assuming no change in the value of \(D_{\text{O}}^{\text{matrix}}\) with the oxygen partial pressure. Values calculated are added to Fig. 6 for comparison.

Oxygen permeability of the matrix (a) and constant A (b) as function of the alloy composition in dry (open symbols) and wet (filled symbols) environment at 1150 and 1100 °C. Note log scale on left hand plot
In general, the oxygen permeability was found to increase with temperature and decrease strongly with nickel content as shown in the semilogarithmic plots of Fig. 6. The oxygen solubility in Fe–Ni alloys was measured by Jullian et al. [17] in RPs at temperatures between 1000 and 1150 °C, and the largest solubility variation measured was one order of magnitude between values in iron and nickel at 1150 °C. However, the oxygen permeability is seen here to vary by two orders of magnitude in all conditions, which indicates that alloy composition has a significant effect also on the oxygen diffusion coefficient.
The presence of water vapour appears to increase the matrix oxygen permeability in iron at 1150 °C, in agreement with Setiawan et al. [7]. However, the products \(\alpha \nu_{\text{eff}} k_{\text{p}}^{{({\text{i}})}}\) calculated for iron at 1,150C are equal for two alloys out of three and additional experiments would be desirable to confirm this result. In addition at this temperature, in nickel-rich alloys [N Fe/(N Fe + N Ni) < 0.5], the oxygen permeability measured under wet conditions is slightly smaller than that measured in hydrogen-free environment. The same trend was observed at 1100 °C for all compositions except for N Fe/(N Fe + N Ni) = 0.2. This reduction in the oxygen permeability under humid condition was in agreement with Guo’s results for pure nickel. The increase of permeability in the presence of humidity in iron at 1150 °C can be explained by an increase of the oxygen solubility and/or diffusivity. Jullian et al. [17] measured the oxygen solubility in iron in hydrogen-free atmosphere and found larger solubility than values measured by Swisher and Turkdogan [18] in H2/H2O gas. On the other hand, the oxygen diffusivity in the matrix might be enhanced by the presence of hydrogen in the iron lattice. Observations of Onink et al. [19], in the Fe–C system, suggested that species in interstitial sites could expand the iron lattice, promoting interstitial diffusion. It has also been reported that metal vacancies have an effect on oxygen diffusion [20, 21], and investigations into interactions between vacancy, O, OH and H species could provide information of use in interpreting permeability values.
Plots of A versus alloy composition (Fig. 6b) show that interface enhancement of oxygen diffusion is weaker in nickel-rich alloys due to the change of oxide particle size with nickel content described in the section on internal oxide morphology. However, the effect of interstitial species on the parameter A for iron and Fe–20Ni base alloys is not clearly understood.
Conclusions
The oxygen permeability of Fe and Fe–Ni alloys was measured at 1150 and 1100 °C in dry and wet conditions using the internal oxidation technique. These experiments showed that the use of H2/H2O gases had a negligible effect on the oxygen permeability. In this material, high permeability is likely due to enhanced oxygen diffusion in the matrix. Varying the alloy Ni/Fe ratio produced a strong, non-ideal effect on oxygen permeability.
References
C. Wagner, Chemie 63, 772 (1959).
R. A. Rapp, Acta Metallurgica 9, 730 (1961).
W.Zhao and B.Gleeson. Oxidation of Metals 83, 607 (2015).
J. Takada, S. Yamamoto, S. Kikuchi, and M. Adachi, Metallurgical Transactions A 17, 221 (1986).
J. Takada and M. Adachi, Journal of Materials Science 21, 2133 (1986).
D. J. Young and O. Ahmed, Materials Science Forum, Vol. 93 (Trans Tech Publication, 2001).
A. R. Setiawan, M. Hanafi bin ani, M. Ueda, K. Kawamura, and T. Maruyama. ISIJ International 50, 259 (2010).
S. Goto, K. Nomaki, and S. Koda, Journal of Japan Institute of Metals 34, 319 (1970).
D. P. Whittle, Y. Shida, G. C. Wood, F. H. Stott, and B. D. Bastow, Philosophical Magazine A 46, 931 (1982).
H. C. Yi, S. W. Guan, W. W. Smeltzer, and A. Petric, Acta Metallurgica andMaterialia 42, 981 (1994).
E. Essuman, G. H. Meier, J. Żurek, M. Hänsel, and W. J. Quadakkers, Oxidation of Metals 69, 143 (2008).
E. Essuman, G. H. Meier, J. Zurek, M. Hänsel, T. Norby, L. Singheiser, and W. J. Quadakkers, Corrosion Science 50, 1753 (2008).
W. Anderson, W. Johnson, and F. Rhines, AIME Transactions 147, 205 (1942).
D. Jullian, A. Prillieux, J. Zhang, and D.J. Young. Materials and Corrosion. doi:10.1002/maco.201508803.
J.G. Duh and M.A. Dayananda, Interdiffusion in Fe–Ni–Cr Alloys at 1,100°C (Defect and Diffusion Forum, 1991), p. 1.
P. Guo, J. Zhang, D. J. Young, and C. H. Konrad, Oxidation of Metals 83, 223 (2015).
D. Jullian, A. Prillieux, J. Zhang, D. Hibbert, and D.J. Young, Oxygen Solubility in Fe–Ni Alloys at High Temperature in Rhines Packs and in Gas Mixtures. Corrosion and Prevention 2015 (Australasian Corrosion Association, Adelaide, 2015).
J. H. Swisher and E. T. Turkdogan, AIME Metal Society Transactions. 239, 426 (1967).
M. Onink, C. M. Brakman, F. D. Tichelaar, E. J. Mittemeijer, and S. van der Zwaag, Scripta Metallurgica and Materialia 29, 1011 (1993).
H. O. Nam, I. S. Hwang, K. H. Lee, and J. H. Kim, Corrosion Science 75, 248 (2013).
S. L. Hang, H. Z. Fang, J. Wang, C. P. Guo, Y. Wang, P. D. Jablonski, and Y. Du, Z.K Liu. Corrosion Science 83, 94 (2014).
Acknowledgements
Financial support from the Australian Research Council under its Discovery Program is gratefully acknowledged.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Prillieux, A., Jullian, D., Zhang, J. et al. Internal Oxidation in Dry and Wet Conditions for Oxygen Permeability of Fe–Ni Alloys at 1150 and 1100 °C. Oxid Met 87, 273–283 (2017). https://doi.org/10.1007/s11085-017-9724-6
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11085-017-9724-6