
Abstract
Role of autophagy in Japanese encephalitis viral (JEV) infection is not well known. In the present study, we reported the role of autophagy flux in microglia activation, neurobehavioral function and neuronal death using a mouse model of JEV. Markers for autophagy (LC3-II/I, SQSTM1/P62, phos-Akt, phos-AMPK), and neuronal death (cleaved caspase 12, H2Ax, polyubiquitin) were investigated by western blot at 1, 3 and 7 days post inoculation. Cathepsin D was measured in cerebral cotex of JEV infected mice spectrophotometrically. Microglia activation and pro-inflammatory cytokines (IL1β, TNF-α, IFNγ, IL6) were measured by immunohistochemistry, western blot and qPCR analysis. In order to determine the neuroinflammatory changes and autophagy mediated neuronal cell death, BV2-microglia and N2a-neuronal cells were used. Autophagy activation marker LC3-II/I and its substrate SQSTM1/P62 were significantly increased while cathepsin D activity was decreased on day 7 post inoculation in cerebral cortex. Microglia in cortex were activated and showed higher expression of proinflammatory mRNA of IL1β, TNF-α, IFNγ and IL6, with increased DNA damage (H2AX) and neuronal cell death pathways in hippocampus and neurobehavioral dysfunction. Similar observations on JEV infection mediated autophagy flux inhibition and neuronal cell death was found in N2a neuronal cell. Collectively, our study provides evidence on the role of autophagy regulation, microglial activation and neurodegeneration following JEV infection.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Autophagy is conserved cellular catabolic mechanism by which cells remove damaged organelle and toxic molecules to maintain the cellular homeostasis. It is initiated by the formation of double membrane vesicle which is called autophagosome that fuses with lysosome and leads to degradation by lysosomal hydolases. Autophagy plays key role in host pathogen relationship and its alteration has been implicated in number of neuroviral infectious diseases [1, 2] such as, relocation of virus is impaired by autophagy pathways whereas some other studies demonstrated that it is used for viral replication. For instance, herpes simplex virus 1 (HSV-1) evades the host autophagy and initiates cytolytic infection in epithelial cells. It has also been reported that dengue and hepatitis C viruses get benefited from autophagy and use the by-product of autophagy to enhance their replication [3,4,5]. Japanese encephalitis (JE) virus has been found to use autophagy pathways as the virus particles are endogenously processed in to autophagosome during viral infection [6], suggesting that host autophagy cellular response is significantly modulated by the virus infection and plays key role in development of disease pathology.
The frequency of cognitive impairment and neurobehavioral dysfunction depends on the severity of JE virus infection to the specific area of the nervous system such as, hippocampus, thalamus and basal ganglia and brainstem [7,8,9]. Progressive neuroinflammation and microglia activation are key components in severe form of JE virus infection and its intervention studies are found to be beneficial for neuronal survival and neurobehavioral functions [9]. In recent study it is observed that complement proteins C3, associated with the inflammatory neurodegeneration pathways is significantly increased in CSF of JE patients [10]. JE virus infection is also associated with increased autophagy markers and activation of neuronal cell death pathways [11,12,13]. However the detailed molecular mechanism needs further evaluation on the role of autophagy in inflammatory response of microglial activation following JE viral infection. In the present study we reported the role of autophagy and microglial activation in neuronal function and its outcome.
Material Methods
Virus
An Indian neurovirulent, GP 78668A (GP78) strain of JEV from Department of Microbiology, Sanjay Gandhi postgraduate institute of medical sciences (SGPGIMS) was used in the study. Virus was propagated in 3–4 days old suckling mice brain. A total of 20 µl from stock was inoculated intracerebrally in 3–4 days old suckling mice. After 4 days of infection; the mice were deeply anesthetized with chloroform and sacrificed. Brain was removed aseptically and homogenized in sterile phosphate buffer saline (PBS) and were centrifuged at 15,000 g for 30 min at 8 °C. The supernatant was collected, aliquoted and stored at − 80 °C till further use. Virus titer was determined by the standard plaque assay [14, 15].
Animal
Two to three weeks old BALB/c mice were used throughout the study. Mice were procured and housed in animal care facility of SGPGIMS, Lucknow. Mice were fed with protein rich diet and water ad libitum. The animals were maintained in an air conditioned room (25 ± 2 °C) with 12 h light (7 a.m.–7 p.m.) and dark cycle. All the experiments were performed during the day light cycle. The study was approved by the institutional ethics committee, and all the experiments were carried out in accordance with the institutional guidelines on the care and use of experimental animals.
JE Virus Inoculation
BALB/c mice were divided into two groups: JEV infected group and mock infected group. In JEV infected group, the mice were inoculated intracerebrally with 3 × 105 plaque forming units (PFU) of JE virus whereas control mice were inoculated with sterile 1XPBS (Sigma, USA) as described before in our previous study [14]. Mice were monitored and eye response, sitting position, body appearances including hair straightening and body weight were measured daily and were sacrificed on days 1, 3 and 7 post-inoculation. The brains were excised aseptically and were processed for biochemical analysis.
