
Abstract
The θ-Al2O3 → α-Al2O3 phase transformation was investigated in thermally grown oxide formed on β-(Ni,Pt)Al bond coats during isothermal exposures at 900–1,200 °C in an argon atmosphere stream with the O2 partial pressure of 1 × 10−5 atm. Local curve fitting was used to evaluate the evolution of the parabolic rate constant, k p , using a general kinetic model (t = A + BΔm + CΔm 2), during the first 5 h of oxidation. All net mass–gain curves exhibited deviations from the classic parabolic model, Δm = k p t ½; a steady state regime was established only after 4 h of exposures, except for the sample oxidized at 1,100 °C.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Improvements in gas turbine efficiency demand an increase in operating turbine entry temperatures (TET) [1]. The current performance of gas turbines in the aeronautic industry is the result of continuous improvements in different areas of engineering, including turbine design, control systems, combustion systems, and materials science. Within materials science the most significant developments have been made in the areas of alloy design, casting technology, and coating methods [2]. High temperature multilayer coatings known as thermal barrier coating (TBC) systems have been used since the early 1980s to protect parts exposed to the highest temperatures (e.g., combustion chamber, rotor blades and stator vanes) [3].
TBC systems are usually designed with four primary constituents: (1) a Ni-base superalloy (SA) as the substrate, providing resistance to static load, creep, and fatigue, (2) an Al-rich bond coat (BC) designed as an Al-reservoir to allow formation of a homogenous oxide scale, (3) a thermally grown oxide (TGO), the result of oxidation of the BC, and (4) a ceramic top coat (TC) as a thermal barrier, typically Y2O3-doped ZrO2 with a composition leading to a “non-transformable” tetragonal phase [4]. The TGO has two principal functions: it binds the ceramic layer to the bond coat, and it acts as a diffusion barrier to limit further substrate oxidation [4, 5]. Notwithstanding that TBC systems have allowed much higher TETs than were previously possible, failures due to a mismatch between thermal expansion coefficients of TBC system layers during operation have limited their lifetime. The failure mechanisms of TBC systems have been classified as extrinsic (e.g., impact damage and molten deposits) and intrinsic (e.g., rumpling, edge-delamination and void formation), as defined by Evans et al. [6]. Most of the intrinsic TBC system failures have involved the BC/TGO/TC interfaces, because of a group of mechanisms associated with strain misfit between layers. These mechanisms are manifested as TC delamination. Because of this, efforts have been made to improve the oxidation lifetimes of the TBC systems by doping the BC with reactive elements [7–14], using different deposition techniques [3, 4, 15] or developing oxidation treatments prior to TC deposition in order to control the TGO properties [16–21].
The control parameters that determine the oxide growth properties are: (1) BC and TC deposition methods, (2) SA, BC, and TC composition, (3) BC surface condition (e.g., grit blasting) before the oxidation treatment, (4) oxygen partial pressure, pO2, (5) temperature, and (6) heat treatment procedure. Regardless of the BC used (e.g., NiPtAl, MCrAlY, etc.), the TGO must comply with the following characteristics to extend the TBC systems’ lifetime: (1) promote growth of α-Al2O3 with the largest possible grain size, (2) present a uniform columnar morphology along the BC surface, (3) have good adhesion with BC and TC, and (4) have slow growth kinetics [18–21]. In 1992 Brumm and Grabke [23] showed that oxidation of the intermetallic B2–NiAl exhibits two polymorphic transitions (γ-Al2O3 → θ-Al2O3 → α-Al2O3) before reaching the stable α-Al2O3 structure. Transformation to the stable α-Al2O3 is accompanied by a volume decrease of the lattice unit cell [16]. For instance, the volume change from θ-Al2O3 to α-Al2O3 (~8 % reduction [22]) can lead to defects and thermal stresses in the BC/TGO interface that may induce failure of the TBC systems.
A transient oxidation stage is generally observed during these phase transitions before reaching a steady state. The γ- to θ-Al2O3 phase transformation leads to a small increase in the parabolic rate constant, k p , while the θ- to α-Al2O3 phase transformation results in a large decrease of k p (almost two orders of magnitude) [23].
