
Abstract
The influence of KCl(s) on the oxidation of Sanicro 28 (35Fe27Cr31Ni) austenitic stainless steel at 600 °C in 5% O2 + 40% H2O and in 5% O2 was investigated. The samples were coated with 0.1 mg/cm2 KCl(s) prior to exposure. The exposure time was 1, 24, 72 and 168 h. Uncoated samples were exposed for reference. The oxidized samples were analyzed by SEM/energy-dispersive X-ray (EDX), X-ray diffraction (XRD), and AES. The amount of chloride and chromate on the samples was analyzed quantitatively by ion chromatography after exposure. KCl(s) is very corrosive towards Sanicro 28. Corrosion is initiated by the formation of potassium chromate through the reaction of KCl(s) with the protective oxide, chloride leaving the sample in the form of HCl. Chromate formation is a sink for chromium in the oxide and leads to a deterioration of its protective properties. In the 5% O2 + 40% H2O environment, there is a stoichiometric relationship between the chromate formed and the chloride consumed. In dry O2 chromate formation is relatively slow, leaving more unreacted KCl(s) on the surface than in 5% O2 + 40% H2O. Once the protective, chromium-rich oxide has been depleted in chromium by chromate formation, the alloy becomes susceptible to direct attack by the remaining KCl(s).
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Power boilers burning biomass and waste contribute to a significant and growing fraction of the environmentally sustainable electricity production in Europe. However, the production of electricity in these plants is restricted by the corrosivity of the fuel. The main problem area is fireside corrosion of the steam superheaters which limits the useful life of the component and results in unplanned outages. In order to circumvent these problems it is necessary to limit steam data, but this result in diminished electricity efficiency and poorer economy.
Presently, superheater tubes for corrosive environments are made mainly of ferritic and austenitic FeCr and FeCrNi steels. These alloys usually form a protective, chromium-rich oxide, (Fe,Cr)2O3, at high temperatures. The alloys may suffer rapid corrosion in environments that tend to destroy the protective oxide. In comparison to coal-fired boilers, the fireside environment in biomass and waste fired boilers is characterized by high concentrations of oxygen, water vapor and alkali- and chlorine-containing species. The high corrosivity of the flue-gas environment is usually attributed to alkali chlorides (KCl and NaCl) in the flue gas, depositing on, e.g., superheater surfaces [1–7]. Some authors have proposed corrosion mechanisms which include the formation of melts [4, 8] or the formation of alkali chromate [8]. However, much previous work in this field is concentrated on the role of chlorine in the corrosion process [1–5, 8].
The corrosivity of alkali chlorides towards metals in oxidizing environments at high temperature is usually attributed to the formation of molecular chlorine which is supposed to generate volatile transition-metal chlorides at the scale/metal interface [1–6, 9, 10]. Several authors use the designation “active oxidation” or “the chlorine cycle” for this hypothetical mechanism [1, 5, 6, 9–11]. However, it may be noted that these mechanisms do not deal with the initiation of corrosion, i.e., the breakdown of the protective oxide which is initially present on the material. In this article the focus is on the early stages of corrosion of the chromia-forming alloy Sanicro 28. Especially, the role of potassium is highlighted.
The present paper investigates the role of solid KCl in the deterioration of the protective scale on the highly alloyed austenitic Sanicro 28 at 600 °C. The focus is on the initial stages of the corrosion process, especially before breakdown of the protective oxide. A mechanism is proposed, focusing on the role of potassium, which explains the initiation of corrosion by the reaction of KCl with the protective oxide to form potassium chromate.