Study 1: Sham and JEV infected mice (n = 6/group) were used for behavioral studies. At 7 days post infection, mice were transcardially perfused with ice-cold 0.9% saline, followed by 4% paraformaldehyde. Brains were removed and post-fixed in 4% paraformaldehyde overnight, and cryoprotected in 30% sucrose and were processed for microglia cell phenotype analysis.
Study 2: Sham and JEV infected (n = 3–4/time point/group) mice were transcardially perfused with ice-cold 0.9% saline at each 1, 3 and 7 day post-infection. Ipsilateral cortical tissue was rapidly dissected for cathepsin D (CTSD) assay and snap-frozen on liquid nitrogen for RNA extraction and western blotting.
Locomotion Functional Analysis
Pattern of motor coordination and locomotion of control and JE virus infected mice were analyzed and recorded using horizontal plane videography. Both JEV infected and control mice were placed on a horizontal plate with a video camera mounted at a distance to capture the image of the animals. The locomotion and its subphases which ranges from braking, and propulsion were recorded. The portions of the paws that were in contact with the treadmill belt were recorded as the stance phase of the stride while the portions that were not in contact with the treadmill belt were recorded as the swing phase of the stride. Kinematic matrix of the gait dynamics were noted in the form of step length (cm).
BV2 Microglia and N2a Neuronal Cell Culture
BV2 microglia (murine microglia cell line) and Neuro2A cells were grown in Dulbecco’s modified Eagle’s medium (DMEM, Invitrogen, Carlsbad, CA) supplemented with 10% fetal equine serum (HyClone, Logan, UT), 1% penicillin and streptomycin (Invitrogen) at 37 °C with 5% CO2 and were treated with mock or JEV (at a multiplicity of infection (MOI) of 10 TCID50/cell) for various time-periods.
Nitric Oxide Assay
Nitric oxide (NO) released into BV2 microglia condition media and blood of control and both JEV infected animals were assayed using a Greiss reagent assay (Invitrogen; G7921), as per the manufacturer’s instructions. NO concentrations were calculated using standard curves generated from a nitrite stock, and results were expressed in micromoles.
Real-Time PCR
Total RNA was extracted from snap-frozen control and JEV infected mice using an RNeasy isolation kit (Qiagen, Valencia, CA) with on-column DNAase treatment (Qiagen, Valencia, CA). cDNA synthesis was performed on 1 µg of total RNA using a Verso cDNA RT kit (Thermo Scientific, Pittsburg, PA); the protocols used were in accordance to the manufacturer’s instructions. Real-time PCR was performed using an ABI 7500 Sequence Detection System (Applied-Biosystems) in the presence of SYBR-green. Standard PCR conditions were used as prescribed in SYBR-Green I core reagent protocol. PCR was performed using nucleotide primers of CD11b, IL1β, TNF-α, IFNγ, IL6 and TLR4. Gene expression was calculated relative to the endogenous control sample (GAPDH) to determine relative expression values, using the 2−ΔΔCt method (where Ct is the threshold cycle).
Western Blotting
Proteins from ipsilateral cortical tissue were extracted using RIPA buffer, equalized, and loaded onto 5–20% gradient gels for SDS PAGE (Bio-Rad; Hercules, CA). Proteins were transferred onto nitrocellulose membranes, and then blocked overnight in 5% milk in 1X TBS containing 0.05% Tween-20 (PBS-T). The membrane was incubated in mouse anti-LC3-I/II (1:1000; BD Transduction Laboratories), anti-SQSTM1/P62 (1:1000; Stem Cell Technologies, Vancouver, BC), anti-Phospho Akt (1:1000; Cell Signaling Technology), rabbit anti-total Akt (1:1000; Cell Signaling Technology), rabbit anti-Phospho AMPK (1:1000; Cell Signaling Technology), rabbit anti-total AMPK (1:1000; Cell Signaling Technology), rabbit anti-doublecortin (1:1000; Cell Signaling Technology), rabbit anti IBA1 (1:1000; BD Transduction Laboratories), mouse anti-caspase 12 (1:1000; Cell Signaling Technology), mouse anti-ubiquitin (1:1000; Cell Signaling Technology), mouse anti-phospho-H2AX (1:1000; Cell Signaling Technology), and rabbit anti-GAPDH (1:2000; Sigma) overnight at 4 °C, then washed three times in TBS-T, and incubated in appropriate HRP-conjugated secondary antibodies for 2 h at room temperature. Membranes were washed three times in TBS-T, and proteins were visualized using Super Signal West Dura Extended Duration Substrate (Thermo Scientific, Rockford, IL). Chemiluminescence was captured via ChemiDocTM XRS + System (Bio-Rad), and protein bands were quantified by densitometric analysis using BioRad Molecular Imaging Software. The data was normalized with endogenous control of GAPDH and expressed in arbitrary units.