It is well known that the growth rate of protective oxide scales is commonly controlled by solid-state diffusion through the oxide scale itself, and it is very well described by the parabolic rate law
where k p is the parabolic rate constant and Δm is the mass–gain per unit area at time t. This behavior was first observed experimentally by Tammann [24] and, independently, by Pilling and Bedworth [25] and subsequently was treated theoretically by Wagner [26]. Nevertheless, important deviations from Tammann’s model have been observed in the growth rates of important oxides like Cr2O3 and Al2O3. Such behavior may arise from different causes, including impurities, grain-boundary diffusion, multilayer scale growth and polymorphism of the oxide (e.g., θ-Al2O3, α-Al2O3, etc.). Different models have been proposed in the literature to consider these deviations based on mixed kinetic behaviors [27–29].
In 1998, Monceau and Pieraggi [27] proposed a methodology to evaluate parabolic rate constants from a local analysis of mass–gain curves for intermetallic materials. They claimed that the use of the classic Δm 2 = k p t model should be replaced by a more general model that considers mixed (diffusion/reaction) control and a transient regime, as well as pure diffusion control.
This method is based on local parabolic fitting of (Δm,t) data over short time intervals to obtain instantaneous values of the parabolic rate constant. This method in general has the advantage over the usual
because it considers the non-idealities associated with oxidation tests. Table 1 summarizes two cases derived from Eq. (2) and shows how the coefficients A, B, and C are related with kinetic parameters. Full details of the model have been reported elsewhere [28].
A power law model is also used to describe the para-linear and pseudo-cubic kinetics of Al2O3 growth, which have been respectively related with phase boundary reaction and grain boundary diffusion (also inversely proportional to the grain size) along the scale: [27, 29]
where k and n are the power law rate constant and rate exponent, respectively.
Important contributions to the understanding of the TGO kinetics for different intermetallics have been reported in the last two decades, some of which suggest that oxidation treatments prior to TC deposition extend the lifetime of TBC systems. However, there is a lack of information about the kinetic behavior of (Ni,Pt)Al bond coats during the first stages of oxidation [4, 5, 23]. The present study aims to contribute to the understanding of the oxidation kinetics of commercial (Ni,Pt)Al bond coats, focusing on the first hours of treatment (t ≤ 5 h) as a function of temperature in an oxidizing atmosphere with a pO2 = 1 × 10−5 atm.
Experimental Procedures
Sample Preparation
All samples used in this work were provided by GE Aircraft Engines (Evendale, OH) as rectangular specimens (1.8 × 1.2 × 0.15 cm), each weighing about 2,900 mg. The specimens consisted of a ∼70 μm thick bond coat deposited onto Rene N5 single-crystal Ni-based superalloy coupons. Bond coats were produced by electroplating a thin layer of Pt onto a grit-blasted substrate. A heat treatment was subsequently performed to diffuse Pt into the substrate, followed by a vapor phase aluminide process (VPA) to introduce Al into the coating. After a second heat treatment the desired β-(Ni,Pt)Al phase was achieved. The resulting bond coat is a bilayer structure consisting of a ∼50 μm thick β-(Ni,Pt)Al and a ∼20 μm thick inter-diffusion zone (IDZ). The substrate and bond coat nominal compositions were determined using inductively coupled plasma atomic emission spectroscopy (ICP-AES, Perkin Elmer Optima DV4300) and electron microprobe analysis (EPMA, JEOL 8900 WD/ED microanalyzer) respectively, and are listed in Table 2. Samples were ultrasonically cleaned using xylene, acetone, methanol–water (1:1), and deionized water for 15 min each to remove surface contamination prior to oxidation treatments.
Oxidation Treatments
Thermogravimetric analyses (TGA) were performed to follow the oxidation kinetics of β-(Ni,Pt)Al BC systems at isothermal temperatures in the range 900–1,200 °C, for 5 h, under a purified argon stream (working gas) with a pO2 = 1 × 10−5 atm. To reach the desired oxygen concentration, pure argon with 1 × 10−4 atm of O2 was passed through a gas purification system (Oxygon Industries OG-120 M). The thermogravimetric analyses were carried out using a thermobalance (Setaram Setsys Evolution 16/18), which has an accuracy of ±0.03 μg. The mass change during TGA experiments was recorded at 5 s intervals. The sample was hung from one end of the beam balance using a 0.4 mm diameter Pt/30 % Rh wire and placed in the hot zone of the vertical furnace.
