
Abstract
In this study, the density functional theory (DFT) is employed to investigate the structural, elastic, optoelectronic, and thermoelectric (TE) properties of K2NaSbZ6 (Z = Br, I) halide double perovskites (HDPs). The evaluated second-order elastic constants and formation energy analysis confirm that the studied HDPs are cubic, stable, and ductile. The electronic band structure calculations disclose that both compounds are determined to be p-type semiconductors with indirect band gaps of 3.20 eV and 2.40 eV for K2NaSbZ6 (Z = Br, I), respectively. The optical features demonstrate the prominent role of the present HDPs in photovoltaic conversion and optoelectronic devices. In the visible region, both HDPs exhibit high optical absorption (> 105 cm−1) with excellent refractive index. Furthermore, the studied TE properties show that for both HDPs, n-type doping is more suitable for improving their TE performances. Therefore, the present study would be beneficial for emerging optoelectronic and TE technologies.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
With the expeditious worldwide human progress, energy demand has become increasingly concerned. Currently, the significant contribution of fossil fuel consumption to the destruction of our ecology is one of the most pressing issues. In this light, widespread efforts are intensifying to look for sustainable alternatives to conventional energy sources (Bildirici and Gökmenoğlu 2017; Mantziaris et al. 2017). Moreover, sunlight is the most prevalent sustainable and clean alternative to conventional energy sources. However, to convert heat from the sun and recover efficiently the unexploited heat from industrial sectors, a variety of cost-effective and environmentally sound technologies are being vastly investigated. In this regard, photovoltaic (PV) solar cells and thermoelectric (TE) devices have sparked considerable interest (Mostafaeipour et al. 2022; Ivanovski et al. 2021; Snaith 2013). Aside from PV, TE technology, with the ability to harvest and convert discarded heat into useful power, is acknowledged as a reliable solution for energy efficiency enhancement (MacDonald 2006; Goldsmid 2010). The pursuit of adequate materials with improved optoelectronic performance and energy conversion efficiency is the key issue (Tarbi et al. 2023a, 2023b, 2022, 2023c, 2023d; Boutramine 2023). In this view, the usefulness of TE materials strongly depends on the TE dimensionless figure of merit (ZT = S2σT/κ). Here S, κ, σ, and T stand respectively for Seebeck coefficient, thermal and electrical conductivities, and absolute temperature (Goldsmid 2021, 2017). Accordingly, for an efficient TE material, high S and σ combined with low κ are required. Concurrently, the full control of these key parameters is an integral yet difficult challenge. In this regard, various techniques have been conducted to further improve ZT by enhancing the power factor (PF = S2σ) as well as reducing κ. Moreover, a TE material with a ZT of unity is considered excellent (Zhao et al. 2014; Aswal et al. 2016; Liu et al. 2016).
In the recent past, double perovskites materials (DPs), generally denoted with the formula A2BB’Z6 (where A = alkaline or rare earth metals, B/B’ = transition or non-transition cations, and Z = oxides or halides), have gathered tremendous curiosity among scientists (Maiti et al. 2019; Tariq et al. 2020; Slavney et al. 2016; Faizan et al. 2016). The excellent optical and TE performances of DPs make them more adequate for solar cells and other clean harvesting energy applications. This is greatly attributed to their appealing features, including high conversion efficiency, adequate band gap tenability, strong optical absorption, long carrier diffusion length, low carrier recombination, and balanced carriers' effective masses. Furthermore, from a crystallographic point of view, the diverse arrangement of elements in DPs is known to provide a large variation in physical properties (Srivastava 2017; Demic et al. 2017). Besides, perovskite-based optoelectronic devices and solar cells offer a promising conception as an emerging low-cost technology with remarkable power conversion efficiency (PCE), which has been enhanced efficiently to impressive values (Singh et al. 2021; Cao and Yan 2021; Kim and Kim 2021; Mirershadi et al. 2016). In this area, single and lead-based hybrid perovskites have been intensively investigated (Zhang et al. 2018; Xiao et al. 2023; Waszkowska et al. 2020; Lu et al. 2022). Their remarkable characteristics and ability to compete with existing technologies are attributed to their low production cost, adequate band gap, and good transport properties. Unfortunately, the expected feasible use of Pb-based perovskites is hindered because of toxicity concerns, structural instability, and their potential degradation with continuous exposure to light and moisture (Ke et al. 2019; Senocrate et al. 2019). Tremendous theoretical and experimental works have been conducted to tackle the toxicity and lack of intrinsic stability of Pb-based perovskites without compromising their conversion efficiency (Shahbazi and Wang 2016; Babayigit et al. 2016a, 2016b; Bouich et al. 2022). Therefore, the focus challenge is to develop a new class of environmentally benevolent and efficient materials to simultaneously handle these issues. Therefore, it has been attempted to exchange the toxic Pb with some non-toxic metals such as Sn and Ge. However, a comprehensive literature review assured that this change in the oxidation state drastically reduces the long-term stability of solar cells (Babayigit et al. 2016a, 2016b; Bouich et al. 2022; Liu et al. 2018; Ghosh et al. 2022; Shao et al. 2018; Kopacic et al. 2018). Alternatively, a new generation perovskites of Pb-free halide DPs (HDPs) has been suggested to deal with this problem for the optoelectronic industry and the clean energy community. In these structures, two divalent Pb2+ cations in APbX3 are exchanged with a pair of trivalent B3+ and monovalent B+ cations, resulting in A2BB’Z6 DPs, which sustain the same crystalline structure and charge balance (Du et al. 2017). Furthermore, these materials provide a wide compositional flexibility, covering a wide spectrum of thermoelectric and solar cell applications (Kung et al. 2020). Interestingly, thousands of researchers worldwide are currently focused on PV and TE materials, and a significant number of research groups are concentrated on Pb-free HDPs. In this regard, several Pb-free HDPs have been evaluated to replace even high Pb-containing perovskites for solar and thermal energy harvesting purposes (Yan et al. 2022; Wu et al. 2021; Bibi et al. 2021; Lu et al. 2020; Chu et al. 2019).
Additionally, an extensive amount of works on the compounds of similar families has been conducted. In this regard, using the first principles method, Waqas et al. investigated the physical properties of M2NaInI6 (M = Rb, Cs) HDPs. They obtained direct band gaps of 1.111 eV and 1.121 eV for Rb2NaInI6 and Cs2NaInI6, respectively. Their results revealed that both studied compounds are appropriate for PV and TE applications (Waqas Mukhtar et al. 2021).
Moreover, Shi et al. assessed the Cs2MBiCl6 (M = Ag, Cu, and Na) and Cs2MBiCl6 (M = K, Rb, and Cs) HDPs. According to the findings, Cs2MBiCl6 (M = Ag, Cu, and Na) exhibited a cubic symmetry along with a relatively low carriers’ effective masses, whereas Cs2MBiCl6 (M = K, Rb, and Cs) displayed a monoclinic structure with a high indirect semiconducting band gap correlated with high carrier effective masses (Shi et al. 2020).
Furthermore, the electronic properties of Tl2NaBiCl6, Cs2NaInBr6, and Cs2NaBiCl6 HDPs have been studied. The findings demonstrated that the compounds have nearest neighbor distances in cation sublattices, and large electro-negativity differences among cations resulted in a narrow discrete band structure (Shi et al. 2015). Similarly, based on a modified Becke-Johnson (mBJ) potential taking into account the spin–orbit coupling (SOC), Khan et al. estimated the properties of Cs2NaMCl6 (M = In, Tl, Sb, and Bi) and revealed that these materials showed direct band gaps for Cs2NaMCl6 (M = In, Tl) and indirect band gaps for Cs2NaMCl6 (M = Sb and Bi) with narrow discrete bands (Khan et al. 2021). Also, S. Zhao et al. evaluated the optical characteristics of Cs2NaBX6 (B = Sb, Bi, and X = Cl, Br, I) and found that these compounds demonstrated exceptional stability and are perfect materials for optoelectronic applications (Zhao et al. 2018). Using the first-principles approach, Yang Gui et al. have systematically investigated Cs2BSbBr6 (B = Li, Na, K, and Rb) HDPs. They reported that Cs2BSbBr6 (B = Li, Na) exhibited a semiconducting indirect band gap, while Cs2BSbBr6 (B = K and Rb) are direct band gap semiconductors (SCs). Moreover, the corresponding reported band gaps are 2.81, 2.96, 3.37, and 3.36 eV, respectively. The significant absorption of visible (VIS) light implies that the materials are suitable for PV applications (Gui et al. 2023).