Immunohistochemistry
Twenty-micrometer coronal brain sections were selected, and standard immunostaining techniques were employed as described before [16]. Briefly, sections were washed three times with 1XPBS, blocked for 1 h in goat serum containing 0.4% Triton X-100, and incubated overnight at 4 °C with primary antibody rabbit anti-Iba-1 (1;1000; Thermo Fisher Scientific). Sections were washed three times with 1XPBS and incubated with appropriate Alexa Fluor-conjugated secondary antibodies (Life Technologies, CA) for 2 h at RT for immunoflurescence staining. Similarly, for immunohistochemistry and for DAB staining, sections were incubated with biotinylated anti-rabbit IgG antibody (Vector Laboratories, Burlingame, CA, USA) for 2 h at room temperature and avidin–biotin-horseradish peroxidase solution (Vectastain elite ABC kit, Vector Laboratories) for 1 h and then reacted with 3, 30-diaminobenzidine (Vector Laboratories) for color development. Images were acquired using a fluorescent Nikon Ti-E inverted microscope, at × 10 (Plan Apo 10 × NA 0.45) or × 20 (Plan APO 20 × NA 0.75) magnification. Exposure times were kept constant for all sections in each experiment.
Cathepsin D Assay
The CTSD/cathepsin D assay was performed by using CTSD assay kit from Abcam (ab65302) as per the manufacturer’s instruction. Briefly, mice were anesthetized, perfused with ice-cold saline, decapitated, and cortical tissue was dissected and homogenized in ice-cold cell lysis buffer provided in the kit. Tissue homogenates were centrifuged at 15,000 ×g for 5 min at 4 °C and were incubated with the CTSD substrate mixture at 37 °C for 1 h and fluorescence was further measured at an excitation wavelength of 328 nm and an emission wavelength of 460 nm. Protein concentration was estimated by the BCA method.
Statistical Analysis
Animal behavior analysis was performed by an investigator blinded to groups. Quantitative data were expressed as mean standard errors of the mean (s.e.m.). Western blot expression were analyzed by one-way analysis of variance (ANOVA), followed by post hoc adjustments using Newman Keuls test. Other gene expressions, animal behavior data were analyzed using Student-t-test. Statistical tests were performed using Graph Pad Prism Program, Version 5.02 for Windows (GraphPad Software, San Diego, CA, USA). A p < 0.05 was considered statistically significant.
Results
Autophagy Flux And Lysosomal Function Altered After JE Virus Infection
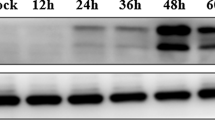
Western blot analysis showed significant increase in the level of autophagy marker LC3-II on day 7 compared to the controls (Fig. 1a, c, d; p < 0.05). This observation suggested that JE virus infection induced autophagosome formation. However, to assess the effect of JE infection on autophagic flux per se we analyzed the levels of autophagy substrate protein SQSTM1/p62 which selectively recognized ubiquitinated cargoes and delivered these to the precursor’s autophagosomes called phagophores, for their degradation. Autophagy substrate protein SQSTM1/p62 significantly increased (p < 0.05) along with LC3-II at 7 day post JEV infection compared to controls (Fig. 1a, e). It was also noted in vitro N2a neuronal cells along with increase in the level of polyubiquitin proteins at 24 h followed by JE viral infection (Fig. 1i). These results represented that autophagy flux was impaired after JE viral infection. Next, to confirm if there was indeed a defect in lysosomal function resulting in the accumulation of autophagy proteins and substrate in cerebral cortex, CTSD enzyme activity was found significantly decreased in cortical extracts on day 7 compared to control (Fig. 1h; p < 0.05). Such low enzymatic activity indicated decreased lysosomal activity after JE viral infection and could be, at least in part, account for the observed impairment in autophagy flux in JE infected mice. We observed that JE virus infection also altered autophagy regulators Akt and AMPK with an increase in Akt (p) (Fig. 1b, f; p < 0.01), and decrease (Fig. 1b, g; p < 0.05) in AMPK (p) expression in cortical tissue at day 7 compared to control.

Autophagy flux inhibited in cortex and autophagy-regulating signaling pathways of Akt (p) and AMPK (p) found significantly altered followed by JE viral infection. a Western blots of autophagy marker LC3-I/II and autophagy substrate SQSTM1/P62 analysis were performed in the cortical tissue lysates of control and JE viral infection animals at the indicated time points as mentioned in this figure. a, c–e Representative western blot and its densitometry analysis showing time dependent expression of autophagy marker of LC3-I&II and autophagy substrate SQSTM1/P62 significantly increased in cortex followed by JE viral infection from day 1 to day 7 in comparison to control. GAPDH serves as a loading control. Data are expressed as mean ± s.e.m.; n = 3, *p < 0.05 vs. control. Along with this, it is also observed that JE viral infection induced autophagy regulating signaling pathways of Akt (p) and AMPK (p) in comparison to total Akt and total AMPK expression in the cortex sub region (b, f, g). Bars represent the mean ± s.e.m.; n = 3, *p < 0.05, **p < 0.01 vs. control. All blots were normalized to their respective total protein concentration. h CTSD enzyme activity determined by in vitro fluorometric assay in the cortical tissue lysates of control and at 7 day JE viral infection animals. n = 6, * p < 0.05. i N2a cells were infected with JE virus for 24 h. Cell lysates were harvested for immunoblotting of LC3-I/II, SQSTM1/P62, cleaved caspase 12, polyubiquitin and GAPDH
Highly Reactive M1 Microglia Activation Phenotypes Predominated the JE Viral Infected Brain
Autophagy flux inhibition was associated with proinflammatory response of microglia activation following JE viral infection. Mouse was infected with JE virus and cortical tissue was collected after 24 h to determine the expression of microglia genes associated with pro-inflammatory (classical/M1) activation. Figure 2a–f showed the relative microglia messenger RNA (mRNA) expression of CD11b, its receptor TLR4 and its associated M1 activation related phenotype genes: IL-1β, tumor necrosis factor-α (TNF-α), IFNγ, and IL6. It was noted that JE virus infected animals had significantly increased mRNA levels of CD11b, TLR4, IL-1 β, TNF-α, IFNγ and IL6 when compared to controls animals (p < 0.05 [CD11b, TNF-α and IFNγ], p < 0.01 [IL-1 β, IL6], p < 0.001 [TLR4]). It was also observed that JE viral infection resulted in reactive microglia morphology in both JEV cortical brain tissue and in vitro BV2-microglia cell culture. It was noted that after JE viral infection, microglia displayed hypertrophic or bushy morphologies which were characterized by larger cell bodies, with thicker, shorter, and highly branched processes in comparison to control cells (Fig. 2g). While in BV2 microglia cells, it was noted that JEV infection increased granularity, cell body fusion at 72 h along with nitrite production at 24 h in JEV infected microglia condition media (p < 0.001) compared to control microglia cells (Fig. 2h).