The reactor furnace was made of dense alumina with an 18-mm inner diameter. The temperature of the furnace was controlled by a Pt–Pt/13 % Rh (S-type) thermocouple placed just below the sample. To minimize any perturbation resulting from gas flow, buoyancy, drag forces or premature sample oxidation before reaching the working temperature, the heating cycle was programmed as follows: (1) evacuation of the analysis chamber to less than 10 Pa, (2) heating to the reaction temperature at a maximum rate of 50 °C/min, (3) back filling the chamber with working gas to atmospheric pressure using a flow rate of 200 mL/min, (4) lowering the flow of the working gas to 20 mL/min and keeping the same at the working temperature for 5 h, and finally (5) cooling the furnace chamber to room temperature at a maximum rate of 50 °C/min with a gas flow rate of 0.3 mL/min.
Characterization
The microstructural evolution of the TGO was characterized by electron microscopy (field emission gun scanning electron microscopy, FEG-SEM, Jeol JSM7401F, and field emission gun scanning transmission electron microscopy FEG-STEM, Jeol JSM7401F). Structural characterization by photo-stimulated luminescence spectroscopy (PSLS) was also done [30–32]. It is desirable that the formed TGO is primarily pure α-Al2O3; however, in practice, Cr 3+ ions tend to be incorporated in the crystal structure of Al2O3. PSLS consists of irradiating Cr-doped Al2O3 with a green (532 nm) or blue (473 nm) laser. Once illuminated, the Cr3+ ions emit a fluorescent radiation due to radioactive decay of the excited electrons to the ground state [33]. The PSLS spectrums of α- and θ-Al2O3 consist of two major bands, with well-characterized frequencies, the R1–R2 and T1–T2 doublets, respectively. The identification of the alumina phase is done on the basis of differences in frequency of caused by different Cr3+-O2 bond distances from the α and θ structures.
PSLS measurements were performed in a micro-Raman mapping spectrometer (Renishaw InVia) connected to a Leica microscope equipped with a 532 nm line-focus laser. The microscope stage was fitted with an automated xyz-motorized stage. Wavelength and laser intensity were internally calibrated using a silicon standard using the peak at 520 cm−1. PSLS spectra were collected in a CCD array using an 1,800 L/mm grating. A 20× microscope objective was used to focus the ∼1 μm spot-sized laser beam and to collect the scattered light. The laser power at the sample was 5 mW, and the acquisition time for each spectrum was in the 0.5–2.0 s range. PSLS spectra were obtained by mapping the sample surface over an ∼1,250 μm2 area with a pitch size of 1 μm.
Results and Discussion
Thermogravimetric Analysis
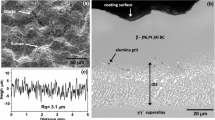
Figure 1 shows representative micrographs of the microstructure of the surface and cross section of the as-received samples. As mentioned previously, those samples were assessed using TGA at isothermal temperatures from 900 to 1,200 °C under a purified argon stream (working gas) with a pO2 = 1 × 10−5 atm. Figure 2 shows the net mass–gain curves of β-(Ni,Pt)Al bond coats systems oxidized for 5 h. The mass–gain rates increase with the temperature from 900 to 1,100 °C, whereas higher temperatures (1,150 and 1,200 °C) led to decrease of the mass–gain rates. This behavior has been observed for many Al2O3 forming alloys [23], and it is associated with the fact that stable α-Al2O3 is controlling the oxidation kinetics at high temperatures [23]. It is also well known that this phase transformation is highly dependent on time, temperature, primary chemical composition (e.g., Al, Pt, Cr), minor additions of other elements (e.g., Y, Zr, Si, Hf) or contaminants (e.g., S), heating procedure (e.g., heating rate), surface preparation (e.g., polishing conditions, grit blasting process, roughness), and pO2 [34–38].