Experimental Procedures
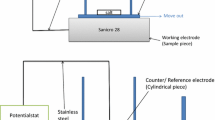
Table 1 shows the composition of the alloy studied. The geometrical area of the sample coupons was 5.56 cm2, (15 × 15 × 2 mm). A hole (ø = 1.5 mm) was drilled for better handling. The exposures were carried out in a horizontal furnace fitted with a 46 mm i.d. SiO2-glass tube. All exposures were isothermal; the temperature was kept at 600 ± 2 °C. The samples were mounted three at a time using an alumina sample holder with dimensions 40 × 50 × 2 mm, featuring three slits for mounting. The samples were positioned parallel to the direction of the flow. The furnace system was fitted with a humidifier producing a 5% O2 + 40% H2O gas mixture (N2 in balance), see Fig. 1. The gas velocity was 3 cm/s. Before exposure the samples were ground to 1,000-grit SiC and polished with 1-μm diamond spray until the surface appeared mirror-like. The polished samples were degreased and cleaned in acetone and ethanol using ultrasonic agitation. Salt was added by spraying the samples with a saturated solution of KCl in water/ethanol (30:70). The samples were dried with cool air and stored in a desiccator prior to exposure in order to prevent electrochemical corrosion. The gravimetric measurements were made using a six-decimal Sartorius™ balance.

Experimental set-up for oxidation studies in dry oxygen and humid environments
X-ray Diffraction, XRD
Crystalline corrosion products were analyzed by X-ray diffraction (XRD) using a Siemens D5000 powder diffractometer, equipped with grazing-incidence-beam attachment and a Göbel mirror. CuKα radiation was used and the angle of incidence was 2°. The detector measured between 20° < 2θ < 65°.
Scanning-Electron Microscopy, SEM/EDX
Analytical scanning-electron microscopy was carried out using an FEI Quanta 200 ESEM in high vacuum, back-scattered electron mode was used for imaging samples exposed with KCl(s). Secondary-electron mode was used for imaging reference samples. The ESEM was equipped with an Oxford Inca energy-dispersive X-ray (EDX) system, which was used for elemental mapping and chemical quantification. The microscope was operated at 10 kV for imaging and 12 kV for EDX analysis.
Auger Electron Spectroscopy, AES
The Auger analyses were performed using a Scanning Auger Microprobe (PHI 660). The electron-beam voltage was 10 kV and the beam current was about 150 nA. The depth profiles were obtained using a differentially pumped ion gun (Ar+) with acceleration voltage 4 kV. The etch rates were calibrated on flat samples of Ta2O5 with a well defined oxide thickness of 1,000 Å. The collected raw data were refined by using MultiPak™ v.6.0 software. The software made it possible to distinguish between the signals from oxidized and metallic iron and chromium, based on the chemical shifts of these elements in oxidized form. Due to the small peak shift in nickel, it was not possible to divide the Ni signal into a metal state and an oxidized state.
Ion Chromatography, IC
To determine the amount of water-soluble chlorides and chromates after exposure the samples were analyzed with a Dionex 1000 with IonPac AS4A-SC analytic column. The samples were leached in 3 mL MilliQ water using ultrasonic agitation for 1 + 10 + 1 min. The flow rate was 2 mL/min and 1.8 mM Na2CO3/1.7 mM NaHCO3 was used for eluation. A Dionex OnGuard II H was used to separate alkali metal ions and transition metals before introducing the sample to the column.
Results
Gravimetry
The great corrosivity of KCl(s) towards alloy Sanicro 28 is illustrated in Fig. 2, showing mass gain as a function of exposure time. In the absence of KCl(s), mass gains are small both in 5% O2 + 40% H2O (wet) and in 5% O2 (dry). In 5% O2 + 40% H2O the mass-gain curve peaks at 24 h indicating that vaporization of chromium(VI) is taking place. In dry 5% O2 the alloy shows protective behavior with sub-parabolic oxidation kinetics. In the presence of KCl(s), mass gain is very rapid, especially in 5% O2. In 5% O2 + KCl(s) the mass gain has a positive slope throughout the exposure while the mass curve peaks at 24 h in 5% O2 + 40% H2O + KCl(s). Again, the negative slope of the mass curve during later stages of the exposure shows that vaporization is taking place. It may be noted that the negative slopes of the curves in 5% O2 + 40% H2O and 5% O2 + 40% H2O + KCl(s) are similar.