Microglia/macrophage activation is robustly increased in cortex and sub-ventricle region followed by JE viral infection. Microglia cell culture followed by JE viral infection demonstrate neurotoxicity. It is observed that JE viral infection-stimulated nitric oxide production in BV2 microglia. a–f qPCR analysis of microglia/macrophage activation genes in the cortex of control and JE viral infected mice at 7 days post infection. JE viral infection-induced expression of CD11b and proinflammatory genes, IL1β, TNF-α, IFNγ, IL6 and TLR4, in JE viral infection mice. Student’s t-test; data = mean ± SEM; n = 4/group; *p < 0.05, **p < 0.01 and ***p < 0.001 versus, control group. g JE viral infection increases the number of highly activated microglia in the cortex and sub-ventricle region of JE viral infected mice compare to control animals. Representative Iba-1 immunohistochemistry images of microglia activation in the cortex of JEV infected and control mice. Scale bars = 50 µm and 100 µm. h BV2 microglia culture were incubated with JEV, microglia activation phenotype were observed by 72 h. h (i) Microscopic analysis showed that microglia got highly activated which can be further characterized by cell body enlargement, increased granularity and cell body fusion (red arrow) followed by JEV infection at 72 h time point compare to controls. (ii) We observed that pro-inflammatory mediator of nitric oxide (NO) was found significantly increased in the supernatant of highly activated microglia 72 hr followed by JEV infection (***p < 0.001 vs. control; Student’s t-test; data = mean ± SEM; n = 6/group)
Autophagy Flux Inhibition was Associated with Decrease in Hippocampus Neurogenesis, Increase in Microglia Activation, Apoptosis and Hippocampus Neuronal Cell Death Followed by JE Viral Infection
It was observed that caspase 12 level were increased in JEV infected mice brain with a peak at 7 day (p < 0.05 compared to control; Fig. 3d). This was associated with increased phos-H2AX, ubiquitin levels in the hippocampus, which peaked at day 7 following JE viral infection (Fig. 3e, f). Along with this, it was also observed that there was significant increase in microglia activation marker, Iba1 at day 3 with a peak at 7 day (p < 0.05 at 3 day and P < 0.01 at 7 day compared to control; Fig. 3c), and a concomitant decrease in hippocampus neurogenesis marker of doublecortin at 7 day post-infection (p < 0.01 compared to control; Fig. 3b). Data suggested that autophagy impairment might contribute to the induction of neuronal apoptosis in hippocampus after JE viral infection.

JE viral infection increased microglia activation, reduced neurogenesis, and neuronal cell death in hippocampus. Hippocampus tissue samples from JE viral infection and control were harvested for western immunoblot analysis of markers of neurogenesis (doublecortin; dcx), microglia activation (Iba1), apoptosis (caspase 12) and neuronal cell death (phos-H2AX, polyubiquitin; ubq). There was significant decrease in dcx (b i,ii) protein expression (**p < 0.01 at 7 days post infection and immunohistochemistry), significant increase in Iba1 (c) protein expression (*p < 0.05, **p < 0.01 at 3 days and 7 days post infection respectively), caspase 12 (d) protein expression (*p < 0.05 at 7 days post infection), H2AX (e) protein expression (*p < 0.05 at 7 days post infection) and ubq (f) protein expression (*p < 0.05, **p < 0.01 and ***p < 0.001 at 1 day, 3 days and 7 days post infection respectively) in the hippocampus tissue samples followed by JE viral infection compare to control (representative western immunoblot shown in a). GAPDH serves as a loading control. Bars represent the mean ± s.e.m.; n = 3, p-values vs control
JE Viral Infection Caused Motor Neuronal Dysfunction And Loss In Body Weight
JE virus-infected mice exhibited progressive to severe form of neurological dysfunction symptoms including motor co-ordination, close eye response, hair straightening, tremors, unstable posture-movement disorders, bradykinesia, hunchback posture, paralysis and loss in body weight at 7 days post-infection compared to control animals (Fig. 4a–d). Pattern of motor coordination during locomotion activity of these animals were analyzed. Lower panels of Fig. 4b showed the lateral view of JEV infected mouse walking on an experimental platform. One complete stride of a mouse was the summation of the stance and swing phases. The stance time is usually characterized in which the paw remains in contact with the belt and included braking and propulsion time period. Whereas, swing phase was the portion of stride phase in which the paws were not in contact with the belt. In locomotor functional analysis, the contributions of stance and swing to stride duration, the paw placement angles of the forelimbs and hind limbs of JEV-treated mice were found altered when compared to control mice. The stride length measurement was compared and found that step length (cm) significantly decreased (p < 0.05) in JE viral infected mice in comparison to control animals (Fig. 4c), and body weight gradually decreased followed by JE viral infected animals compared to control animals (Fig. 4d).