Microstructure of the as-received samples: a surface and b cross section

Experimental net mass–gain curves for β-(Ni,Pt)Al bond coats during isothermal oxidation in a purified argon stream with a pO2 = 1 × 10−5 atm (Color figure online)
Figure 3 shows the net mass–gain curves using a Δm versus t ½ plot, which is useful to illustrate deviations from the straight line expected from the classic parabolic model (Eq. 1). Based on these plots, one can conclude that all net mass–gain curves exhibit slight deviations from the ideal behavior. This can be explained by inferring that initially a mixed (diffusion/reaction) control stage predominated followed by a transient oxidation stage that preceded the establishment of steady state. In the present case, the transient oxidation stage is associated with the polymorphic transition of the alumina (e.g., γ-Al2O3 → θ-Al2O3 → α-Al2O3). This behavior is discussed in detail in the following sections.

Δm versus t ½ plots showing the deviations from the classic parabolic model for the net mass–gain curves of β-(Ni,Pt)Al bond coats during isothermal oxidation (Color figure online)
Microstructural and Structural Analysis
The TGO morphology after oxidation treatments was studied using FEG-SEM over the surface of the oxidized samples. Figure 4 shows clear changes in TGO microstructure as a result of different mechanisms controlling the growth of alumina phases. Samples treated from 900 to 1,050 °C (Fig. 4a–d) exhibited grains in whisker and platelet morphology, which is typical of the Al2O3 metastable phases. The reason of this morphology is that γ- and θ-Al2O3 phases grow by outward cation diffusion (p-type oxide) [39–41]. Samples treated at 1,100 and 1,150 °C (Fig. 4e) showed clear evidence of the first α-Al2O3 grains in combination with the microstructure of metastable Al2O3 phases. Finally, the sample treated at 1,200 °C showed a homogeneous microstructure, which is characteristic of the pure α-Al2O3 phase following inherent BC surface roughness (Fig. 1a). α-Al2O3 growth is predominantly controlled by inward anion diffusion (n-type oxide) and outward cation diffusion (p-type oxide) mainly along grain boundaries. This type of growth mechanism is manifested by characteristic ridges in the TGO/gas and BC/TGO interfaces [42]. However, well developed ridge structures were not observed in these samples. The explanation of this effect is associated with the low pO2 used in these experiments (1 × 10−5 atm), as has been recently shown by Wada et al. [43].

Surface microstructure of samples treated for 5 h at: a 900 °C, b 950 °C, c 1,000 °C, d 1,050 °C, e 1,100 °C and f 1,150 °C
The cracks observed in the sample treated at 950 °C (Fig. 4b) are probably associated with the θ-Al2O3 → α-Al2O3 transformation and will be discussed below. The sample treated at 1,000 °C shows imprints resulting from the inward growth of α-Al2O3 grains, as well as faceted voids at locally spalled areas (Fig. 4c). The formation of these interfacial voids occurs during the first minutes of oxidation, as confirmed by the works of Zimmerman et al. [44]. and Smialek [45] who report the presence of faceted voids after 6 and 4.5 min, respectively, in NiAl alloys. The presence of such voids has been attributed to different causes. For instance, the Kirkendall effects promoted by the unbalanced fluxes of aluminum (outward diffusion) and Ni/Pt (inward diffusion) can result in interfacial void formation [46]. Vacancy injection and evaporation of the Al from the void surface can act as void growth mechanisms [47]. The presence of third elements (e.g., Pt, Cr), reactive elements (e.g., Hf, Y, Zr) and minor amounts of alloy impurities (e.g., S) can also affect void nucleation and growth [48]. It has been shown that a grit-blasting process after the aluminizing process in β-(Ni,Pt)Al alloys mitigate the interfacial void formation in service conditions [49].
Figure 5 shows a cross-sectional view of a sample oxidized at 1,200 °C, which was prepared using a focused ion beam technique (FIB, Jeol JEM-9320) and observed using FEG-STEM. A uniform scale of α-Al2O3 grains is visible along the surface with a vertical length of 0.62 ± 0.2 μm and a lateral length of 1.4 ± 0.15 μm.