Mass gain versus exposure time for Sanicro 28 exposed at 600 °C in 5% O2 + 40% H2O and 5% O2 in the absence and in the presence of KCl(s)
Quantitative Analysis of Water-Soluble Anions
Table 2 show the amount of water-soluble chloride found by ion chromatography after exposure. After 1 h of exposure only 45% and 66% of the added chloride was found in 5% O2 + 40% H2O and 5% O2 environments, respectively. After 24 h, less than 1% and 6% of the added chloride was accounted for in the analysis. Because chloride on the sample surface is expected to be in water-soluble form, the results imply that chloride has vaporized from the sample. Interestingly, the analysis also shows the formation of significant amounts of chromate (CrO4 2-), see Table 3 and Fig. 3. The amount of chromate increases with exposure time, especially in the 5% O2 + 40% H2O environment.

Ion concentration versus exposure time for Sanicro 28 exposed at 600 °C in 5% O2 + 40% H2O and 5% O2 in the presence of KCl(s)
Crystalline Phases Detected by XRD
The crystalline phases detected by XRD are listed in Table 4. Diffraction from the substrate was observed from all samples, irrespective of exposure time and environment, indicating that the oxide scales were thin. In the absence of KCl(s), the corrosion products detected were corundum-type α-(FeCr)2O3 and spinel-type oxide, irrespective of exposure time. In the salt-containing exposures, KCl(s) is detected after 1 h but not after longer exposure times. This is in accordance with the quantitative analysis for chloride (compare Table 2). Potassium chromate was detected in all KCl(s) runs, see Fig. 4, except after 168 h in 5% O2. In addition to K2CrO4, α-(FeCr)2O3 and spinel-type oxide also formed in the KCl(s) runs.

XRD diffractogram of Sanicro 28 exposed at 600 °C for 24 h in 5% O2 + 40% H2O with KCl(s). The symbols indicate: K2CrO4 (▲), α-(FeCr)2O3 (■), M3O4 (♦), and substrate (•)
Characterization of the Oxide Formed in the Absence of KCl(s)
Figure 5 shows an SEM image and an AES depth profile after 24 h of exposure in 5% O2 + 40% H2O. The network of dark lines in the smooth oxide corresponds to the alloy grain boundaries. AES depth profiling indicated an oxide thickness of about 120–140 nm. The inner part of the oxide is enriched in chromium. The situation is very similar after 168 h exposure in the same environment, but the oxide has become marginally thicker, about 140–150 nm. The oxide formed in 5% O2 is initially similar to the oxide formed in 5% O2 + 40% H2O (see Fig. 6). After 24 h in 5% O2 the oxide is slightly thinner than in the wet environment, about 80–100 nm. This is in fair agreement with the mass-gain results that correspond to an average oxide thickness of 63 nm. Nickel is enriched at the outer part of the oxide formed in 5% O2. After 168 h of exposure in the same environment the oxide has become about twice as thick (190–240 nm, according to AES). It may be noted that the chromium content in the oxide is consistently somewhat higher in 5% O2 compared to in 5% O2 + 40% H2O.

SEM image and AES depth profile of Sanicro 28 exposed at 600 °C for 24 h in 5% O2 + 40% H2O without KCl(s). The bar corresponds to 50 μm

SEM image and AES depth profile of Sanicro 28 exposed at 600 °C for 24 h in 5% O2 without KCl(s). The bar corresponds to 50 μm
Characterization of the Scale Formed in the Presence of KCl(s)
Figure 7 shows an SEM image of a sample before exposure with KCl(s) added to the sample. Cubic KCl crystals with typical sizes of 10–50 μm are distributed over the surface. Exposure in the presence of KCl(s) causes rapid corrosion. The corroded surface can be described in terms of three characteristic morphologies: more or less reacted KCl crystals, the oxide between the KCl crystals and facetted particles on top of the oxide (see Fig. 8). The oxide between the KCl particles may be relatively smooth (termed the base oxide) or rather uneven depending on exposure time and environment.

