
Abstract
Andrographolide from leaves of Andrographis paniculata has been known to possess various bioactivities. In the present study, we aimed to explore the neuroprotection of andrographolide against inflammation-mediated injury and oxidative damage. In initial studies, our findings showed that pretreatment with andrographolide could effectively reduce neuronal cell death caused by LPS-induced conditioned supernatants. The further results indicated that this neuroprotective effect may be mainly due to the inhibition on the production of NO, TNF-α, IL-6, ROS, iNOS and enhancement of expression of anti-inflammatory marker CD206. Moreover, mechanism study revealed that the anti-inflammatory activity of andrographolide may be related to the suppression of nuclear translocation of NF-κB as well as the activation of Nrf2 and HO-1. Our study also showed that andrographolide could scavenge ROS and protect PC12 cells against H2O2- and 6-OHDA-mediated oxidative damage. In addition, several derivatives of andrographolide were prepared for evaluating the role of 3, 14, 19-hydroxy group on anti-inflammatory effect and cytoprotection of andrographolide. In conclusion, andrographolide protected neurons against inflammation-mediated injury via NF-κB inhibition and Nrf2/HO-1 activation and resisted oxidative damage via inhibiting ROS production. Our results will contribute to further exploration of the therapeutic potential of andrographolide in relation to neuroinflammation and neurodegenerative diseases.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Though the average age of the world’s population continues to gradually rise, more people than ever will be afflicted with age-related neurodegenerative diseases and functional deterioration. The previous reports have shown that neuronal dysfunction and death are attributed to pathogenesis of neurodegenerative disorders. For example, Alzheimer’s disease (AD) is related to the death of hippocampal and cortical neurons [1]; substantia nigral and midbrain dopaminergic neurons are responsible for Parkinson’s disease (PD) [2]; stroke involves the death of cortical and striatal neurons [3].
Microglia, the primary immune effector cells in the central nerves system (CNS), play a critical role in immune regulation [4] and neuronal homeostasis [5]. Responding for danger signals, microglia become activated by changing morphology from resting ramified to amoeboid phagocytic cells and then produce inflammatory cytokines and chemokines, phagocytizing the toxic cellular debris or invading pathogens [6, 7]. However, over-activated microglia could release various cytotoxic mediators, such as NO, TNF-α, interleukin-1β (IL-1β), IL-6, ROS, and cyclooxygenase-2 (COX-2) [8], leading to neuronal dysfunction and cell death. Therefore, pharmacological inhibition of microglia-mediated neuroinflammation may represent a promising therapeutic strategy to protect neuronal cells and alleviate the progression of those neurological diseases.
ROS are important in regulating diverse signaling pathways involved in cell differentiation, proliferation, and apoptosis [9]. Oxidative stress, arising from uncontrolled production of ROS, causes damage to lipids, proteins, and DNA in neural cells [10]. Due to a high oxygen consumption ratio [11] and a low content of antioxidant defenses in the brain, neuronal cells are particularly vulnerable to oxidative stress. Accumulating evidence indicated that oxidative stress plays a key role in various neurodegenerative processes [9]. Therefore, the use of antioxidants may be one possible strategy for the development of neuroprotective drugs.
Andrographolide, the major labdane diterpene isolated from the leaves or whole plant of Andrographis paniculata, was reported to exhibit a wide spectrum of biological activity, such as anti-tumor [12], anti-inflammatory [13], antimalarial [14], antibacterial [15], and antiviral [16]. In a previous study, andrographolide could inhibit ROS production in neutrophils and the production of NO, TNF-α and IL-6 secretion in macrophages [17, 18]. Further, pretreatment by andrographolide could protect dopaminergic neurons in mesencephalic neuron-glia cultures against LPS-induced neurotoxicity and the protective effect was glia-dependent [13]. In addition, andrographolide protected H2O2-treated PC12 cells cultured in astrocyte-conditioned medium [19]. However, the underlying mechanism is largely unclear. To further expand on the types of neuronal damage maybe attenuated by andrographolide, we studied the microglia-mediated neuroinflammation insult and oxidative damage. The working mechanism was also involved. Moreover, the anti-neuroinflammatory and neuroprotective properties of andrographolide derivatives 1–5 were evaluated.