JE viral infection causes motor neuronal dysfunction and loss in body weight. a–d Mice appearance and body orientation for JE infected and control animals were analyzed on plane horizontal surface by open field analysis and footprint analysis. JE viral-infected mice exhibited progressive and severe form of neurological dysfunction symptom which includes resting tremors, unstable posture-movement disorders (a), bradykinesia, close eye response (i), hunchback posture (ii), hair straighten (iii), and paralysis at 7 days post-infection. Pattern of motor coordination during locomotion activity of JE infected and control animals were analyzed. Representative images of a step cycle, which is performed by a JEV infected mouse, shown in five consecutive frames (iv). Lateral view of JEV infected animals represented the altered response of stride, stance, and swing for hind limb and braking, propulsion for fore limb at 7 days with disease progression. Step length were analyzed on plane horizontal surface for JE infected and control animals by footprint analysis. Bar graph shows the step length (in cm; c) and progressive loss in body weight (gm; d) in control (n = 6) and JEV infected mice (n = 9). Data are expressed as mean ± s.e.m. *p < 0.05, ***p < 0.001 vs. control mice, Student’s t-test and one way ANOVA. (e) Schematic diagram for neuroinflammation and neuronal cell death in developing brain followed by JE viral infection
Discussion
In our study it is highlighted that JE viral infection promotes autophagy while inhibiting autophagy flux as the disease progresses, and is associated with proinflammatory response of microglia activation, cytokine production and behavioral changes, suggesting that autophagy function plays key role in brain dysfunction and abnormality in JE (Summarized in Fig. 4e).
Our results are in consistence with other viral diseases where the importance of autophagy has been demonstrated and autophagy flux inhibition being used for viral replication, involved in disease pathology. For instance, human immunodeficiency virus (HIV) used autophagy machinery during early stage of viral replication by inhibiting the conversion of autophagosomes to autolysosomes [17]. Similarly, RNA virus human parainfluenza virus inhibits autophagosomes maturation and autophagosomes-lysosome fusion which further lead to viral release and causes increase in apoptotic cell death of infected cells [18, 19]. Our results are in agreement with these observations and confirm that autophagy activation with autophagy flux inhibition is general response rather than JE specific. It is noted that JE virus infected mice exhibited severe neurological damage. These results are in agreement with, human and mouse model studies, in which JE virus antigen affinity in brain were observed and found associated with severe neurological dysfunction and neuronal cell death [20,21,22]. In continuation to that, in the current study it was explored further, the underlying molecular mechanism of these severe neurological sequel and cell death pathways in detailed manner with time scale following JE infection. It was evident that neuronal cell death following JE viral infection was propagated by strong inflammatory response and autophagy played key role in determining both neuroinflammatory response as well as survival of neurons [7]. Conversion of MAP1LC3B or LC3 (microtubule-associated protein 1 light chain 3) from LC3-I to another isoform LC3-II is the representation of autophagosome formation. However LC3-II is also degraded during the process of lysosomal degradation of autophagic cargo. Therefore at a steady state the amount of LC3-II represented net difference between the rate of autophagosome formation and its degradation during the course of autophagic flux. Along with this it is also observed that when autophagy flux increases autophagy substrate-SQSTM1/p62 level decreases with the process and when autophagy flux decreases their level increases with other autophagy substrates [23, 24]. Therefore the increased accumulation of these two proteins of LC3-II and p62 might be suggestive of a non functional autophagy at later stages of autophagy progression wherein autophagosomes were accumulated but could not merge with lysosomes or failed to degrade cargo due to lysosomal dysfunction [25, 26]. The significant decrease in cathepsin D activity, which is a lysosomal proteinase, at 7 day in the cortex of JEV infection compared to control animals, also suggests that lysosomal function is impaired at late stage and associated with JEV inducing a late block in autophagic flux at the lysosomal step which in turn results in reduced turnover of both LC3-II and p62. In our study it was also observed that autophagy impairment associated with proinflammatory markers (classical; M1 like) of microglia activation following JE viral infection. It was found that, microglia showed altered phenotype and increased number of highly activated ones displaying a bushy and hypertrophic cellular morphology compared to control mice. In addition to that, the expression levels of M1 (classical) activation genes (IL-1β, TNFα, IFN-γ, and IL6) were found to be significantly higher in JE virus infected mice brain compared to the control ones. It has been comprehended that proinflammatory mediators, such as IL-1b, TNF-α, and IFNγ, produced by M1-polarized microglia can react with superoxide free radicals and are involved in generation of reactive oxygen species (ROS) and other neurotoxic substances that might contribute to neuronal dysfunction and cell death [27, 28]. Notably, in infected mice, microglia activation in hippocampus was localized at sites of increased neurodegeneration and this was associated with decrease in marker of hippocampus neurogenesis-doublecortin, significant increase in accumulation of poly-ubiquitinated protein and increase of H2AX protein expression, markers of oxidative stress-induced DNA damage at 7 days in JE virus infected mice brain compared to the control animals [29]. These results further support the previous finding of human autopsy study in which grey matter areas of the brain including thalamus and hippocampus are found to be the primary JEV affected brain region with presence of activated microglia and significant neuronal damage [30]. However, at present in the study it is not possible to define the complete mechanism of autophagy flux regulation in the neuronal protection and improvement of the functional outcome followed by JE viral infection. It cannot be determined whether enhanced autophagy flux control the neuroinflammatory response, microglia activation, apoptotic neuronal cascade or it delays its progression [31]. We can only hypothesize that in our JE viral infection model, autophagy is a key mechanism for the regulation of the neuroinflammatory response and autophagy flux pathways could be important target to preserve neuronal survival and neurobehavioral function.