SEM image of Sanicro 28 with KCl(s) before exposure. The bar corresponds to 20 μm

SEM images of Sanicro 28 exposed at 600 °C for 1 h in (a) 5% O2 + 40% H2O and (b) 5% O2 with KCl(s). The bars correspond to 100 μm
One Hour
Exposure in 5% O2 + 40% H2O
The SEM image in Fig. 9 shows a partly reacted KCl crystal and the base oxide with particles on top. A network of thin dark lines can be seen that correspond to the alloy grain boundaries. EDX mapping shows that chromium and iron are depleted at the alloy grain boundaries while nickel is enriched. Much of the surface is covered by the thin and smooth base oxide, presumably corresponding to the mixture of chromium-rich, corundum-type oxide found by XRD (see Table 4). Facetted particles that appear dark in the SEM image have nucleated all over the base oxide. EDX point analysis shows that the particles are rich in chromium, potassium and oxygen. It is concluded that the particles correspond to K2CrO4 identified by XRD. The amount of potassium chromate varies from grain to grain. In certain areas, mostly in the vicinity of former KCl particles, the potassium chromate forms a semi-continuous layer on top of the oxide (see Fig. 8a). In these regions the underlying oxide is porous. In many cases the KCl particles have become completely covered by iron-rich oxide at this stage. The oxide forms a shell corresponding to the shape of the original crystals. IC shows that 45% of the added chloride remains on the samples, see Table 3. EDX analyses of the sample showed that chlorine is associated with potassium, showing that unreacted KCl(s) is still present. In addition, there were indications of traces of chlorine along the alloy grain boundaries.

SEM image and EDX elemental maps of Sanicro 28 exposed at 600 °C for 1 h in 5% O2 + 40% H2O with KCl(s). The bar corresponds to 50 μm
Exposure in 5% O2
Exposure in 5% O2 with KCl(s) for 1 h produced the same crystalline corrosion products as the corresponding wet environment (see Table 4). However, the morphology of the corroded surface is quite different. In this case the oxide between the former KCl particles consists partly of a coarse and flaky oxide and partly of a smooth oxide similar to that seen in the corresponding wet run, see Fig. 8b. The potassium-chromate particles are associated with the smooth oxide. The chromate particles are less abundant than in the corresponding exposure in water vapor. This is in accordance with the quantitative analysis for chromate in Table 3. An iron-rich oxide covers most of the reacted KCl particles. This oxide tends to be thicker in comparison to the corresponding wet run and it spreads out over the surface in the immediate vicinity of the KCl particles. In many cases hematite whiskers form on the former KCl particles.
24 hours
Exposure in 5% O2 + 40% H2O
Figure 10 shows the surface morphology after 24 h. The oxide on the grain centers has become rougher and more iron rich in comparison to in the 1-hour exposure (see Fig. 10a). Close to the grain boundaries the oxide is still smooth. The increased surface roughness is in line with the continuing corrosion between 1 and 24 h indicated by gravimetry (see Fig. 2). The mass gain after 24 h corresponds to an average oxide thickness of 1.0 μm. This is in agreement with results from Auger depth profiling on the alloy grain centers, showing an oxide thickness of about 1.0 μm, see Fig. 11. The potassium-chromate particles now agglomerate over the alloy grain boundaries while they are less occurring in the scale over the alloy grain centers (see Figs. 10 and 11) The K2CrO4 particles have grown in size compared to after 1 h. At this stage all KCl particles have been replaced by iron-rich oxide shells, EDX analysis giving no indication at all for chlorine. This is in agreement with IC that showed that less than 1% of the added Cl remained on the samples, see Table 3.

SEM images of Sanicro 28 exposed at 600 °C for 24 h in (a) 5% O2 + 40% H2O and (b) 5% O2 with KCl(s). The bars correspond to 100 μm

SEM image and AES depth profile of Sanicro 28 exposed at 600 °C for 24 h in 5% O2 + 40% H2O with KCl(s). The bar corresponds to 100 μm
Exposure in 5% O2
Similar to the 1-hour exposures, the oxide formed in 5% O2 is rougher than that formed in the wet environment. Compared to 1 h in dry O2, the area covered by flaky oxide has increased after 24 h (see Fig. 10b). Many iron-rich oxide nodules have formed, especially in the vicinity of the former KCl particles. K2CrO4 particles are found both on the flaky oxide and on the smooth oxide. The chromate particles do not decorate the alloy grain boundaries as they do in the wet environment. Similar to the corresponding 5% O2 + 40% H2O exposure, there is no indication of unreacted KCl(s) on the surface. However, IC analysis reveals that about 6% of the chloride added remains on the sample at this stage. Large, iron-rich-oxide islands have formed at the position of the former KCl particles. In comparison to the wet exposure, the area around the former KCl particles is more affected by corrosion and is covered by a thick, cracked scale. Similarly to the corresponding 1-hour exposure, numerous hematite whisker formations can be seen.