Interestingly, potassium-based HDPs have generated a great deal of interest due to their exceptional optoelectronic properties, which include superb growth uniformity, long diffusion length, high optical quality, and good carrier mobility. K-based HDPs have been studied by several researchers. Recently, A. Mera et al. have investigated the electronic, elastic, optical, and TE properties of K2NaTlX6 (X = Cl, Br, and I) using the ab initio method. The results highlighted the potential of these materials for clean energy conversion applications (Mera et al. 2023). In another study, Al-Qaisi et al. analyzed the physical characteristics of K2CuSbX6 (X = Cl, Br, and I). They reported that the indirect band gaps of these compounds are 1.12, 0.80, and 0.43 eV, respectively. Furthermore, at room temperature, these HDPs showed an ultralow value of lattice conductivity and a high ZT. The results also certified their extensive applicability for optoelectronic and TE devices (Al-Qaisi et al. 2023).
In view of a comprehensive literature review, it is believed that K2NaSbZ6 (Z = Br, I) HDPs have not yet been studied. Therefore, the purpose of this study is to use the DFT approach to conduct an ab initio evaluation of HDPs as suited for green energy applications. With the use of the generalized gradient approximation (GGA) and modified Becke-Johnson (mBJ) potential, the thermoelectric, electrical, optical, and structural properties of K2NaSbZ6 (Z = Br, I) are identified in this work. It is anticipated that the current study will provide important new understanding for upcoming theoretical and experimental surveys.
2 Computational method
In the present contribution, the structural, elastic, optoelectronic, and TE properties of K2NaSbZ6 (Z = Br, I) HDPs are elaborated through the density functional theory (DFT) as it is properly designed in the WIEN2K simulation package (Schwarz et al. 2002).The optimal method for figuring out the electronic properties of solids is the first-principles-dependent FP-LAPW scheme, which was used in this study's computation procedure. The exchange–correlation functional wisely adopted here is Perdew Burke Ernzerhof's (PBE) Generalized Gradient Approximation (GGA) (Perdew et al. 1996). This approximation efficiently gives the ground state properties but underestimates the accuracy of the band gap and electronic properties that are required to outline the optical and TE properties. Therefore, to overcome this underestimate, the trans-Balaha modified Becke-Johnson exchange potential (TB-mBJ) has been employed (Tran and Blaha 2009; Aslam et al. 2021).
In the FP-LAPW technique, the primitive cell volume is disintegrated into two sides: the interstitial side and non-overlapping muffin tin spheres (MT) of radius RTM. Within the MT spheres, the eigenfunctions with spherical harmonics are expanded, whereas the plane wave functions are developed over the interstitial space. In a reciprocal lattice, the basis set is controlled by the input cut-off parameters ℓmax = 10, Gmax = 12, and KmaxRMT = 7. Here, ℓmax, Gmax, Kmax, and RMT stand for the highest angular momentum, the Gaussian factor, the maximum k-vector, and the smallest MT sphere radius, correspondingly. For an accurate conversion, (17 × 17 × 17) k-points were used for structural optimization and electro-optical outcomes, but (49 × 49 × 49) k-points were utilized for TE characteristics. The convergence criterion during the geometry optimization was obtained consistently by 10−5 Ry. Under constant relaxation time approximation, TE characteristics were investigated using the Boltz-Trap code incorporated with the semi-classical Boltzmann theory (Madsen et al. 2006a).
3 Results and discussion
3.1 Structural properties
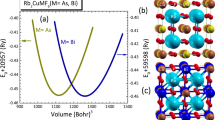
In practical design and simulation, the structural features and stabilities of materials are relevant and strongly correlated with almost all their physical properties. Typically, by optimizing the ground energy versus volume (E–V), the material's structural stability as well as its ground-state elastic and thermodynamic properties can be perfectly achieved. The studied K2NaSbZ6 (Z = Br, I) HDPs have updated database entries with preliminary information in the Materials Project Web Dataset (Materials Project 2023a; Materials Project 2023b). The full optimization of the titled HDPs is conducted within the framework of the PBE-GGA treatment. Therefore, the obtained ground energy versus volume (E–V) was carefully added to the Birch-Murnaghan equation of state (EOS) (Murnaghan 1944). The optimized crystal structures are plotted in Fig. 1a–d. Thus, K2NaSbI6 is the most optimized compound as compared to K2NaSbBr6, owing to its smaller ground-state energy. Table 1 presents an inventory of the ground-state structural properties of K2NaSbZ6 (Z = Br, I) that have been observed and compared to the previously published results.

Crystal structures (b, d) and energy versus volume curves (a, c) for K2NaSbZ6 (Z = Br, I) compounds
The computed equilibrium lattice constants are 11.42 Å and 12.30 Å for K2NaSbBr6 and K2NaSbI6, respectively. The observed increase in the equilibrium lattice constant is supported by shifting the univalent anion Br to I, attributed to a lager radius of I than that of Br. Also, the reported densities of 3.13 g/cm3 and 3.52 g/cm3 are consistent with lattice constant changes. The results obtained are listed in Table 2. As expected, the reported lattice constants fit well with the available results (Materials Project 2023a; Materials Project 2023b). Next, to investigate the thermodynamic stability of both HDPs, their formation energy (ΔEf) is determined through the formula described in Al-Muhimeed et al. (2022); Al-Qaisi et al. 2021). Consequently, the present HDPs appear to be thermodynamically stable, as certified by negative ΔEf, and can be experimentally synthesized. Moreover, K2NaSbBr6 is thermodynamically more favorable than K2NaSbI6. Additionally, the compound's ion compatibility factor guarantees the structural stability of A2BB′Z6 DPs. Meanwhile, this compatibility is inferred using the Goldschmidt empirical tolerance factor \(\tau = \frac{({r}_{A}+{r}_{Z})}{\sqrt{2}\left({r}_{Z}+{r}_{BB{\prime}}\right)}\),where \({r}_{Z}\) and \({r}_{A}\) stand for the Z and A ionic radii and \(r_{{BB^{\prime } }}\) is the average of the ionic radii of B and B’ (Bartel et al. 2019; Fedorovskiy et al. 2020). The ideal tolerance factor for cubic stable perovskites is unity, while the optimal range of 0.81 < τ < 1.11 is also classified as a stable region (Aslam et al. 2021). The calculated τ values of the studied K2NaSbZ6 (Z = Br, I) are all within the optimal range, as shown in Table 1, confirming a stable cubic HDPs structure.