Materials and Methods
Chemicals and Reagents
Andrographolide (purity ≥ 98%) was purchased from J&K Scientific (Beijing, China). Dulbecco’s modified Eagle’s medium (DMEM) and horse serum (HS) were from Gibco Ltd. (Grand Island, NY, USA). Fetal bovine serum (FBS) was purchased from Hyclone (Logan, UT, USA). LPS from Escherichia coli 0111:B4, poly-l-lysine (PLL) solution and Hoechst 33342 were obtained from Sigma-Aldrich Co. (St. Louis, MO). MTT was from Solarbio Technology Company (Beijing, China). LDH assay kit, NO assay kit, and 2,7-dichlorofluorescein diacetate (DCFH-DA) were purchased from Beyotime Institute of Biotechnology (Jiangsu, China). TNF-α and IL-6 enzyme-linked immunosorbent assay (ELISA) kits were purchased from Boster Biological Technology (Wuhan, China). RevertAid First Strand cDNA Synthesis kit and TRIzol™ reagent were from Thermo Fisher Scientific. Rabbit polyclonal antibody against iNOS, NF-κB p65 and Nrf2 were purchased from Proteintech (Wuhan, China). 6-OHDA was supplied by Santa Cruz Biotechnology (Santa Cruz, CA). All the chemicals and reagents were of analytical grade.
Cell Culture
Murine BV-2 microglia cell line was purchased from Cell Resource Centre of Peking Union Medical College (Beijing, China) and cultured in a humidified atmosphere containing 5% CO2 at 37 °C using DMEM supplemented with 10% heat-inactivated FBS and 1% penicillin/streptomycin.
Rat pheochromocytoma PC12 cell line was purchased from the Shanghai Institute of Biochemistry and Cell Biology (Chinese Academy of Sciences), and maintained in DMEM containing 10% heat-inactivated HS and 5% FBS with 1% penicillin/streptomycin at 37 °C in a humidified 5% CO2 atmosphere.
Microglia-Conditioned Medium and Treatment
BV-2 cells were plated in 6-well plates (3.0 × 105 cells/ml) and allowed to attach overnight. After treatment with andrographolide (0.5, 5, 12.5, 25 μM) and LPS for 24 h, the supernatants were collected as microglia-conditioned medium (CM). Moreover, the supernatants from treatment with 0.1% DMSO (CMC) or 1 μg/ml LPS (CML) were collected to be used in later experiments.
PC12 cells were seeded in 96-well plates coated with PLL at a density of 2.5 × 105 cells/ml for 24 h and treated for 24 h according to the following methods: (1) directly incubated with CM; (2) incubated with andrographolide and CMC; (3) incubated with andrographolide and CML. The viability of PC12 cells was analyzed by the MTT assay.
MTT Assay
Briefly, PC12 cells (2.5 × 105 cells/ml) were treated for 24 h in triplicate in 96-well microplates. After treatment, MTT solution (10 μl of 5 mg/ml) was added to each well and incubated for 4 h at 37 °C. Then culture media were discarded and DMSO was added to dissolve the formazan dye. The optical density was measured at 570 nm on a microplate reader (Mode 680, Bio-Rad, Tokyo, Japan).
LDH Release Assay
After treatment (in Microglia-conditioned Medium and Treatment), the supernatants of BV-2 cells were collected and incubated with PC12 cells for 24 h. LDH released into the culture medium was detected using LDH assay kit following the manufacturer’s instructions. The absorbance was measured at 490 nm by a microplate reader. The optical density of the supernatants from BV-2 cells was used as control. LDH release (%) = LDH activity in supernatants from PC12 cells (% of control)—LDH activity in supernatants from BV-2 cells (% of control).
Hoechst 33342 Staining
Cells were seeded on the climbing slices coated with PLL. After treatments, cells were fixed using 4% of paraformaldehyde for 10 min at 4 °C. Then Hoechst 33342 was subsequently added to a final concentration of 5 μg/ml to stain the nuclei. The cells were visualized and photographed on a fluorescence microscopy (Olympus BX53).
Nitrite Quantification
NO production was determined indirectly from the supernatants by a NO assay kit based on the Griess reaction. Briefly, 50 μl of BV-2 culture supernatants were collected and put into the counterpart well of another plates. Then, an equal volume of Griess reagents I and II was added and the absorbance was measured at 540 nm. Nitrite concentrations were calculated using a sodium nitrite standard curve.