STEM micrographs of the cross section view showing the α-Al2O3 grain morphology for the sample treated at 1,200 °C for 5 h
The α-Al2O3 content in the TGO was determined by PSLS based on the following equation
where A R1, A R2, A T1 and A T2 are the integrated intensities corresponding to the characteristic R1–R2 and T1–T2 doublets for the α- and θ-Al2O3, respectively, and ϕ is a correction factor that considers the weaker luminescence signal for θ-Al2O3. Tolpygo and Clarke [50] reported that θ-Al2O3 luminescence is 10–12 times weaker than α-Al2O3. Based on this, a constant value of ϕ = 12 was used in this work. Fitting of the spectra was performed using Renishaw’s WiRE v3.2 software to unambiguously determine the contributing intensity and thus the position of the R1–R2 and T1–T2 peaks.
Figure 6 shows the evolution of the TGO phases as obtained from PSLS analysis. A sigmoidal behavior as a function of the treatment temperature is observed, reaching a plateau at 1,150 °C, where θ-Al2O3 was no longer detected in the scanned area. Thus, one can conclude that for 900 and 950 °C the TGO growth kinetics are mainly controlled by the growth of θ-Al2O3, though the growth of γ-Al2O3 cannot be ruled out, especially during heating and very early oxidation stages. However, γ-Al2O3 was not observed using PSLS after 5 h of treatment at any temperature. From 1,000 to 1,100 °C the oxidation kinetics exhibit θ-Al2O3 as the initial phase, followed by a θ-Al2O3 → α-Al2O3 transition stage where both phases coexist. This claim is clearly confirmed at 1,100 °C based on the microstructural analysis, where both θ- and α-Al2O3 were observed (Fig. 4e).

Evolution of the α-Al2O3 phase transformation as a function of temperature after 5 h of oxidation treatment of (Ni,Pt)Al bond coats. These results were obtained by image analysis of PSLS mappings. A typical PSLS spectrum for a TGO composed of both α- and θ-Al2O3 phases is also shown
Figure 6 also compares the fraction of α-Al2O3 obtained in the present work at 1,100 °C in a purified argon stream with a pO2 = 1 × 10−5 atm with the fraction of α-Al2O3 grown on a diamond polished (001) face of a β-NiAl(Zr) sample oxidized in air at 1,100 °C, as reported by Veal et al. [51]. A higher transformation percent (from θ-Al2O3 → α-Al2O3) is observed in the β-(Ni,Al)Pt BC system, 68.4 %, in comparison with the 34.3 % observed in the β-NiAl(Zr) after 5 h of oxidation treatment. A comparable transformation rate was reached only after 7.68 h of treatment. These differences are mainly associated with chemical composition because it has been reported that larger ions such as Zr (Y, La and Hf) slow down the θ-Al2O3 → α-Al2O3 transformation [52, 53], whereas the presence of Cr (and Fe) accelerates the transformation [54, 55].
The growth and transformation of an alumina scale at high temperatures on Al2O3-forming alloys (e.g., NiAl and NiPtAl) can be divided into three regions of transformation, as has been proposed by Veal et al. [51]. (1) In the early stages, the alumina scale is controlled by the growth of metastable phases (γ- and/or θ-Al2O3) via outward Al diffusion [39]. After an incubation period, the stable α-Al2O3 nucleates at the metal–oxide (M/O) interface, and its grains grow laterally more rapidly than they grow vertically [51]. (2) A uniform layer of α-Al2O3 is established along the M/O interface blocking outward transport of Al and stifling the growth of θ-Al2O3. The growth of new oxide is now mainly controlled by the slow growth kinetics of α-Al2O3. This stage is characterized by θ-Al2O3 → α-Al2O3 transformation along the scale-alloy interface. (3) Once the transformation stage has been completed, α-Al2O3 is the only phase present in the TGO, and it controls the scale growth kinetics. The duration of the different stages is highly dependent on the chemical composition, surface conditions, temperature, pO2, etc. [23, 50, 51].
The θ-Al2O3 → α-Al2O3 transformation involves an approximately 8 % volume reduction [22]. Therefore, radial tensile cracks can develop in the scale during transformation (Fig. 4b). Both the stress state to which the alumina scale is subjected during the transformation process and the thermal stresses developed during cooling and formation of interfacial voids can lead to cracking and spallation of the TGO scale.