3.2 Elastic properties
To overcome the challenge in the manufacturing engineering of excellent materials for PV and optoelectronic applications, mechanical properties and second-order elastic constants (SOECs) must be considered. In this study, the Cubic Elastic Package (Jamal et al. 2014) was adopted to elucidate the elastic behavior of the investigated materials. The elastic constants (Cij) were assessed in the PBE-GGA treatment for the equilibrium cubic structure at zero external pressure. Since both K2NaSbZ6 (Z = Br, I) HDPs demonstrate the cubic crystal structure, three different independent SOECs (C11, C12, and C44) are required for the dimensional structure strength determination. The elastic stability is validated by executing the Born-Huang elastic stability criteria for cubic symmetry: C11 + 2C12 > 0, C44 > 0, C11 − C12 > 0, and C12 < B < C11 (Wang et al. 1993). Hence, the reported positive values of Cij, tabulated in Table 2, ensure that K2NaSbZ6 (Z = Br, I) HDPs are mechanically stable in cubic crystal structure. As we can see, both HDPs have significant C11 values as compared to the other two shear SOECs (C12 and C44); accordingly, these HDPs possess high resistance to deformation in the (100) plan and < 100 > direction. Also, due to its greater C11, K2NaSbBr6 is stiffer as compared to K2NaSbI6. Furthermore, by considering the Voigt-Reuss-Hill averaging scheme (Hill 1952), various averages of elastic constant-based moduli such as bulk modulus (B), shear modulus (G), Young modulus (Y), Poisson’s ratio (ν), and anisotropy factor (A) were predicted using the calculated SOECs (Hill 1952; Li et al. 2012; Luan et al. 2018; Ranganathan and Ostoja-Starzewski 2008). Therefore, due to its greater B value, K2NaSbBr6 is much more resistant to volumetric changes than K2NaSbI6. In addition, the values of G suggest that K2NaSbBr6 has a higher resistance to plastic deformation than K2NaSbI6. Concurrently, B > G illustrates that both compounds have higher resistance to volumetric than shape strains. As evaluated from the Y modulus, the K2NaSbBr6 compound is resistive to longitudinal deformation. The A factor describes the elastic anisotropy of a crystal (Li et al. 2012). A = 1 is determined for isotropic compounds, and its deviation from unity provides the degree of anisotropic character. The observed anisotropy can be ascribed to several bond orientations in various crystallographic planes and it is associated with a significant difference in the shear and longitudinal elastic constants. It is obvious from Table 2 that K2NaSbI6 is highly anisotropic compared to K2NaSbBr6. Moreover, the Pugh (B/G) and Poisson (ν) ratios are critical parameters in describing whether the material is ductile (B/G > 1.75, ν > 0.26) or brittle (B/G < 1.75, ν < 0.26) (Li et al. 2012; Pugh 1954). The recorded values reveal that both K2NaSbZ6 (Z = Br, I) HDPs are ductile; consequently, they can be applied to malleable PV and optoelectronic devices. Also, owing to the higher values of B/G and υ, it is clear from Table 2 that K2NaSbBr6 is more malleable than K2NaSbI6. The ionic bonding character of K2NaSbZ6 (Z = Br, I) compounds is accounted for by their positive Cp = C12–C44 (Pettifor 1992).
3.3 Thermodynamic properties
Next, the Navier–Stokes equations are used to inspect the studied HDPs average sound velocity (vm) with reference to its longitudinal (vl) and transverse (vt) components, expressed as: \({v}_{l}= {\left(\frac{3B+ 4G}{3\rho }\right)}^{1/2},{v}_{t}= {\left(\frac{G}{\rho }\right)}^{1/2},{v}_{m}= {\left[\frac{1}{3}\left(\frac{2}{{{v}_{t}}^{3}}+ \frac{1}{{{v}_{l}}^{3}}\right)\right]}^{-1/3}\). Moreover, the Debye temperature (θD) depends upon vm as follows: \({\theta }_{D}= \frac{h}{{k}_{B}}{\left[\frac{3n}{4\pi }\left(\frac{{N}_{A}\rho }{M}\right)\right]}^{1/3}{v}_{m}\) (Schreiber et al. 1975; Jasiukiewicz and Karpus 2003). Here, h, ρ, kB, and NA stand for Planck constant, material density, Boltzmann constant, and Avogadro number. As a result, the reported θD is higher for K2NaSbBr6 than for K2NaSbI6, which demonstrates that the lattice vibration sustainability of Br-based DP is greater than that of I-based DP.
Additionally, the directional elastic constant C11 has been used to estimate the melting temperature of present HDPs by using the standard equation (Dar et al. 2019): \({T}_{m}(K)=(553+\frac{5.91}{GPa}{C}_{11})\pm 300\). The obtained results, summarized in Table 2, show that Tm is greater for K2NaSbBr6 as compared to K2NaSbI6.This outcome agrees with the reported Y modulus, indicating stronger bonding and retention in Br-based DP compared to I-based DP. This difference is attributed to the larger ionic size of I ions. Besides, this validates the practical use of the present materials for relatively elevated-temperature applications.
3.4 Electronic properties
3.4.1 Electronic band structure
The electronic bands structure of materials is the primary powerful tool used to predict the optoelectronic and TE properties. However, a suitable optoelectronic material must be acquainted with its electronic, optical features and the width of the band gap, as the maximum optical absorption occurs in this region. Indeed, from a practical point of view, evaluating the electronic and optical characteristics accurately delivers convenient evidence concerning their synthesis and device fabrication. In the present contribution, the electronic bands structure of the studied K2NaSbZ6 (Z = Br, I) HDPs was assessed using both PBE-GGA and mBJ-GGA schemes. Since the PBE-GGA method is noted to underestimate the experimental band gaps, we employed the mBJ-GGA, which has been adopted successfully in several recent studies (Aslam et al. 2021; Chen et al. 2020).
The Fig. 2a–f depicts the computed bands structure of the K2NaSbZ6 (Z = Br, I) HDPs under focus along the (W → L → Γ → X → W → K) high-symmetry k-space path in the Brillouin zone (BZ). Also shown is the corresponding total density of states (TDOS). In this figure, the Fermi level (EF = 0) is selected as the energy reference. As presented here, the conduction band minima (CBM) and the valence band maxima (VBM) are positioned, respectively, at X and L symmetric k-points, leading to an indirect (X-L) band gap. Moreover, the EF level seems to be unoccupied, indicating that K2NaSbZ6 (Z = Br, I) exhibits a semiconducting character. The recorded electronic band gaps for K2NaSbBr6 and K2NaSbI6 with the application of PBE-GGA are 2.47 eV and 1.89 eV, respectively. It is worth noting that these results are consistent with the GGA findings reported earlier (Chen et al. 2020). However, when TB-mBJ exchange potential was assisted by GGA, the fundamental band gap magnitudes are enhanced to be 3.20 eV and 2.40 eV for K2NaSbBr6 and K2NaSbI6, respectively. This observed enhancement can be attributed to the fact that the TB-mBJ exchange potential slightly pushes the CBs, predominated by Sb-5p and Br/I-4p/5p electronic states, away from the EF. Also, as reported in Table 1, the ground state band gap values of the present HDPs decrease with the order of increasing halide ionic size from Br to I. Therefore, owing to the fact that Br has a lower ionic radius, as in Fig. 1b, d, substituting Br with I causes a red-shift in the band gap within the VIS spectrum. Hence, this ensures that the studied HDPs can be used in solar cells, optoelectronic devices, and energy-harvesting devices.

Electronic band structure and total density of state of K2NaSbZ6 (Z = Br, I) using TB-mBJ-GGA and PBE-GGA methods
TDOS patterns reveal consistent densities and band gaps in the electronic band structure of the examined HDPs. Remarkably, the TDOS below the EF is found to be richer than in the CB, indicating a p-type conductivity. Moreover, the observed notable dispersion in the highest VB along (L- Γ- X) path, lead to a small holes’ effective mass with a high mobility allowing a p-type conductivity character. Furthermore, the states in the highest VB are flat along (X-W-K) path confirming the low occupied orbital near EF. Also, this can be affiliated to the relatively large holes’ effective mass.
3.4.2 Density of states
The partial densities of states (PDOS) are conducted to gain further insight regarding the K, Na, Sb, and Br/I elemental contributions to the VBs and CBs, which helps to comprehensively estimate the electronic structure influence on the device performance. The obtained results achieved by TB-mBJ-GGA treatment are depicted in Fig. 3a–j. The PDOS contour reveals that the VBs and CBs edges are mostly composed of Sb-5s, Sb-5p, and Br/I-4p/5p orbitals. Moreover, K-4s and Na-3s contributions occurred in the CB deep region, which does not contribute to the electronic behavior. This means that K+ and Na+ cations have a non-resistant function in the formation of the frontier bands and only act as charge donors to maintain structural stability. In this regard, the bottom of the CB is attributed predominantly to Sb-5p and partially to p-orbitals of halogens, while the top of the VB involves Sb-5s and Br/I-4p/5p states. From Fig. 3, it can be seen that the contribution of the Sb-5p decreases ongoing from Br to I. Interestingly, the changing of halide ions causes a red-shift of I-5p and Sb-5p states in the CB with the substitution of Br with I, which is the main cause of the observed red-shifted band gap. Moreover, the noticed weak PDOS at EF in the VBM comes from Br/I-4p/5p and Sb-5s states. Our reported results regarding the role of different halogen orbitals are consistent with the recently reported contributions (Al-Qaisi et al. 2021; Anbarasan et al. 2021).