Measurement of ROS
Cells were seeded on the climbing slices coated with PLL. After treatments, the supernatant were removed and fixed using 4% of paraformaldehyde for 10 min. Then, the cells were incubated with fluorescence dye DCFH-DA (10 μM) in the darkness for 30 min at 37 °C and photographed under a fluorescence microscopy. For quantitative analysis, cells were seeded in 6-well plates and treated for indicated time. The cells were harvested and then re-suspended in 10.0 μM DCFH-DA solution at 37 °C for 30 min. Intracellular ROS were quantified in PBS. Fluorescence absorbance was detected on a fluorescence plate reader with excitation/emission wavelengths of 485 nm/538 nm (Molecular Devices, CA). The ROS level is expressed as the percentage of treated cells compared to the control.
Immunofluorescence
BV-2 cells were seeded on the climbing slices coated with PLL in 24-well plates (1.2 × 105 cells/ml) for 24 h. After treatments, the cells were fixed with cold 4% paraformaldehyde for 15 min at room temperature and permeabilized with 0.5% Triton X-100 for 20 min. After blocking with 10% normal goat serum for 30 min, cells were incubated with rabbit polyclonal anti-iNOS (1:50), rabbit polyclonal anti-NFκB/p65 (1:50) and rabbit polyclonal anti-Nrf2 (1:50) in a humidified chamber overnight at 4 °C. Then, the cells were incubated with a secondary TRITC-conjugated goat anti-rabbit IgG antibody diluted to 1:64 in PBST at 37 °C for 1 h. The nucleus were stained by 4′-6-diamidino-2-phenylindole (DAPI) and analyzed by the fluorescence microscopy (OLYMPUS, Japan).
Determination of TNF-α and IL-6
BV-2 cells were treated with LPS (1 μg/ml) and andrographolide for 6 h (TNF-α) or 24 h (IL-6). Then the culture medium was collected for proinflammatory mediator assay by ELISA kit according to the manufacturer’s instructions.
Reverse Transcription-Polymerase Chain Reaction (RT-PCR)
BV-2 cells were treated with LPS and/or andrographolide for 24 h. Total RNA was isolated using TRIzol reagent according to the manufacturer’s instruction. cDNA was synthesized using RevertAid First Strand cDNA Synthesis kit. RT-generated cDNA encoding CD206, Arginase-1 (Arg-1), iNOS, HO-1 and GAPDH genes were amplified using PCR. Primer sequences used in this study were as follows: for CD206: 5′-CTT-CGG-GCC-TTT-GGA-ATA-AT-3′ and reverse 5′-TAG-AAG-AGC-CCT-TGG-GTT-GA-3′ [20]; for Arg-1: 5′-TGA-AAG-GAA-AGT-TCC-CAG-ATG-T-3′ and reverse 5′-TGA-AGG-TCT-CTT-CCA-TCA-CCT-T-3′ [21]; for iNOS: 5′-CCC-TTC-CGA-AGT-TTC-TGG-CAG-CAG-C-3′ and reverse 5′-GGC-TGT-CAG -AGC-CTC-GTG-GCT-TTG-G-3′ [22]; for HO-1: 5′-GAG-AAT-GCT-GAG-TTC -ATG-3′ and reverse 5′-ATG-TTG-AGC-AGG-AAG-GC-3′ [23]; for GAPDH: 5′-AAC-TTT-GGC-ATT-GTG-GAA-GG-3′ and reverse 5′-GGA-TGC-AGG-GAT -GAT-GTT-CT-3′ [20]. Changes in transcriptional level of indicated gene were calculated with an Image J analyzer using GAPDH as an internal control.
Western Blotting
Western blotting was used for detection of protein expression. BV-2 cells were seeded at 3.0 × 105 cells/ml in 100 mm2 cell culture dishes. After treatment, nuclear and cytosolic fractions were prepared using nuclear and cytoplasmic extraction reagents (Beyotime, Jiangsu, China) according to the manufacturer’s instructions. Equal amounts of protein were separated on SDS-PAGE and transferred to polyvinylidenefluoride (PVDF) membranes. After that, membranes were blocked with 5% nonfat milk for 2 h at room temperature and cells were incubated with primary antibody against polyclonal anti-NFκB/p65, anti-Nrf2, anti-lamin B1 and anti-β-actin (1:1000) overnight at 4 ◦C. Following three washes in Tris-buffered saline-Tween (TBS-T), horseradish peroxidase (HRP)-conjugated IgG secondary antibody (diluted 1:1000) was applied for 2 h at room temperature. Immunoreactive bands were visualized using the electrochemiluminescence (ECL) detection system.