168 hours
Exposure in 5% O2 + 40% H2O
After 168 h the oxide-scale morphology is similar to that described for the 24 h samples with the conspicuous iron-rich-oxide islands at the position of the former KCl particles (see Fig. 12a). The main difference is that the decoration of the alloy grain boundaries by potassium-chromate particles has become less pronounced. The chromate particles now tend to cluster, forming agglomerates (~ 10–30 μm in size) mainly along the alloy grain boundaries. At this stage few smooth oxide areas remain, most of the surface is covered by rough oxide.

SEM images of Sanicro 28 exposed at 600 °C for 168 h in (a) 5% O2 + 40% H2O and (b) 5% O2 with KCl(s). The bars correspond to 100 μm
Exposure in 5% O2
After 168 h in 5% O2, the oxide between the former KCl particles has become rougher compared to the corresponding 24 h exposure, see Fig. 12b. The whole scale is strongly undulated and cracked. In some areas the outermost part of the scale has spalled, revealing a porous, underlying oxide. The number of iron-rich-oxide nodules has increased, especially in the vicinity of the former KCl particles. In comparison to the corresponding exposure in 5% O2 + 40% H2O, the surface is more heavily attacked. In accordance with the XRD results (Table 4), K2CrO4 particles were not detected by SEM/EDX. This is the only KCl run where K2CrO4 was not identified.
Discussion
The results show that when alloy Sanicro 28 is exposed to dry or wet oxygen at 600 °C in the absence of KCl(s), it forms a protective, chromia-rich oxide of the corundum type. Exposure to water vapor does not compromise the alloy’s excellent ability to resist corrosion (Fig. 2). It has been shown that exposure of FeCr alloys to atmospheres containing water vapor and oxygen at high temperature results in the vaporization of chromic acid: [12–15]
The mass loss (Fig. 2) and the decrease in chromium content in the oxide (Fig. 5 and Fig. 6) registered in wet oxygen clearly indicate that chromia is lost from the protective oxide by vaporization also in the present case. Apparently, the supply of chromium to the oxide by diffusion in the alloy (primarily by grain boundary diffusion) is sufficient to maintain protective properties all over the surface in this environment. This is in contrast to austenitic alloys with lower chromium content, (e.g., alloy 304L) that tend to suffer a partial breakdown of the protective properties in wet oxygen at 600 °C [16].
The present results show that KCl(s) is quite corrosive towards alloy Sanicro 28 (Figs. 2, 8, and 10). Figure 3 and Table 3 provides an important clue as to what is happening, showing that significant amounts of chromate, CrO4 2-, form on the surface. For further evidence for chromate formation, see also Fig. 4 and Table 4. In the presence of water vapor and oxygen, chromium(III) oxide reacts with KCl(s) to form potassium chromate according to:
In the absence of water vapor the corresponding reaction produces elemental chlorine instead of HCl:
Thermodynamic calculations show that, under our experimental conditions, the formation of chlorine dominates only in extremely dry conditions (< 0.1 ppm H2O) [17]. The water-vapor concentration in the “dry” runs in the present study is about 10 ppm, implying that HCl is the dominant gaseous product in both exposure environments studied.
In analogy to reaction (1), chromate formation consumes chromia from the oxide. A comparison of samples exposed with and without KCl(s) shows that, in the present case, the reaction of chromia in the protective oxide with KCl to form chromate (reaction (2)) is much faster than the loss of chromia by vaporization of CrO2(OH)2 (reaction (1)). The SEM images and the analysis for chromate (Figs. 3, 8, and 10, Tables 3 and 4) show that a large fraction of the added KCl(s) has formed potassium chromate already after 1 h and that little unreacted KCl(s) is left on the surface after 24 h. The analysis of soluble chloride on the surface shows the same trend (Table 2).