Figure 7 shows the surface of the oxidized samples after cooling, where spallation regions can be clearly identified only for the samples treated from 950 to 1,100 °C. The sample treated at 900 °C presented no evidence of spallation. Based on this result it can be concluded that only the samples in the second region of transformation exhibited TGO scale spallation after cooling.

Surface morphology of the treated samples, showing the spallation regions observed after cooling: a 950 °C, b 1,000 °C, c 1,050 °C, d 1,100 °C, e 1,150 °C (no spallation) and f 1,200 °C (no spallation)
Kinetics Analysis
In order to study the parabolic rate constant, k p , during transient and steady state regimes, the net mass–gain curves where analyzed using the local fitting procedure to a general parabolic law (Eq. 2), as proposed by Monceau and Pieraggi [27]. Figure 8 shows the time dependence of k p for the oxidized samples. The steady state regimes are shown in Fig. 8b. According to Fig. 8b a steady state regime was reached for all samples after 4 h of treatment, except for the sample treated at 1,100 °C, in which k p still shows a decreasing tendency after 4.5 h. A similar result was reported by Cadoret et al. [56] for low sulfur (S < 1 ppmw) Ni40Pt10Al50 alloys oxidized in air at 1,100 °C for 5 h, where the effect of Pt-additions on the growth of Al2O3 phases was discussed. In particular, Pt favors the growth of θ-Al2O3 and consequently delays its transformation. It was proposed that this effect helps to relax the stresses during the θ-Al2O3 to α-Al2O3 transformation. This effect was attenuated in the presence of high sulfur contents (S = 90 ppmw), and it was suggested that sulfur segregation to the BC/TGO interface limits Al segregation. However, it was reported that the transient stage never exceeded 1 or 2 h; whereas, in the present work, this stage was extended up to 4 h. These variations can be associated with differences in the surface preparation (as-VPA process versus surface polished to 5 μm finish) and chemical composition (e.g., sulfur content).

Log k p plotted as a function of time for β-(Ni,Pt)Al bond coats during isothermal oxidation in a purified argon stream with a pO2 = 1 × 10−5 atm: a transient and steady state regimes and b steady state regime (Color figure online)
Figure 9 shows an Arrhenius plot of the k p estimated in this work along with the Brumm and Grabke [23] data for a single crystal NiAl. A good agreement is observed between the growth kinetics during the first 5 min and the slope obtained by Brumm and Grabke for θ-Al2O3. Based on this, it is confirmed that the metastable θ-Al2O3 controls the growth kinetics during early stages (red dots) from 900 to 1,100 °C, whereas that at 1,150 and 1,200 °C α-Al2O3 is the dominant phase. The striped zone represents the transition regime before reaching the steady state regime after 4 h (blue dots). Finally, a comparison between our k p values and those reported by Cadoret et al. [56]. for single crystals (oriented along [001] direction) of Ni50Al50 and Ni40Pt10Al50 alloys (oxidized under 1 atm of pure oxygen) doped with different concentration of sulfur is shown in Fig. 10. Based on this, it is observed that at 900 °C the \( k_{p}^{\theta } \) is about 3 times lower that the reported by Cadoret et al. [56]. for NiPtAl alloys, while at 1,100 °C it is only 1.5 times lower for NiPtAl alloys. This difference is directly associated with the different pO2 conditions used during the oxidation treatments. The decrease observed at 1,100 °C is also associated with a pretreatment performed on the samples treated at 1,100 °C to grow a 500 nm thick oxide scale at 900 °C.