Analysis of H2O2 and 6-OHDA-Induced PC12 Cell Death
PC12 cells were seeded in 96-well plates (1.5 × 105 cells/ml) coated with PLL. After 24 h, andrographolide in fresh medium containing 2% HS and 1% FBS was added to each well followed by incubation for an additional 24 h at 37 °C. Then the supernatants were removed and the cells were incubated with 1.0 mM H2O2 or 0.9 mM 6-OHDA for another 24 h. After treatment, cells were incubated for 4 h with a solution of MTT (final concentration = 0.5 mg/ml). An extraction buffer (100 μl, 10% SDS, 5% isobutanol, 0.01 M HCl) was added and the cells were incubated overnight at 37 °C. The optical density of absorbance at 570 nm was measured.
Chemical Synthesis
Compounds 1–5 were synthesized according to a reported procedure [24]. Andrographolide was reacted with excess acetic anhydride at 80 °C for 1 h to afford compounds 1–4 in 18.4%, 13.5%, 22.5% and 22.7% yields (total 77.1%), respectively. tert-Butyldimethylsilyl ether group was obtained in basic condition and de-protected using formic acid/water in THF for the preparation of new compound 5 as a white solid (14.9%). 5: 1H NMR (500 MHz, CDCl3) δ 8.06 (1H, s), 7.00 (1H, m), 5.92 (1H, m), 4.91 (1H, s), 4.63 (1H, d, J = 15 Hz), 4.53 (2H, m), 4.49 (1H, d, J = 10 Hz), 4.26 (1H, d, J = 10 Hz), 4.21 (1H, d, J = 10 Hz), 2.45 (3H, m), 2.12 (3H, s), 2.05 (3H, s), 1.05 (3H, s), 0.75 (3H, s); 13C NMR (125 MHz, CDCl3) δ 170.71, 170.69, 169.23, 161.24, 150.27, 146.60, 124.34, 109.22, 79.74, 71.82, 68.01, 64.34, 56.03, 55.45, 41.49, 39.18, 38.02, 37.15, 25.40, 24.81, 24.47, 22.89, 21.36, 20.95, 14.75; HRMS (ESI) m/z calcd for C25H35O8+ (M + H)+ 463.23264, found 463.23254.
Computer Docking
The crystal structure of NF-κB p50 (PDBID: 1NFK) was used for docking simulations, and the three dimensional structures of studied molecules were constructed using Moden5.7 software package [25]. The docking simulations were carried out using Autodock 4.2 software package [26]. The lamarckian genetic algorithm (LGA) was selected for the ligand conformational search. 100 runs were carried out for each simulation, and other docking parameters were remained as their default values. The generated 100 conformations were used for clustering analysis for each studied molecules.
Statistical Analysis
All results above were presented as mean ± S.D. Unless otherwise stated, each experiment was repeated at least three separate experiments run at least in triplicate. The data were analyzed by one-way ANOVA following by Dunnett’s multi-comparison test or Student’s t test, using SPSS program (version 12.0).
Results
Effect of Andrographolide on PC12 Neuronal Death from Microglia-mediated Neuronal Damage
To investigate the neuroprotective effect of andrographolide against LPS-induced neurotoxicity, we first determined the cell viability of PC12 cells using MTT assay. Exposure of PC12 cells directly to CM (Fig. 1a) led to an increased population of viable cells in a dose-dependent manner (Fig. 1b). Then, PC12 cells were incubated simultaneously with andrographolide and CMC or CML. The results showed that post-treatment by andrographolide did not protect PC12 cells from damage induced by LPS-induced microglia (Fig. 1c, d). LDH is released into the culture medium following loss of membrane integrity resulting from cell damage. Similarly, decrease of LDH release was also observed in PC12 cells exposed to CM (Fig. 1e). Moreover, morphological changes of PC12 cells subjected to CM were also determined (Fig. 1f). The controls were in normal forms with no fragmentation and the conditional medium from LPS stimulated BV-2 cells resulted in increased cell body area, more cell debris, and irregular shape. However, these changes were reversed by the administration of andrographolide. While at 25 μM, the change almost disappeared comparing to control.