The ubiquitin–proteasome system (UPS) is the major intracellular protein degradation machinery and plays pivotal role in cellular processes such as the cell cycle, apoptosis, the host immune response, endocytosis, and signal transduction [32]. Role of UPS in flavivirus (for eg; DENV, WNV) replication, regulation and cellular internalization has been noted in the recent studies [33, 34]. Moreover, in other study it is noted that protein ubiquitination is involved in entry and cellular trafficking of JE viral infection [35]. In agreement to this, it was also noted that there was accumulation of polyubiquitin protein with increased JE viral replication in both hippocampus of JE viral infected mice and in vitro neuronal (N2a) cells. This result represented that JE virus utilized host cellular machinery for their own benefit, causing impaired autophagy flux and associated with accumulation of polyubiquitin protein.
Furthermore, to gain detailed insight into the molecular regulation of autophagy followed by JE viral infection, we assessed the activation status of Akt (serine/threonine-specific protein kinase) and AMPK (5′-AMP-activated protein kinase) expression by western blot in cortex. In previous study, it is noted that Akt regulates several cellular processes including proliferation, survival, and plays a major role in metabolism [36]. It is observed that Akt activation may stimulate protein synthesis through TOR activation and thus inhibit autophagy. In contrast, AMPK is activated under metabolically stressed conditions and brings about cellular homeostasis by switching on energy-generating catabolic processes like fatty acid oxidation and glycolysis along with autophagy initiation [37]. However, our western blot analysis represent that there is cross talk between AMPK and Akt pathways and represented the condition for substrate accumulation (similar condition are observed when autophagy flux inhibited) in cortical tissue. However, the detailed metabolic mediated molecular mechanism for the Akt/AMPK signaling pathways followed by JE viral infection is not known. Therefore more studies are needed to investigate this relationship after the JE viral infection.
The limitation of the study is that, the pathomechanism of JE viral infection disease had been observed for only up to 7 days and not beyond that due to significant rise in mice mortality as the disease progressed. The study confirmed the role of autophagy regulation in JE virus pathogenesis. We clearly demonstrated that JE viral infection led to impaired autophagy flux at 7 day post infection, reflected a deleterious effect and skewed microglia activation towards proinflammatory/M1 phenotype that further contributed to neuronal cell death. These observations provide a mechanistic link between JEV-induced imbalance in autophagy flux pathways, neuroinflammation and neurodegeneration.