Total (TDOS) and partial (PDOS) density of states of K2NaSbZ6 (Z = Br, I) using the TB-mBJ-GAA method
3.5 Optical properties
The light-matter interaction and the nature of the band gap are the main factors determining the optical performance of investigated materials. In this study, the optical properties of K2NaSbZ6 (Z = Br, I) HDPs are comprehensively described in terms of the dielectric function using the TB-mBJ potential. Generally, the dielectric function, also called the permittivity of a material has a complex form as follows: ε (ω) = ε1 (ω) + iε2 (ω) (Hilal et al. 2016). The real part ε1 (ω) describes the induced polarization due to the light-material interaction, while the imaginary part ε2 (ω) reflects the capability to attenuate the transmitted light through a crystal (Albanesi et al. 2005; Bobrov et al. 2010). The frequency/energy dependence of other correlated linear optical properties such as optical absorption coefficient α (ω), refractive index n (ω), optical energy loss function L (ω), reflectivity R (ω), and optical conductivity σ (ω) are addressed using ε (ω) components (Gajdoš et al. 2006; Yu and Cardona 1999; Dreesel and Gruner 2002).
3.5.1 Dielectric function
The computed frequency/energy-dependent ε1 (ω) of K2NaSbZ6 (Z = Br, I), displayed in Fig. 4a, depicts a weak energy dependence character at low energy region. With the order of increasing halides ionic radii from Br to I, the enhanced static value, ε1 (0), from 2.96 to 3.95 confirm the fact that heavy halogens are optically more polarizable. The noticed higher ε1 (0) favors a weak charge carrier recombination rate improving the performance of optoelectronic devices. Therefore, this parameter is inversely linked to electronic band gap energy demonstrated by the Penn model: ε1(0) = 1 + (ħωp/Eg)2, where Eg, ħ and ωp are the band gap energy, Plank constant, and electron’s plasma frequency, respectively (Penn 1962). In the VIS range, the primary peaks of ε1(ω), associated with the maximal dispersion of light, occur at the resonance frequencies. These frequencies shift to higher energy upon going from I to Br. Above the resonance positions, the ε1(ω) starts to drop sharply with many relaxation peaks attributed to the electronic inter-band transitions. The relaxation time can be extracted from the peak frequency as follows: τr = 1/ω = ħ/E. It is worth noting that, owing to its relatively smaller band gap energy, K2NaSbI6 exhibits the highest peak in the VIS region as compared to K2NaSbBr6. Furthermore, beyond the energy values of 9.51 (11.41 eV), where ε1(ω) < 0, the present K2NaSbI6 (K2NaSbBr6) HDPs exhibit metallic behavior with a total reflection. In this energy range, the frequency of the incident radiation is greater than the plasma frequency.

Optical properties of (a–h) K2NaSbZ6 (Z = Br, I) HDPs calculated using the TB-mBJ-GGA method
Figure 4b shows that for both compounds, ε2 (ω) remains zero for all photons with energy lower than the band gap energy, which indicates that the energy is not sufficient for electronic inter-band transition. Accordingly, from the threshold points of ε2 (ω), the extracted optical band gaps of K2NaSbBr6 (K2NaSbI6) of 3.19 (2.36 eV), listed in Table 3, agree extremely with the computed electronic band gaps. Moreover, the ε2 (ω) increases from the threshold points reaching its highest values of 4.36 (4.01 eV) and 5.55 (3.33 eV) for K2NaSbBr6 and K2NaSbI6, respectively. The main peaks of ε2 (ω) are located at 5.62 eV and 3.33 eV for K2NaSbBr6 and K2NaSbI6, respectively. We note that, the multi-peak phenomenon observed in the VIS and UV regions can be attributed to the different rates of conceivable electronic transition from Sb-5p and Br/I-4p/5p states of the VB to the empty Sb-5p states in the CB. Therefore, this shows that K2NaSbZ6 (Z = Br, I) HDPs possess different optical absorption abilities in a broad range of the electromagnetic spectrum. Subsequently, for K2NaSbI6 the observed highest peak magnitudes typically cause the polarized electrons to extend easily into conducting states, enhancing the PV conversion efficiency. Also, it is clear from Fig. 4b that the absorption bands red-shifted by substituting Br with I. Moreover, the optimum working regions within VIS and UV ranges make the interested HDPs highly adequate for PV conversion and optoelectronic devices.
3.5.2 Refraction index
The optical transparency of a material is mainly determined by the optical refractive index n (ω). Furthermore, n (ω) refers to the ratio of the light speed in vacuum to that in the considered crystal. It provides the degree of bending of light when passing through a material. Evaluating this parameter is of great importance for the accurate usage of materials in optical applications. As depicted in Fig. 4c and listed in Table 3, on going from Br to I, the zero-frequency limit of refractive index increases from 1.72 to 1.99 in correspondence with the red-shifted band gaps. Moreover, these values are connected to ε1 (0) as follows: n2(0) = ε1(0). Furthermore, beyond n (0), the patterns of n (ω) closely follow ε1(ω) reaching its maximum values of 2.64 at 2.76 eV and 2.35 at 3.44 eV, respectively for K2NaSbI6 and K2NaSbBr6. In the UV spectrum, the highest refractive index of K2NaSbI6 is 2.32 at 7.20 eV followed by 2.07 noticed at 9.05 eV for K2NaSbBr6. Similarly to the above-mentioned observation on ε1 (ω); at energies of 10 and 11.93 eV, the n (ω) values of K2NaSbI6 and K2NaSbBr6 are less than unity indicating that these HDPs become optically opaque.
3.5.3 Extinction coefficient
The extinction coefficient, k (ω), regarded as the imaginary component of the complex refractive index, represents another substantial optical function directly linked to the photonic energy’s optical absorption capacity. This can be checked from Fig. 4d, where the k (ω) followed a similar trend as ε2 (ω) spectral. One can see that, in a broad electromagnetic spectrum range, the value of k (ω) increases on going from K2NaSbBr6 to K2NaSbI6. Furthermore, the values of k (ω) are 0.10 at 3.23 eV for K2NaSbBr6 and 0.11 at 2.48 eV for K2NaSbI6 around the dispersion principal absorption edge. Additionally, the k (ω) shows the first set of peaks at 3.36 eV (4.07 eV) and the second set of peaks at 4.45 eV (5.70 eV) for K2NaSbI6 (K2NaSbBr6).
3.5.4 Optical reflectivity
The optical reflectivity R (ω) refers to the material’s capability to reflect the incident photons impinging on its surface. Accordingly, the R (ω) improves considerably as the impinging photons are absorbed less. Figure 4e depicts the optical reflectivity of K2NaSbZ6 (Z = Br, I) HDPs. For both compounds, the values of the static optical reflectivity are 10.93% and 7.00% for K2NaSbI6 and K2NaSbBr6, respectively, as recorded at zero energy. These provide insight into the materials’ surface quality. In the VIS spectrum, the maximum values of reflectivity are noticed to be roughly 25.24% and 14.48% for K2NaSbI6 and K2NaSbBr6, respectively. Moreover, in the UV region, K2NaSbI6 and K2NaSbBr6 exhibit high reflectivity beyond 9.51 eV and 11.41 eV, where ε1(ω) < 0, highlighting that in this region, these HDPs can be used as shielding materials.
3.5.5 Energy loss factor
The energy loss factor L (ω) is another important optical function. Other than absorption and reflection, this parameter provides fruitful information about the loss of photonic energy during the scattering process when the electron passes through a material or is depleted by heating. The computed L (ω) for K2NaSbZ6 (Z = Br, I) HDPs is depicted in Fig. 4f. One can see that no scattering is observed for photonic energy less than band gaps, which means that no energy loss occurs. Furthermore, in all cases, L(ω) is negligible for the first peaks, while it reveals the highest values of 1.14 and 2.06 for the second peaks observed at 5.29 eV and 6.57 eV for K2NaSbI6 and K2NaSbBr6 attributed to the Plasmon losses. However, from a practical point of view the first peaks are typically useful. It is noteworthy that in the VIS region, the photonic energy loss is very negligible. Therefore, fewer R (ω) and L (ω) are beneficial to a good electronic transport and enhance the photoelectric effect efficiency of the studied materials for PV and optoelectronic devices.