Neuroprotection of andrographolide against microglia-mediated neuronal damage. a Microglia-conditioned medium were prepared. Cell survival rate of PC12 cells after treatment with CM (b), andrographolide and CMC (c), andrographolide and CML (d) for 24 h was evaluated by MTT assay. e The content of LDH in the medium of PC12 cells after treatment with CM was analyzed by LDH assay kit. LDH activity in supernatants from BV-2 cells (%) was used as control. f Morphological changes in PC12 cells were observed by phase contrast microscopy (10 ×). Nuclei were stained with Hoechst 33342 and examined by fluorescence microscopy (20 ×). Apoptotic cells are marked with an asterisk. All the data are presented as the mean ± SD of three independent experiments. *P < 0.05 versus control; **P < 0.01 versus control; &&P < 0.001 versus control
Effect of Andrographolide on the Production of NO, the Protein and mRNA Expression of iNOS in Microglia
NO is widely used as an indicator of microglial activation among many inflammatory mediators. To study the anti-inflammatory characteristics of andrographolide, we firstly evaluated the inhibitory effect against LPS-induced NO production in microglia. Treatment of BV-2 cells with andrographolide and LPS concentration dependently reduced NO production (Fig. 2a). To exclude the possibility that the NO inhibition of andrographolide was due to the cytotoxicity, MTT assay was used. Andrographolide did not affect cell viability of BV-2 cells under this experimental condition (Data not shown), while could rescue the morphological changes of BV-2 cells in a dose-dependent manner (Fig. 2b). iNOS was generated in activated microglial cells and mediated the synthesis of NO. As shown in Fig. 2c, the level of LPS-induced iNOS expression was gradually decreased with increased concentration of andrographolide. Moreover, the gene expression of iNOS was also decreased after exposure to andrographolide (Fig. 2d).

Effect of andrographolide on the production of NO, the protein and mRNA expression of iNOS in LPS-induced BV-2 cells. BV-2 cells were treated with different concentrations of andrographolide (Andro) in the presence or absence of LPS for 24 h. a The NO production was determined by Griess reagents. b Morphological changes of BV-2 cells were observed by phase contrast microscopy (10 ×). c The iNOS expression was evaluated by immunofluorescence using confocal microscopy (20 ×). d The mRNA level of iNOS was measured using RT-PCR. The data are presented as the mean ± SD of three independent experiments. **P < 0.01 versus control; &&P < 0.001 versus control
Effect of Andrographolide on the Production of Inflammatory Mediators and mRNA Expression of Anti-inflammatory Mediators in Microglia
Changes in the level of inflammatory mediators TNF-α and IL-6 were also determined in LPS-induced BV-2 cells (Fig. 3a, b). Generally, the production of TNF-α and IL-6 was significantly inhibited by andrographolide. In particular, IL-6 secretion decreased to near basal levels in BV-2 cells exposed to the highest concentration of andrographolide (25 μM). Intracellular ROS can trigger a cascade of deleterious events in the inflammatory process. As shown in Fig. 3c, pretreatment of andrographolide diminished LPS-induced intracellular ROS production in a dose-dependent manner. Andrographolide also markedly increased the expression of anti-inflammatory mediator CD206 (Fig. 3d), while no significant effect on the Arg-1 expression (Fig. 3e).

Effect of andrographolide on the production of inflammatory mediators and mRNA expression of anti-inflammatory mediators in LPS-induced BV-2 cells. BV-2 microglia cells were incubated with andrographolide in the presence or absence of LPS for 6 h (TNF-α) or 24 h (IL-6), TNF-α (a) and IL-6 (b) concentrations were detected by ELISA kit. c After treatment for 24 h, the cells were collected and the determination of ROS level was carried out at the 485 nm excitation and 538 nm emission on a fluorescence plate reader. Total RNA was isolated using TRIzol reagent at three times in different days and the mRNA level of anti-inflammatory genes CD206 (d) and Arg-1 (e) was measured using RT-PCR. The data are presented as the mean ± SD of three independent experiments. *P < 0.05 versus control; **P < 0.01 versus control; &&P < 0.001 versus control
Effect of Andrographolide on the Activation of NF-κB and Nrf2 in Microglia
As NF-κB is an important transcription factor that regulates proinflammatory mediators, we next determined the effect of andrographolide on LPS-induced translocation of NF-κB by confocal microscopy. As shown in Fig. 4a, LPS exposure resulted in significant NF-κB p65 accumulation (red fluorescence marked with an arrow) in nucleus. However, LPS-induced nuclear translocation of NF-κB was significantly inhibited by the presence of andrographolide especially at 25 μM. The results were further confirmed by Western blotting (Fig. 4b). To further investigate the anti-inflammatory mechanism of andrographolide, activation of Nrf2 were also analyzed. Immunocytochemical analysis and western blotting clearly revealed that LPS alone has little influence on Nrf2 expression, but Nrf2 protein was significantly upregulated in the presence of andrographolide especially at 25 μM (Fig. 4c, d). We next examined the effect of andrographolide on HO-1 expression, which is directly linked to Nrf2-dependent activation. Using RT-PCR analysis, we found that HO-1 mRNA level was markedly increased in the presence of andrographolide at 12.5 and 25 μM compared with LPS alone (Fig. 4e).