Arrhenius plot summarizing the k p values of β-(Ni,Pt)Al bond coats during isothermal oxidation with a pO2 = 1 × 10−5 atm. Bold lines correspond to the γ-Al2O3, θ-Al2O3 and α-Al2O3 lines refer Brumm and Grabke [23] (Color figure online)

Conclusions
All net mass–gain curves exhibited deviations from the classic parabolic model proposed by Tammann because a transient oxidation regime (including θ-Al2O3 phase growth, α-Al2O3 grain nucleation, θ- to α-Al2O3 phase transformation, and grain coarsening) controls the initial oxidations kinetics at all temperatures tested (900–1,200 °C). The growth kinetics behavior of the thermally grown oxide on the (Ni,Pt)Al bond coats can be well described by the general parabolic law t = A + BΔm + CΔm 2 using a local fitting procedure. A steady state regime was reached after 4 h of treatment (except for the sample treated at 1,100 °C), unlike the results reported in the literature [56] for similar systems where a steady state regime was reached in less than 2 h. This difference is primarily associated with the surface preparation, because in the present study the samples were oxidized after the VPA process where the characteristic ridges of the BC grains are exposed (Fig. 1), while in the reported data the samples were previously polished removing the BC ridges and decreasing the species diffusion.
References
J. R. Nicholls, MRS Bulletin 28, 659 (2003).
D. R. Clarke and C. G. Levi, Annual Review of Materials Research 33, 383 (2003).
R. C. Reed, The Superalloys Fundamentals and Applications (Cambridge University Press, Cambridge, 2006), p. 283.
S. Bose, High Temperature Coatings (Elsevier, Amsterdam, 2007), p. 155.
W. Gao and Z. Li, Developments in High-Temperature Corrosion and Protection of Materials (Woodhead Publishing, Cambridge, 2008), p. 476.
A. G. Evans, D. R. Clarke, and C. G. Levi, Journal of the European Ceramic Society 28, 1405 (2008).
P. Y. Hou, Annual Review of Materials Research 38, 275 (2008).
B. Pieraggi and R.A. Rapp, Journal de Physique IV, Colloque C9, supplément au Journal de Physique III, 275 (1993).
B. A. Pint, Oxidation of Metals 45, 1 (1996).
B. M. Warnes, Surface & Coatings Technology 146–147, 7 (2001).
B. A. Pint, Proceedings of the John Stringer Symposium (ASM, Materials Park, OH, 2001).
B. A. Pint, Journal of the American Ceramic Society 86, 686 (2003).
J. A. Haynes, B. A. Pint, K. L. More, Y. Zhang, and I. G. Wright, Oxidation of Metals 58, 513 (2002).
D. Toma, W. Brandl, and U. Koster, Oxidation of Metals 53, 125 (2000).
V. K. Champagne, The Cold Spray Materials Deposition Process Fundamentals And Applications (Woodhead, Cambridge, 2007), p. 245.
M. Matsumoto, Surface & Coatings Technology 202, 2743 (2008).
S. Kitaoka, T. Kuroyama, M. Matsumoto, R. Kitazawa, and Y. Kagawa, Corrosion Science 52, 429 (2010).
I. Spitsberg and K. Moreb, Materials Science and Engineering A A417, 322 (2006).
V. K. Tolpygo and D. R. Clarke, Surface & Coatings Technology 200, 1276 (2005).
L. M. He, Y. F. Su, L. F. Allard, M. J. Lance, and W. Y. Lee, Metallurgical and Materials Transactions A: Physical Metallurgy and Materials Science 35A, 1113 (2004).
A. Hesnawi, L. Hefei, Z. Zhaohui, G. Shengkai, and X. Huibin, Surface & Coatings Technology 201, 6793 (2007).
A. H. Heuer, D. B. Hovis, J. L. Smialek, and B. Gleeson, Journal of the American Ceramic Society 94, S146 (2011).
M. W. Brumm and H. J. Grabke, Corrosion Science 33, 79 (1992).
T. G. Tammann, Zeitschrift für Anorganische und Allgemeine Chemie 111, 78 (1920).
N. B. Pilling and R. E. Bedworth, Journal Institute of Metals 29, 529 (1923).
C. Wagner, Journal of the Electrochemical Society 99, 369 (1952).
D. Monceau and B. Pieraggi, Oxidation of Metals 50, 477 (1998).
B. Pieraggi, Oxidation of Metals 27, 177 (1987).
W. J. Quadakkers, D. Naumenko, E. Wessel, V. Kochubey, and L. Singheiser, Oxidation of Metals 61, 17 (2004).
Q. Ma and D. R. Clarke, Journal of the American Ceramic Society 76, 1433 (1993).
D. M. Lipkin and D. R. Clarke, Oxidation of Metals 45, 267 (1996).
D. M. Lipkin, H. Schaffer, F. Adar, and D. R. Clarke, Applied Physics Letters 70, 2550 (1997).
X. Lu, S. Venugopalan, Kim Hyunjung, M. Grimsditch, S. Rodriguez, and A. K. Ramdas, Physical Review B: Condensed Matter 79, 5204 (2009).