3.5.6 Absorption coefficient
The optical absorption coefficient α (ω) provides the material's potentiality to absorb a specific photonic energy. Especially, for PV conversion, the materials should reveal a higher (> 105 cm−1) optical absorption coefficient in the entire VIS spectrum region. The α(ω) spectra, depicted in Fig. 4g, reveal that K2NaSbZ6 (Z = Br, I) exhibits a broadband absorption spectrum concentrated within VIS-UV regions, which are highly suitable for PV and optoelectronic devices. The absorption edges are pivotal since they refer to the critical points before which there is no absorption. It is customary to realize that the corresponding energies red-shifted, on going from Br to I, showing good coherence between electronic and optical band gaps. The inset of Fig. 4g displays the optical band gap energies of 2.31 (3.07 eV) for K2NaSbI6 (K2NaSbBr6) extracted using the Tauc’s plots. A slight overestimation has been outlined as compared to the extracted optical values from ε2 (ω). Moreover, the transition from Sb-5p and Br/I-4p/5p valence states to the unoccupied Sb-5p conduction states gets a noticeable peak values of 4.15 × 105 cm−1 and 4.68 × 105 cm−1 in the VIS region for K2NaSbI6 and K2NaSbBr6, respectively. Furthermore, in all cases, the absorption coefficient drastically increases in the UV spectrum. Accordingly, considering the band gap energy value and the optical absorption spectrum, the K2NaSbI6 is highly adequate for PV applications.
3.5.7 Optical conductivity
The optical conductivity σ (ω) refers to the bonds breaking process when the incident photons are absorbed and reveals fruitful information about the generated free charge carriers. Moreover, as depicted in Fig. 4h, the frequency/energy-dependent σ (ω) trend is directly connected to α (ω). In the VIS region, K2NaSbI6 (K2NaSbBr6) show maximum σ (ω) of 2276.32 (206.58 (Ω.cm)−1), indicating a higher absorption capability of the interested materials. Besides, the highest optical conductivities were observed at the UV region with magnitudes of 5721.26 (5713.56 (Ω.cm)−1) for K2NaSbI6 (K2NaSbBr6). We note that the energy-dependent σ (ω) trends of K2NaSbI6 are alike but lager than that of K2NaSbBr6. Also, in both VIS and UV ranges, the highest peak values of K2NaSbI6 are larger than that of K2NaSbBr6, which may be attributed to the carrier’s density of these materials. Thus, the present HDPs are optically active with spectrum energy, which promotes their route towards PV and optoelectronic technologies even at high frequencies.
3.6 Transport properties
Using the mathematical formalism reported elsewhere (Allen et al. 1996; Ziman 2001; Hurd 1972; Madsen et al. 2006b), we have comprehensively evaluated the electronic transport and TE performance of K2NaSbZ6 (Z = Br, I) HDPs in terms of Seebeck coefficient (S), electric (σ/τ) and electronic thermal (κe/τ) conductivities, figure of merit (ZT = σS2T/κ), and TE power factor (PF = S2σ/τ). In the present calculations, the relaxation time we have used is τ = 1014 s. The computed electronic transport and TE features of K2NaSbBr6 and K2NaSbI6 against the chemical potential µ (eV) at four selected temperatures (300, 500, 700, and 900 K) are illustrated in Figs. 5a–e and 6a–e, respectively. The negative and positive µ differentiate between the p-type and n-type doping regions. The µ (eV) = µF (eV) = 0 for a pristine compound is indicated by the vertical dotted lines.

Thermoelectric properties of K2NaSbBr6 HDPs with respect to μ − μF at various selected temperatures computed using the TB-mBJ-GGA method

Thermoelectric properties of K2NaSbI6 HDPs with respect to μ − μF at various selected temperatures computed using the TB-mBJ-GGA method
3.6.1 Seebeck coefficient
The Seebeck coefficient (S) is the pivotal parameter used for evaluating the TE conversion efficiency of materials. It measures the magnitude of the potential difference induced by the temperature gradient pending the energy transfer across the ends of a conductor. However, positive S (negative S) suggests that the conduction is mainly due to holes (electrons). The S (μV/K) against μ (eV) is demonstrated in Figs. 5a and 6a for K2NaSbBr6 and K2NaSbI6, respectively, at 300, 500, 700 and 900 K. In the p-type doping region, the magnitude of S remains almost zero. Nevertheless, for n-type doping, S exhibits prominent peaks and valleys throughout the entire considered μ region. The observed high values at 300 K are 2984.8 (− 2826.05 μV/K) and 2965.75 (− 2782.97 μV/K) for K2NaSbBr6 and K2NaSbI6, respectively. Furthermore, the peak values gradually decline as the temperature rises reaching 1678.15 (− 1716.55 μV/K) and 1255.43 (− 1287.03 μV/K), respectively, for K2NaSbBr6 and K2NaSbI6 at 900 K. The positive values show that holes are the predominant carriers, confirming that both compounds are p-type SCs. For pristine compounds (μ = μF = 0), it is found that S decreases linearly from 180.88 (183.84 μV/K) to 145.62 (155.87μV/K) for K2NaSbBr6 (K2NaSbI6) with an increase in temperature from 300 to 900 K. This behavior is ascribed to the fact that with the increased temperature, the bound electrons were thermally excited inducing electron–hole pairs. Also, on comparing the reported S magnitudes over the whole temperature range, it can be concluded that K2NaSbI6 shows a good TE response as compared to K2NaSbBr6.
3.6.2 Electrical conductivity
Next, a good TE material also possesses a sufficiently high electrical conductivity (σ/τ) directly linked to ZT. The variation of σ/τ as a function of μ for K2NaSbBr6 and K2NaSbI6 at 300, 500, 700 and 900 K is plotted in Figs. 5b and 6b, respectively. We can see that, the μ-dependent σ/τ is similar for selected temperatures. Moreover, in the p-type doping region (μ < μF), σ/τ reaches its highest peak with maximum values of 8.47 × 1019 (Ωms)−1 observed at − 1.64 eV for K2NaSbBr6 and 9.79 × 1019 (Ωms)−1 at − 0.83 eV for K2NaSbI6, indicating the highest available density of states. Furthermore, the present HDPs show some insulating response due to the disappearance of the density of states. Nevertheless, beyond this interval, σ/τ reaches its highest value of 5.26 × 1019 (Ωms)−1 at 3.88 eV for K2NaSbBr6 and 6.14 × 1019 (Ωms)−1 at 3.21 eV for K2NaSbI6. Furthermore, at μ = μF = 0, with increasing temperature, σ/τ demonstrates smooth behavior. The observed smaller increase with temperature suggests the typical SC character of these compounds. Throughout, the considerable σ/τ peak intensity observed for K2NaSbI6 is ascribed to the fact that the band gap shrinks on going from Br to I. Therefore, requiring less excitation energy, the free carrier density rises, giving rise to the increased σ/τ.
3.6.3 Electronic thermal conductivity
The total thermal conductivity (κ) consists of a lattice component assigned to lattice phonons (κL) and an electronic component (κe) attributed to the motion of charge carriers.
In this work, we have only considered the electronic component of κ against chemical potential and temperature for K2NaSbZ6 (Z = Br, I) HDPs as depicted in Figs. 5c and 6c. The κe/τ and σ/τ parameters depend mainly on the carrier density. So, they follow a related behavior with respect to μ. However, κe/τ rises abruptly with temperature compared to σ/τ, indicating that κe/τ is mainly attributed to the free carriers. This behavior fits well with the Wiedemann–Franz law which reports the proportionality between them expressed as follows: κ = σLT, where L = 2.44 × 10−8 J2K−2C−2 is the Lorentz number for free electrons. Furthermore, the ratio of σ/τ to κe/τ is extremely considerable (105), which ensure the significant TE performance of the studied HDPs. Moreover, in the p-type doping region (μ < μF), the observed highest peak values are 17.23 × 1014 (W/mKs) at − 1.03 eV and 13.76 × 1014 (W/mKs) at − 1.80 eV for K2NaSbI6 and K2NaSbBr6, respectively. Similarly, in both regions, we examined the same behavior observed in σ/τ; the κe/τ peak values of K2NaSbI6 are larger than those of K2NaSbBr6.