The formation of potassium chromate depletes the protective, corundum-type oxide in chromium, leaving behind an iron-rich oxide with relatively poor protective properties:
Initially, reaction (4) results in the destruction of the protective properties only locally, in the immediate vicinity of the KCl crystals. It is argued that this is because the supply of KCl is greatest there. The reaction continues until all available KCl has been consumed (or vaporized).
The greater thermodynamic driving force for reaction (2) in humid oxygen compared to dry oxygen corresponds to a more rapid formation of chromate in the 5% O2 + 40% H2O environment. In fact, there is a stoichiometric relationship (within the errors of the analysis) between the loss of chloride from the surface and chromate formation in the 5% O2 + 40% H2O environment (Table 3). This proves that chloride is lost primarily from the surface in the form of HCl by reaction (4), and that the loss of chloride by other routes is of minor importance. In contrast, chromate formation only corresponds to a fraction of the chloride lost after 24 h in dry oxygen. This shows that, in the latter case, other chlorine-containing species are also being lost from the surface, in addition to HCl. The equilibrium vapor pressure of KCl(g) at 600 °C is 3 · 10−6 atm (calculated from Barin [17]) making it a probable candidate. It is suggested that the greater loss of chlorine-containing species other than HCl in dry O2, compared to wet O2, is due to KCl(s) vaporization. Because reaction (4) is relatively slow in dry O2 (Table 2), KCl(s) is expected to be present on the surface for a longer time in dry O2 in comparison to the 5% O2 + 40% H2O environment. It may be noted that while KCl(g) is the most probable vaporizing species, the vaporization of transition-metal chlorides cannot be ruled out. To summarize, the results imply that KCl(s) on the surface either reacts to form chromate and HCl or vaporizes as KCl(g). The rapid consumption of KCl(s) explains why the corrosion rate in the presence of KCl decreases with time, especially in the 5% O2 + 40% H2O environment.
Considering that the distribution of KCl(s) before exposure is quite uneven (Fig. 7) it is notable that K2CrO4 particles initially form over the whole surface (Fig. 8). This implies that KCl is quite mobile on the sample surface. In principle, KCl has to be transported from the KCl crystals either in gaseous form or as an adsorbate:
Because of the relatively low equilibrium vapor pressure of KCl(g) over solid KCl at the exposure temperature (see above), it is suggested that KCl is transported mainly in adsorbed form on the sample surface. The even distribution of chromate in the 1 h exposures also indicates that the reaction is not influenced by lateral variations in the availability of chromium. The amount of chromate formed after 1 h in 5% O2 + 40% H2O + KCl(s) and the 5% O2 + KCl(s) environments corresponds to 40–45 and 15–20 nm of pure chromium oxide, respectively (Table 3). This corresponds roughly to the amount of chromium present in the oxide film formed in the absence of KCl(s). The tendency for K2CrO4 to be concentrated along the alloy grain boundaries after longer exposure times in 5% O2 + 40% H2O environment is considered significant. It is argued that, after the initial stage, most of the easily available chromium in the oxide and in the alloy immediately beneath it has been consumed. As the chromate formation proceeds, the transport of chromium to the surface by diffusion along the alloy grain boundaries becomes the rate-determining step for the reaction. This interpretation is supported by the chromium depletion at the alloy grain boundaries seen in the plane-view images (Fig. 9). It is suggested that the chromate particles that initially form over the alloy grain centers may be overgrown by oxide after 24 h in 5% O2 + 40% H2O. Potassium chromate does not aggregate along the grain boundaries in dry O2 after 24 h. This is attributed to the slower formation of K2CrO4 in that environment (see Fig. 3 and Table 3).