G. C. Rybicki and J. L. Smialek, Oxidation of Metals 31, 275 (1989).
T. F. An, H. R. Guan, X. F. Sun, and Z. Q. Hu, Oxidation of Metals 54, 301 (2000).
D. Monceau, K. Bouhanek, R. Peraldi, A. Malie, and B. Pieraggi, Journal of Materials Research 15, 665 (2000).
V. K. Tolpygo, D. R. Clarke, and K. S. Murphy, Metallurgical and Materials Transactions A: Physical Metallurgy and Materials Science 32A, 1467 (2001).
L. Xie, Y. Sohn, E. H. Jordan, and M. Gell, Surface & Coatings Technology 176, 57 (2003).
J. Jedlinski and G. Borchardt, Solid State Ionics 50, 67 (1992).
J. Doychak, J. L. Smialek, and T. E. Mitchell, Metallurgical and Materials Transactions A: Physical Metallurgy and Materials Science 20A, 499 (1989).
B. A. Pint, J. R. Martinans, and L. W. Hobbs, Solid State Ionics 78, 99 (1995).
J. Doychak, J. L. Smialek, and C.A. Barret, NASA Technical Memorandum, 101455 (1988).
M. Wada, T. Matsudaira, and S. Kitaoka, Journal of the Ceramic Society of Japan 119, 832 (2011).
D. Zimmerman, M. Bobeth, M. Rühle, and D. R. Clarke, Zeitschrift fuer Metallkunde 95, 84 (2004).
J. L. Smialek, Metallurgical and Materials Transactions A 9A, 309 (1978).
M. W. Brumm and H. J. Grabke, Corrosion Science 34, 547 (1993).
H. Svensson, M. Christensen, P. Knutsson, G. Wahnstrom, and K. Stiller, Corrosion Science 51, 539 (2009).
J. A. Haynes, B. A. Pint, K. L. More, Y. Zhang, and I. G. Wright, Oxidation of Metals 58, 513 (2002).
J. A. Haynes, Scripta Materialia 44, 1147 (2001).
V. K. Tolpygo and D. R. Clarke, Materials at High Temperature 17, 59 (2000).
B. W. Veal, A. P. Paulikas, and R. C. Birtcher, Applied Physics Letters 89, 161916 (2006).
B. A. Pint, M. Treska, and L. W. Hobbs, Oxidation of Metals 47, 1 (1997).
D. Renusch, M. Grimsditch, I. Koshelev, B. W. Veal, and P. Y. Hou, Oxidation of Metals 4, 471 (1997).
X. F. Zhang, K. Thaidigsmann, J. Ager, and P. Y. Hou, Journal of Materials Research 21, 1409 (2006).
W. C. Hagel, Corrosion 21, 316 (1965).
Y. Cadoret, D. Monceau, M. P. Bacos, P. Jasso, V. Maurice, and P. Marcus, Oxidation of Metals 64, 185 (2005).
Acknowledgments
The authors thank B. Gleeson at the University of Pittsburgh, USA, for stimulating discussions. This project was funded by Conacyt FOMIX-QRO-2008-C02-10599. The authors also thank Wilber Antunez Flores at the Laboratorio Nacional de Nanotecnologia (CIMAV) for the technical support during samples characterization by SEM.
The authors express their grateful acknowledgment to the technical support from Wilber Antunez Flores.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Alvarado-Orozco, J.M., Morales-Estrella, R., Boldrick, M.S. et al. First Stages of Oxidation of Pt-Modified Nickel Aluminide Bond Coat Systems at Low Oxygen Partial Pressure. Oxid Met 78, 269–284 (2012). https://doi.org/10.1007/s11085-012-9305-7
Received:
Revised:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11085-012-9305-7