3.6.4 Thermoelectric power factor
The power factor (PF = S2σ/τ) is generally used to evaluate the TE performance of materials without incorporating the lattice vibration and current effects. A material which efficiently extracts heat has higher PF values. The plots of the PF for K2NaSbZ6 (Z = Br, I) materials concerning μ are depicted in Figs. 5d and 6d. Therefore, the PF values for both materials are relatively higher for the n-type doping region than the p-type and increased with temperature. At μ = μF = 0, the room temperature PF for K2NaSbBr6 and K2NaSbI6 materials are 1.59 × 1011 W/mK2s and 1.51 × 1011 W/mK2s, respectively.
3.6.5 Figure of merit
The dimensionless TE figure of merit ZT = S2σT/κ, encompassing all these parameters, is the main factor employed to scrutinize the TE conversion efficiency of materials. The enhanced TE efficiency can be obtained with high σ/τ and low κe/τ. The ZT values of K2NaSbBr6 and K2NaSbI6 against μ are plotted in Figs. 5e and 6e. It has been noticed that, in all circumstances, ZT achieves the highest value for n-type doping. Also, both considered HDPs materials possess the highest ZT close to 1 for the μ regions corresponding to the highest S values. For pristine compounds, the ZT values are nearly constant under the temperature effect and decrease from approximately 0.723 to 0.721 ongoing from K2NaSbBr6 to K2NaSbI6. Hence, it is predicted that the HDPs under investigation have a good TE response.
4 Conclusion
Lead-free double perovskites have become an emerging aspirant for photovoltaic conversion and thermoelectric applications. In the present article, structural, elastic, electronic, optical, and TE properties of K2NaSbZ6 (Z = Br, I) have been investigated. The elastic constants, tolerance factors and negative formation energy confirm the mechanical, structural, and thermodynamic stabilities. The Poisson and Pugh ratios confirm the studied compounds' ductile nature, while large values of Debye and melting temperature ensure their existence at high temperatures. The double perovskites containing Br and I have band gaps of 3.20 and 2.40 eV, respectively. Their absorption regions extend from visible to ultraviolet light, which enhances their suitability for optoelectronic applications such as solar cells. Finally, the large values of the figure of merit are also important for thermoelectric generators. Therefore, the broad absorption bands in the visible region and large figures of merit provide significant motivation for the researchers to realize them for energy harvesting.
References
Albanesi, E.A., Blanca, E.P., Petukhov, A.G.: Calculated optical spectra of IV–VI semiconductors PbS, PbSe and PbTe. Comput. Mater. Sci. 32, 85–95 (2005)
Allen, P.B.: Boltzmann theory and resistivity of metals. In: Chelikowsky, J.R., Louie, S.G. (eds.) Quantum Theory of Real Materials, pp. 219–250. Klüwer, Boston (1996)
Al-Muhimeed, T.I., Aljameel, A.I., Mera, A., Saad, S., Nazir, G., Albalawi, H., Mahmood, Q.: First principle study of optoelectronic and mechanical properties of lead-free double perovskites Cs2SeX6 (X= Cl, Br, I). J. Taibah Univ. Sci. 16(1), 155–162 (2022)
Al-Qaisi, S., Rai, D., Haq, B.U., Ahmed, R., Vu, T.V., Khuili, M., Tahir, S.A., Alhashim, H.H.: First-principles investigation of structural, elastic, thermodynamic, electronic and optical properties of lead-free double perovskites halides: Cs2LiYX6 (X= Br, I). Mater. Chem. Phys. 258, 123945–123954 (2021)
Al-Qaisi, S., Mahmood, Q., Kattan, N.A., Alhassan, S., Alshahrani, T., Sfina, N., Brini, S., Hakamy, A., Mera, A., Amin, M.A.: Tuning of band gap by variation of halide ions in K2CuSbX6 (X= Cl, Br, I) for solar cells and thermoelectric applications. J. Phys. Chem. Solids 174, 111184–111191 (2023)
Anbarasan, R., Sundar, J.K., Srinivasan, M., Ramasamy, P.: First principle insight on the structural, mechanical, electronic and optical properties of indirect band gap photovoltaic material Cs2NaBiX6 (X= Cl, Br, I). Comput. Condens. Matter. 28, e00581 (2021)
Aslam, F., Sabir, B., Hassan, M.: Structural, electronic, optical, thermoelectric, and transport properties of indium-based double perovskite halides Cs2InAgX6 (X= Cl, Br, I) for energy applications. Appl. Phys. A 127, 1–12 (2021)
Aswal, D.K., Basu, R., Singh, A.: Key issues in development of thermoelectric power generators: high figure-of-merit materials and their highly conducting interfaces with metallic interconnects. Energ. Conver. Manage. 114, 50–67 (2016)
Babayigit, A., Thanh, D.D., Ethirajan, A., Manca, J., Muller, M., Boyen, H.G., Conings, B.: Assessing the toxicity of Pb- and Sn- based perovskite solar cells in model organism danio rerio. Sci. Rep. 6, 1–11 (2016a)
Babayigit, A., Ethirajan, A., Muller, M., Conings, B.: Toxicity of organometal halide perovskite solar cells. Nat. Mater. 15, 247–251 (2016b)
Bartel, C.J., Sutton, C., Goldsmith, B.R., Ouyang, R., Musgrave, C.B., Ghiringhelli, L.M., Scheffler, M.: New tolerance factor to predict the stability of perovskite oxides and halides. Sci. Adv. 5, eaav0693 (2019)
Bibi, A., Lee, I., Nah, Y., Allam, O., Kim, H., Quan, L.N., Kim, D.H.: Lead-free halide double perovskites: toward stable and sustainable optoelectronic devices. Mater. Today 49, 123–144 (2021)
Bildirici, M.E., Gökmenoğlu, S.M.: Environmental pollution, hydropower energy consumption and economic growth: evidence from G7 countries. Renew. Sust. Energ. Rev. 75, 68–85 (2017)
Bobrov, V.B., Trigger, S.A., Van Heijst, G.J.F., Schram, P.P.J.M.: Kramers-Kronig relations for the dielectric function and the static conductivity of Coulomb systems. EPL 90, 10003–10007 (2010)
Bouich, A., Marí-Guaita, J., Sahraoui, B., Palacios, P., Marí, B.: Tetrabutylammonium (TBA)-doped methylammonium lead iodide: high quality and stable perovskite thin films. Front. Energy Res. 10, 840817 (2022)
Boutramine, A.: Temperature-and composition-dependent band gap energy and electron-phonon coupling in InAs1−xSbx semiconductors alloys for infrared photodetection. J. Electron. Mater. 52, 6031–6041 (2023)
Cao, J., Yan, F.: Recent progress in tin-based perovskite solar cells. Energy Environ. Sci. 14(3), 1286–1325 (2021)