References
Ke PY (2018) The multifaceted roles of autophagy in flavivirus-host interactions. Int J Mol Sci 19(12):3940. https://doi.org/10.3390/ijms19123940
Sharma M, Sharma KB, Chauhan S, Bhattacharyya S, Vrati S, Kalia M (2018) Diphenyleneiodonium enhances oxidative stress and inhibits Japanese encephalitis virus induced autophagy and ER stress pathways. Biochem Biophys Res Commun 502(2):232–237. https://doi.org/10.1016/j.bbrc.2018.05.149
Jin R, Zhu W, Cao S, Chen R, Jin H, Liu Y, Wang S, Wang W, Xiao G (2013) Japanese encephalitis virus activates autophagy as a viral immune evasion strategy. PLoS ONE 8(1):e52909. https://doi.org/10.1371/journal.pone.0052909
Lee YR, Lei HY, Liu MT, Wang JR, Chen SH, Jiang-Shieh YF, Lin YS, Yeh TM, Liu CC, Liu HS (2008) Autophagic machinery activated by dengue virus enhances virus replication. Virology 374(2):240–248. https://doi.org/10.1016/j.virol.2008.02.016
Sir D, Chen WL, Choi J, Wakita T, Yen TS, Ou JH (2008) Induction of incomplete autophagic response by hepatitis C virus via the unfolded protein response. Hepatology 48(4):1054–1061. https://doi.org/10.1002/hep.22464
Li JK, Liang JJ, Liao CL, Lin YL (2012) Autophagy is involved in the early step of Japanese encephalitis virus infection. Microbes Infect 14(2):159–168. https://doi.org/10.1016/j.micinf.2011.09.001
Ghoshal A, Das S, Ghosh S, Mishra MK, Sharma V, Koli P, Sen E, Basu A (2007) Proinflammatory mediators released by activated microglia induces neuronal death in Japanese encephalitis. Glia 55(5):483–496. https://doi.org/10.1002/glia.20474
Kalita J, Misra UK (2000) Comparison of CT scan and MRI findings in the diagnosis of Japanese encephalitis. J Neurol Sci 174(1):3–8
Saxena V, Mathur A, Krishnani N, Dhole TN (2008) An insufficient anti-inflammatory cytokine response in mouse brain is associated with increased tissue pathology and viral load during Japanese encephalitis virus infection. Arch Virol 153(2):283–292. https://doi.org/10.1007/s00705-007-1098-7
Bodea LG, Wang Y, Linnartz-Gerlach B, Kopatz J, Sinkkonen L, Musgrove R, Kaoma T, Muller A, Vallar L, Di Monte DA, Balling R, Neumann H (2014) Neurodegeneration by activation of the microglial complement-phagosome pathway. J Neurosci 34(25):8546–8556. https://doi.org/10.1523/JNEUROSCI.5002-13.2014
Das S, Basu A (2008) Japanese encephalitis virus infects neural progenitor cells and decreases their proliferation. J Neurochem 106(4):1624–1636. https://doi.org/10.1111/j.1471-4159.2008.05511.x
Mukherjee S, Singh N, Sengupta N, Fatima M, Seth P, Mahadevan A, Shankar SK, Bhattacharyya A, Basu A (2017) Japanese encephalitis virus induces human neural stem/progenitor cell death by elevating GRP78, PHB and hnRNPC through ER stress. Cell Death Dis 8(1):e2556. https://doi.org/10.1038/cddis.2016.394
Sharma M, Bhattacharyya S, Nain M, Kaur M, Sood V, Gupta V, Khasa R, Abdin MZ, Vrati S, Kalia M (2014) Japanese encephalitis virus replication is negatively regulated by autophagy and occurs on LC3-I- and EDEM1-containing membranes. Autophagy 10(9):1637–1651. https://doi.org/10.4161/auto.29455
Shukla V, Shakya AK, Shukla M, Kumari N, Krishnani N, Dhole TN, Misra UK (2016) Circulating levels of matrix metalloproteinases and tissue inhibitors of matrix metalloproteinases during Japanese encephalitis virus infection. Virusdisease 27(1):63–76. https://doi.org/10.1007/s13337-015-0301-9
Yang KD, Yeh WT, Chen RF, Chuon HL, Tsai HP, Yao CW, Shaio MF (2004) A model to study neurotropism and persistency of Japanese encephalitis virus infection in human neuroblastoma cells and leukocytes. J Gen Virol 85(Pt 3):635–642. https://doi.org/10.1099/vir.0.19426-0
Kumar A, Barrett JP, Alvarez-Croda DM, Stoica BA, Faden AI, Loane DJ (2016) NOX2 drives M1-like microglial/macrophage activation and neurodegeneration following experimental traumatic brain injury. Brain Behav Immun 58:291–309. https://doi.org/10.1016/j.bbi.2016.07.158
Kyei GB, Dinkins C, Davis AS, Roberts E, Singh SB, Dong C, Wu L, Kominami E, Ueno T, Yamamoto A, Federico M, Panganiban A, Vergne I, Deretic V (2009) Autophagy pathway intersects with HIV-1 biosynthesis and regulates viral yields in macrophages. J Cell Biol 186(2):255–268. https://doi.org/10.1083/jcb.200903070
Ding B, Zhang G, Yang X, Zhang S, Chen L, Yan Q, Xu M, Banerjee AK, Chen M (2014) Phosphoprotein of human parainfluenza virus type 3 blocks autophagosome-lysosome fusion to increase virus production. Cell Host Microbe 15(5):564–577. https://doi.org/10.1016/j.chom.2014.04.004
Faure M (2014) The p value of HPIV3-mediated autophagy inhibition. Cell Host Microbe 15(5):519–521. https://doi.org/10.1016/j.chom.2014.04.014
Chauhan PS, Khanna VK, Kalita J, Misra UK (2017) Japanese Encephalitis virus infection results in transient dysfunction of memory learning and cholinesterase inhibition. Mol Neurobiol 54(6):4705–4715. https://doi.org/10.1007/s12035-016-9963-6