The results clearly show that KCl(s) causes corrosion of alloy Sanicro 28 through potassium-chromate formation that depletes the protective oxide in chromium as discussed above. However, the results also show that corrosion is faster in dry O2 than in the wet environment, while more potassium chromate forms in the latter environment. Because it is unlikely that water vapor inhibits corrosion, this implies that KCl also causes corrosion by another mechanism. Once the protective, chromium-rich oxide has become depleted in chromium by chromate formation, the alloy becomes susceptible to direct attack by the remaining KCl. We believe that chloride ions can penetrate the iron-rich oxide and form transition-metal chlorides at the alloy/scale interface. The transition-metal chlorides at the interface are suggested to enhance corrosion attack by destroying scale adhesion. This type of chloride-enhanced corrosion is expected primarily in the vicinity of the former KCl crystals (Fig. 12b) where a thick, convoluted and cracked oxide forms, especially in dry O2. The penetration of chloride to the scale/metal interface on alloy Sanicro 28 will be treated in a coming paper.
Conclusions
KCl(s) accelerates the corrosion of alloy Sanicro 28 at 600 °C, the mass gains being more than 20 times greater than in the absence of KCl(s). Corrosion is initiated by the formation of potassium chromate through the reaction of KCl(s) with the protective oxide, chloride leaving the sample in the form of HCl. In 5% O2 + 40% H2O environment, quantitative analysis revealed a stoichiometric relationship between the chromate formed and the chloride consumed. Chromate formation is a sink for chromium in the oxide and will lead to a deterioration of its protective properties. In dry O2, chromate formation is relatively slow, leaving more unreacted KCl(s) on the surface than in the wet environment. Once the protective, chromium-rich oxide has been depleted in chromium by chromate formation, the alloy becomes susceptible to direct attack by the remaining KCl. It is suggested that the chloride ions can penetrate the iron-rich oxide, forming transition-metal chlorides at the alloy/scale interface. Corrosion attack is especially rapid in the vicinity of the former KCl crystals where a thick convoluted and cracked oxide forms.
References
H. J. Grabke, E. Reese, and M. Spiegel, Corrosion Science 37(7), 1023 (1995).
H. P. Michelsen, et al., Fuel Processing Technology 54(1–3), 95 (1998).
M. Montgomery and A. Karlsson, Materials and Corrosion 50(10), 579 (1999).
H. P. Nielsen, F. J. Frandsen, and K. Dam-Johansen, Energy & Fuels 13(6), 1114 (1999).
T. Valente, Journal of Materials Engineering and Performance 10(5), 604 (2001).
M. A. Montgomery, A. Karlsson, and O. H. Larsen, Materials and Corrosion 53(2), 121 (2002).
H. P. Nielsen et al., Progress in Energy and Combustion Science 26(3), 283 (2000).
Y. Shinata, Oxidation of Metals 27(5–6), 315 (1987).
M. J. McNallan et al., International Corrosion Conference Series NACE-6, 316 (1983).
Y. Y. Lee and M. J. McNallan, Metallurgical Transactions A—Physical Metallurgy and Materials Science 18(6), 1099 (1987).
J. M. Abels and H. H. Strehblow, Corrosion Science 39(1), 115 (1997).
H. Asteman et al., Oxidation of Metals 52(1/2), 95 (1999).
H. Asteman et al., Oxidation of Metals 54(1/2), 11 (2000).
H. Asteman, J. E. Svensson, and L. G. Johansson, Corrosion Science 44(11), 2635 (2002).
K. Segerdahl, J. E. Svensson, and L. G. Johansson, Materials and Corrosion 53(4), 247 (2002).
J. Pettersson et al., Oxidation of Metals 64(1–2), 23 (2005).
I. Barin, Thermochemical Data of Pure Substances, 3rd edn. (VCH, 1995).
Acknowledgment
This work was carried out within the High Temperature Corrosion Centre (HTC) at Chalmers University of Technology.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Pettersson, C., Johansson, LG. & Svensson, JE. The Influence of Small Amounts of KCl(s) on the Initial Stages of the Corrosion of Alloy Sanicro 28 at 600 °C. Oxid Met 70, 241–256 (2008). https://doi.org/10.1007/s11085-008-9118-x
Received:
Revised:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11085-008-9118-x