Chen, X., Wang, C., Li, Z., Hou, Z., Yin, W.J.: Bayesian optimization based on a unified figure of merit for accelerated materials screening: A case study of halide perovskites. Sci. China Mater. 63, 1024–1035 (2020)
Chu, L., Ahmad, W., Liu, W., Yang, J., Zhang, R., Sun, Y., Yang, J., Li, X.: Lead-free halide double perovskite materials: a new superstar toward green and stable optoelectronic applications. Nano-Micro Letters. 11, 1–18 (2019)
Dar, S.A., Sharma, R., Srivastava, V., Sakalle, U.K.: Investigation on the electronic structure, optical, elastic, mechanical, thermodynamic and thermoelectric properties of wide band gap semiconductor double perovskite Ba2InTaO6. RSC Adv. 9, 9522–9532 (2019)
Demic, S., Ozcivan, A.N., Can, M., Ozbek, C., Karakaya, M.: Recent progresses in perovskite solar cells. In: Das, N. (ed.) Nanostructured Solar Cells, pp. 277–304. Rijeka, Intech (2017)
Dreesel, M., Gruner, M.G.: Electrodynamics of Solids: Optical Properties of Electrons in Matter. Cambridge University Press, Cambridge (2002)
Du, K.Z., Meng, W., Wang, X., Yan, Y., Mitzi, D.B.: Bandgap engineering of lead-free double perovskite Cs2AgBiBr6 through trivalent metal alloying. Angew. Chem. Int. Ed. 56(28), 8158–8162 (2017)
Faizan, M., Murtaza, G., Khan, S.H., Khan, A., Mehmood, A., Khenata, R., Hussain, S.: First-principles study of the double perovskites Sr2XOsO6 (X= Li, Na, Ca) for spintronics applications. Bull. Mater. Sci. 39, 1419–1425 (2016)
Fedorovskiy, A.E., Drigo, N.A., Nazeeruddin, M.K.: The role of Goldschmidt’s tolerance factor in the formation of A2BX6 double halide perovskites and its optimal range. Small Methods. 4, 1900426–1900432 (2020)
Gajdoš, M., Hummer, K., Kresse, G., Furthmüller, G.J., Bechstedt, F.: Linear optical properties in the projector-augmented wave methodology. Phys. Rev. B 73, 045112–045120 (2006)
Ghosh, S., Shankar, H., Kar, P.: Recent developments of lead-free halide double perovskites: a new superstar in the optoelectronic field. Mater. Adv 3, 3742–3765 (2022)
Goldsmid, H.J.: Introduction to Thermoelectricity. Springer, Berlin (2010)
Goldsmid, H.J.: Improving the thermoelectric figure of merit. Sci. Technol. Adv. Mater. 22, 280–284 (2021)
Goldsmid, H.J.: The seebeck and peltier effects In The Physics of Thermoelectric Energy Conversion IOP Concise Physics, 2053–2571 (2017)
Gui, Y., Yao, Y., Geng, H., Zhang, J., Yuan, D.: Systematic investigation of the mechanical, electronic structure and optical properties of halide double perovskite Cs2BSbBr6 (B= Li, Na, K, Rb) with first-principles method. Int. J. Mod. Phys. C 05, 2350062 (2023)
Hilal, M., Rashid, B., Khan, S.H., Khan, A.: Investigation of electro-optical properties of InSb under the influence of spin-orbit interaction at room temperature. Mater. Chem. Phys. 184, 41–48 (2016)
Hill, R.: The elastic behaviour of a crystalline aggregate. Proc. Phys. Soc. a. 65, 349 (1952)
Hurd, C.M.: The Hall Effect in Metals and Alloys. Plenum Press, New York-London (1972)
Ivanovski, K., Hailemariam, A., Smyth, R.: The effect of renewable and non-renewable energy consumption on economic growth: non-parametric evidence. J. Clean. Prod. 286, 124956–124970 (2021)
Jamal, M., Asadabadi, S.J., Ahmad, I., Aliabad, H.R.: Elastic constants of cubic crystals. Comput. Mater. Sci. 95, 592–599 (2014)
Jasiukiewicz, C., Karpus, V.: Debye temperature of cubic crystals. Solid State Commun. 128, 167–169 (2003)
Ke, W., Stoumpos, C.C., Kanatzidis, M.G.: “Unleaded” perovskites: status quo and future prospects of tin-based perovskite solar cells. Adv. Mater. 31(47), 1803230 (2019)
Khan, I., Shahab, I.U., Haq, A., Ali, Z., Ali, I.: Ahmad, Elastic and optoelectronic properties of Cs2NaMCl6 (M = In, Tl, Sb, Bi). J. Electron. Mater. 50(2), 456–466 (2021)
Kim, G.H., Kim, D.S.: Development of perovskite solar cells with> 25% conversion efficiency. Joule 5, 1033–1035 (2021)
Kopacic, I., Friesenbichler, B., Hoefler, S.F., Kunert, B., Plank, H., Rath, T., Trimmel, G.: Enhanced performance of germanium halide perovskite solar cells through compositional engineering. ACS Appl. Energy Mater. 1(2), 343–347 (2018)
Kung, P.K., Li, M.H., Lin, P.Y., Jhang, J.Y., Pantaler, M., Lupascu, D.C., Chen, P.: Lead free double perovskites for perovskite solar cells. Sol. RRL 4(2), 1900306–1900337 (2020)
Li, X.Q., Zhao, J.J., Xu, J.C.: Mechanical properties of bcc Fe-Cr alloys by first-principles simulations. Front. Phys. 7, 360–365 (2012)
Liu, W., Kim, H.S., Jie, Q., Ren, Z.: Importance of high power factor in thermoelectric materials for power generation application: a perspective. Scr. Comput. Sci. Appl. Math. Materialia 111, 3–9 (2016)
Liu, C., Li, W., Fan, J., Mai, Y.: A brief review on the lead element substitution in perovskite solar cells. J. Energy Chem. 27(4), 1054–1066 (2018)
Lu, L., Pan, X., Luo, J., Sun, Z.: Recent advances and optoelectronic applications of lead-free halide double perovskites, Chemistry–A. Eur. J. 26, 16975–16984 (2020)
Lu, R., Liu, Y., Zhang, J., Zhao, D., Guo, X., Li, C.: Highly efficient (200) oriented MAPbI3 perovskite solar cells. J. Chem. Eng. 433, 133845 (2022)
Luan, X., Qin, H., Liu, F., Dai, Z., Yi, Y., Li, Q.: The mechanical properties and elastic anisotropies of cubic Ni3Al from first principles calculations. Crystals 8, 307 (2018)
MacDonald, D.K.C.: Thermoelectricity: An Introduction to the Principles, Dover Publications, Inc. Mineola, New York (2006)
Madsen, G.K., Singh, D.J.: BoltzTraP. A code for calculating band-structure dependent quantities. Comput. Phys. Commun. 175, 67–71 (2006a)
Maiti, T., Saxena, M., Roy, P.: Double perovskite (Sr2B′B′′O6) oxides for high temperature thermoelectric power generation—a review. J. Mater. Res. 34, 107–125 (2019)
Mantziaris, S., Iliopoulos, C., Theodorakopoulou, I., Petropoulou, E.: Perennial energy crops versus durum wheat in low input lands: economic analysis of a Greek case study. Renew. Sust. Energ. Rev. 80, 789–800 (2017)
Materials Project, Data retrieved from the Materials Project for K2NaSbI6 (mp-1110961) from database version v2022.10.28. (2023).