Kalita J, Misra UK, Srivastava A (2009) Cognitive impairment in encephalitis: P3 and MRI correlation. Electromyogr Clin Neurophysiol 49(1):27–33
Misra UK, Kalita J (1997) Anterior horn cells are also involved in Japanese encephalitis. Acta Neurol Scand 96(2):114–117
Bjorkoy G, Lamark T, Johansen T (2006) p62/SQSTM1: a missing link between protein aggregates and the autophagy machinery. Autophagy 2(2):138–139. https://doi.org/10.4161/auto.2.2.2405
Ichimura Y, Kominami E, Tanaka K, Komatsu M (2008) Selective turnover of p62/A170/SQSTM1 by autophagy. Autophagy 4(8):1063–1066. https://doi.org/10.4161/auto.6826
Bjorkoy G, Lamark T, Brech A, Outzen H, Perander M, Overvatn A, Stenmark H, Johansen T (2005) p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J Cell Biol 171(4):603–614. https://doi.org/10.1083/jcb.200507002
Zhou J, Sinha RA, Lesmana R, Yau WWY, Yen PM (2018) Pharmacological inhibition of lysosomal activity as a method for monitoring thyroid hormone-induced autophagic flux in mammalian cells in vitro. Methods Mol Biol 1801:111–122. https://doi.org/10.1007/978-1-4939-7902-8_11
Kumar A, Loane DJ (2012) Neuroinflammation after traumatic brain injury: opportunities for therapeutic intervention. Brain Behav Immun 26(8):1191–1201. https://doi.org/10.1016/j.bbi.2012.06.008
Kaushik DK, Gupta M, Kumawat KL, Basu A (2012) NLRP3 inflammasome: key mediator of neuroinflammation in murine Japanese encephalitis. PLoS ONE 7(2):e32270. https://doi.org/10.1371/journal.pone.0032270
Silva AR, Santos AC, Farfel JM, Grinberg LT, Ferretti RE, Campos AH, Cunha IW, Begnami MD, Rocha RM, Carraro DM, de Braganca Pereira CA, Jacob-Filho W, Brentani H (2014) Repair of oxidative DNA damage, cell-cycle regulation and neuronal death may influence the clinical manifestation of Alzheimer’s disease. PLoS ONE 9(6):e99897. https://doi.org/10.1371/journal.pone.0099897
Sips GJ, Wilschut J, Smit JM (2012) Neuroinvasive flavivirus infections. Rev Med Virol 22(2):69–87. https://doi.org/10.1002/rmv.712
Yang S, Qiang L, Sample A, Shah P, He YY (2017) NF-kappaB signaling activation induced by chloroquine requires autophagosome, p62 protein, and c-Jun N-terminal kinase (JNK) signaling and promotes tumor cell resistance. J Biol Chem 292(8):3379–3388. https://doi.org/10.1074/jbc.M116.756536
Glickman MH, Ciechanover A (2002) The ubiquitin-proteasome proteolytic pathway: destruction for the sake of construction. Physiol Rev 82(2):373–428. https://doi.org/10.1152/physrev.00027.2001
Choy MM, Zhang SL, Costa VV, Tan HC, Horrevorts S, Ooi EE (2015) Proteasome inhibition suppresses dengue virus egress in antibody dependent infection. PLoS Negl Trop Dis 9(11):e0004058. https://doi.org/10.1371/journal.pntd.0004058
Krishnan MN, Ng A, Sukumaran B, Gilfoy FD, Uchil PD, Sultana H, Brass AL, Adametz R, Tsui M, Qian F, Montgomery RR, Lev S, Mason PW, Koski RA, Elledge SJ, Xavier RJ, Agaisse H, Fikrig E (2008) RNA interference screen for human genes associated with West Nile virus infection. Nature 455(7210):242–245. https://doi.org/10.1038/nature07207
Wang S, Liu H, Zu X, Liu Y, Chen L, Zhu X, Zhang L, Zhou Z, Xiao G, Wang W (2016) The ubiquitin-proteasome system is essential for the productive entry of Japanese encephalitis virus. Virology 498:116–127. https://doi.org/10.1016/j.virol.2016.08.013
Vivanco I, Sawyers CL (2002) The phosphatidylinositol 3-Kinase AKT pathway in human cancer. Nat Rev Cancer 2(7):489–501. https://doi.org/10.1038/nrc839nrc839[pii]
Hopkins TA, Dyck JR, Lopaschuk GD (2003) AMP-activated protein kinase regulation of fatty acid oxidation in the ischaemic heart. Biochem Soc Trans 31(Pt 1):207–212. https://doi.org/10.1042/bst0310207
Acknowledgements
Authors acknowledge Ms. Swati Singh, Mr. Firoz Ahmed for technical assistant in animal experiments.
Funding
This study was supported by the Ramalingaswami re-entry fellowship (BT/RLF/Re-entry/13/2014) from Department of Biotechnology, Ministry of Science and Technology, Govt. of India to A.K. and ICMR (59/05/2019/ONLINE/BMS/TRM) and SERB (SRG/2019/000398) to RAS.
Author information
Authors and Affiliations
Contributions
AK: Conceptualization, Methodology, Validation, Formal analysis, Investigation, Data curation, Visualization. AK and UK: Writing—original draft, RAS, GS, AB, MS: Methodology, Validation, JK, RAS, ST, TND: Resources, Supervision. All authors reviewed the manuscript.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Kumar, A., Kalita, J., Sinha, R.A. et al. Impaired Autophagy Flux is Associated with Proinflammatory Microglia Activation Following Japanese Encephalitis Virus Infection. Neurochem Res 45, 2184–2195 (2020). https://doi.org/10.1007/s11064-020-03080-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11064-020-03080-5