Materials Project, Data retrieved from the Materials Project for K2NaSbBr6 (mp-1110891) from database version v2022.10.28.(2023)
Mera, A., Almeshal, A., Rouf, S.A., Zelai, T., Aljameel, A.I., Hakami, O., Mahmood, Q.: Modification of band gaps by changing anions to optimize the double perovskites K2NaTlX6 (X= Cl, Br, I) for solar cells and transport applications. Chem. Phys. Lett. 56, 140754–140762 (2023)
Mirershadi, S., Ahmadi-Kandjani, S., Zawadzka, A., Rouhbakhsh, H., Sahraoui, B.: Third order nonlinear optical properties of organometal halide perovskite by means of the Z-scan technique. Chem. Phys. Lett. 647, 7–13 (2016)
Mostafaeipour, A., Bidokhti, A., Fakhrzad, M.B., Sadegheih, A., Mehrjerdi, Y.Z.: A new model for the use of renewable electricity to reduce carbon dioxide emissions. Energy 238, 121602–121618 (2022)
Murnaghan, F.: The compressibility of media under extreme pressures. Proc. Natl. Acad. Sci. u.s.a. 30, 244 (1944)
Penn, D.R.: Wave-number-dependent dielectric function of semiconductors. Phys. Rev. 128, 2093–2097 (1962)
Perdew, J.P., Burke, K., Ernzerhof, M.: Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865–3868 (1996)
Pettifor, D.: Theoretical predictions of structure and related properties of intermetallics. Mater. Sci. Technol. 8, 345–349 (1992)
Pugh, S.: XCII Relations between the elastic moduli and the plastic properties of polycrystalline pure metals. Lond. Edinb. Dublin Phil. Mag. J. Sci. 45, 823–843 (1954)
Ranganathan, S.I., Ostoja-Starzewski, M.: Universal elastic anisotropy index. Phys. Rev. Lett. 101, 055504–055507 (2008)
Schreiber, E., Anderson, O.L., Soga, N., Bell, J.F.: Elastic constants and their measurement. J. Appl. Mech. 42, 747–748 (1975)
Schwarz, K., Blaha, P., Madsen, G.K.: Electronic structure calculations of solids using the WIEN2k package for material sciences. Comput. Phys. Commun. 147, 71–76 (2002)
Senocrate, A., Kim, G.Y., Gratzel, M., Maier, J.: Thermochemical stability of hybrid halide perovskites. ACS Energy Lett. 4(12), 2859–2870 (2019)
Shahbazi, M., Wang, H.: Progress in research on the stability of organometal perovskite solar cells. Sol. Energy 123, 74–87 (2016)
Shao, S., Liu, J., Portale, G., Fang, H.-H., Blake, G.R., ten Brink, G.H., Koster, L.J.A., Loi, M.A.: Highly reproducible Sn-based hybrid perovskite solar cells with 9% effciency. Adv. Energy Mater. 8, 1702019–1702028 (2018)
Shi, H., Du, M.H.: Discrete electronic bands in semiconductors and insulators: potential high-light-yield scintillators. Phys. Rev. Appl. 3(5), 054005–054014 (2015)
Shi, W., Cai, T., Wang, Z., Chen, O.: The effects of monovalent metal cations on the crystal and electronic structures of Cs2MBiCl6 (M= Ag, Cu, Na, K, Rb, and Cs) perovskites. J. Chem. Phys. 153, 141101–141108 (2020)
Singh, B.P., Goyal, S.K., Kumar, P.: Solar PV cell materials and technologies: Analyzing the recent developments. Mater. Today Proc. 43, 2843–2849 (2021)
Slavney, A.H., Hu, T., Lindenberg, A.M., Karunadasa, H.I.: A bismuth-halide double perovskite with long carrier recombination lifetime for photovoltaic applications. J. Am. Chem. Soc. 138, 2138–2141 (2016)
Snaith, H.J.: Perovskites: the emergence of a new era for low-cost, high-efficiency solar cells. J. Phys. Chem. Lett. 4, 3623–3630 (2013)
Srivastava, R.: Perovskite as light harvester: Prospects, efficiency, pitfalls and roadmap. In: Das, N. (ed.) Nanostructured Solar Cells, pp. 245–276. Rijeka, Intech (2017)
Tarbi, A., Chtouki, T., Bouich, A., Elkouari, Y., Erguig, H., Migalska-Zalas, A.: Prediction of mechanical properties of In1-x GaxAsyP1-y lattice-matched to different substrates using artificial neural network (ANN). Adv. Mater. Process. 9, 1437–1447 (2022)
Tarbi, A., Chtouki, T., Sellam, M.A., Elkouari, Y., Erguig, H., Migalska-Zalas, A.: Predicting the bandgap energy of distorted GaSbxAs1-x and InSbxAs1-x using design of experiment (DoE) and artificial intelligence (AI): A comparative study. J. Phys. Chem. Solids 175, 111180 (2023a)
Tarbi, A., Chtouki, T., Erguig, H., Migalska-Zalas, A., Aissat, A.: Modeling and optimization of Sb and N resonance states effect on the band structure of mismatched III-NV alloys using artificial neural networks. Mater. Sci. Eng. B 290, 116312 (2023b)
Tarbi, A., Chtouki, T., Sellam, M.A., Benahmed, A., El Kouari, Y., Erguig, H., Migalska-Zalas, A., Goncharova, I., Taboukhat, S.: Optimization of ultra-thin CIGS-based solar cells by strained In1− xGaxAs absorption layer: 1D SCAPS modeling. J. Comput. Electron. 12, 1–8 (2023c)
Tarbi, A., Chtouki, T., Sellam, M.A., Benahmed, A., Kouari, Y.E., Erguig, H., Migalska-Zalas, A., Goncharova, I., Taboukhat, S., Tlemçani, M.: Deformed ternary phosphides III-P for efficient light control in optoelectronic applications. Lasers Manuf. Mater. Process. 10, 471–484 (2023d)
Tariq, M., Ali, M.A., Laref, A., Murtaza, G.: Anion replacement effect on the physical properties of metal halide double perovskites Cs2AgInX6 (X= F, Cl, Br, I). Solid State Commun. 314, 113929–113936 (2020)
Tran, F., Blaha, P.: Accurate band gaps of semiconductors and insulators with a semilocal exchange-correlation potential. Phys. Rev. Lett. 102, 226401–226405 (2009)
Wang, J., Yip, S., Phillpot, S.R., Wolf, D.: Crystal instabilities at finite strain. Phys. Rev. Lett. 71, 4182–4185 (1993)
Waqas Mukhtar, M., Ramzan, M., Rashid, M., Naz, G., Imran, M., Fahim, F., AlObaid, A.A., Al-Muhimeed, T.I., Mahmood, Q.: New lead-free double perovskites A2NaInI6 (A = Cs, Rb) for solar cells and renewable energy; first principles analysis. Mater. Sci. Eng. B 273, 115420–115425 (2021)
Waszkowska, K., Guichaoua, D., Jędryka, J., Syvorotka, I., Syvorotka, N.Y., Kityk, A.V., Sahraoui, B.: Second-and third-order nonlinear optical response of perovskite LiTaO3. In 22nd International Conference on Transparent Optical Networks (ICTON). IEEE. 1–4. (2020)
Wu, Y., Li, X., Zeng, H.: Lead-free halide double perovskites: structure, luminescence, and applications. Small. Stru. 2, 2000071 (2021)
Xiao, X., Liang, S., Si, J., Xu, Q., Zhang, H., Ma, L., Yang, C., Zhang, X.: Performance of LiTaO3 crystals and thin films and their application. Cryst. 13, 1233 (2023)
Yan, L., Zhao, L., Zhao, C., Lin, S.: Theoretical understanding of thermoelectric energy conversion efficiency in lead-free halide double perovskites showing intrinsic defect tolerance. Appl. Therm. Eng. 215, 119024–119034 (2022)
Yu, Y.P., Cardona, M.: Fundamental of Semiconductor Physics and Materials Properties. Springer, Berlin (1999)
Zhang, L., Dai, X., Li, T., Li, J., Li, H.: CH3NH3PbX3 (X= I, Br) encapsulated in silicon carbide/carbon nanotube as advanced diodes. Sci. Rep. 8, 15187 (2018)
Zhao, L.D., Lo, S.H., Zhang, Y., Sun, H., Tan, G., Uher, C., Wolverton, C., Dravid, V.P., Kanatzidis, M.G.: Ultralow thermal conductivity and high thermoelectric figure of merit in SnSe crystals. Nature 508, 373–377 (2014)
Zhao, S., Yamamoto, K., Iikubo, S., Hayase, S., Ma, T.: First-principles study of electronic and optical properties of lead-free double perovskites Cs2NaBX6 (B= Sb, Bi; X= Cl, Br, I). J. Phys. Chem. Solid 117, 117–121 (2018)
Ziman, J.M.: Electrons and Phonons: The Theory of Transport Phenomena in Solids. Oxford University Press, London (2001)
Acknowledgements
The authors extend their sincere appreciation to Princess Nourah bint Abdulrahman University Researchers Supporting Project number (PNURSP2024R71), Princess Nourah bint Abdulrahman University, Riyadh, Saudi Arabia. This study is supported via funding from Prince Sattam bin Abdulaziz University project number (PSAU/2023/R/1445).
Author information
Authors and Affiliations
Contributions
AB: Conceptualization, formal analysis, methodology, writing—original draft, writing—review & editing. SA: Data curation, writing—review & editing, visualization, investigation, software. SS: Data curation, writing—review & editing. NI: Data curation, writing—review & editing. TAA: Data curation, writing—review & editing. SB: Data curation, writing—review & editing. ASV: Writing—review & editing, methodology. SB: Visualization, data curation. RS: Software, writing—review & editing.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Conflict of interests
The authors have no conflict of interest to declare that are relevant to the content of this article.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Boutramine, A., Al-Qaisi, S., Samah, S. et al. Optoelectronic and thermoelectric properties of new lead-free K2NaSbZ6 (Z = Br, I) halide double-perovskites for clean energy applications: a DFT study. Opt Quant Electron 56, 417 (2024). https://doi.org/10.1007/s11082-024-06344-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11082-024-06344-4