Effect of andrographolide on the activation of NF-κB p65 and Nrf2 expression in BV-2 cells. BV-2 microglia cells were incubated with andrographolide in the presence or absence of LPS for 24 h. After treatment, the NF-κB p65 and Nrf2 expression were evaluated by the fluorescence microscopy (a and c). BV-2 microglial cells were seeded in 100 mm2 cell culture dishes and treated with andrographolide in the presence or absence of LPS for 24 h. Then the nuclear and cytosolic fractions were prepared and the levels of NF-κB p65, Nrf2, β-actin, and lamin B1 were measured by Western blot analysis (b and d). Total RNA was isolated using TRIzol reagent at three times in different days and the mRNA level of HO-1 was measured using RT-PCR (e). The data are presented as the mean ± SD of three independent experiments. **P < 0.01 versus control
Effect of Andrographolide on H2O2 and 6-OHDA-Induced PC12 Cell Death
The evaluation of toxicity indicated that andrographolide up to 50 μM did not show significant toxic effect to PC12 cells (Data not shown), we then selected 50, 25, 12.5 and 5 μM for the follow-up studies. As shown in Fig. 5a, andrographolide enhanced the cellular resistance against H2O2 (1.0 mM) toxicity in a concentration-dependent manner. Similarly, the cell growth suppression caused by 6-OHDA (0.9 mM) was also restored by andrographolide at 5 and 12.5 μM (Fig. 5b). However, higher concentrations (25 and 50 μM) seemed to block this protection to some extent, probably due to the synergy toxic effect of andrographolide and 6-OHDA. Moreover, pretreatment of the cells with andrographolide remarkably decreased the population of apoptotic nuclei. ROS, regulated by the antioxidant defense system, are uncontrolled by cells under oxidative stress or cellular damage, finally causing neuronal cell damage and death. In the present study, we also found that pretreatment of the cells with andrographolide remarkably reduced the ROS accumulation in PC12 cells in the presence of H2O2 and 6-OHDA (Fig. 5c, d).

Effect of andrographolide on cell viability and ROS level in H2O2 and 6-OHDA-induced PC12 cells. PC-12 cells were incubated with andrographolide for 24 h and then treated with H2O2 (a) or 6-OHDA (b) for another 24 h. After treatment, nuclei were stained with Hoechst 33342 and cell viability was measured by MTT assay. For measurement of ROS, PC-12 cells were incubated with andrographolide for 24 h and then treated with H2O2 (1.0 mM) for 1 h or 6-OHDA (0.9 mM) for 6 h. The cells were visualized and photographed under a confocal microscopy. The data are presented as the mean ± SD of three independent experiments. *P < 0.05 versus control; **P < 0.01 versus control; &&P < 0.001 versus control
Effect of Andrographolide Derivatives on the Production of NO in LPS-Induced BV-2 Cells and H2O2- or 6-OHDA-Induced PC12 Cell Death
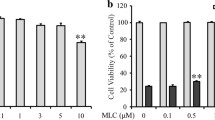
In order to understand the role of 3, 14, 19-hydroxy group, compounds 1–5 were prepared (Scheme 1) and the inhibitory effect on LPS-induced NO production was first evaluated by Griess assay (Fig. 6a). Compound 1 exhibited moderate activity similar to andrographolide, suggesting that introduction of acetyl group to C-19 is not critical to NO inhibitory activity. Interestingly, compound 3 was superior to compound 1 against NO production, indicating that the presence of an ester functionality at C-3 is beneficial for anti-inflammatory effect. Moreover, compound 3 also showed better inhibitory activity than compound 2 having the ester functionality at C-14. However, introduction of acetyl groups both to C-14 and C-3 of andrographolide (compound 4 and 5) was found to have no significant effect on inhibitory activity. In addition, acetyl group contributed to the greater log P (Table 1), with compound 4 showing the highest lipophilicity. This probably led to the weak cytotoxicity of 4 (Fig. 6b) and more potent inhibitory effect on NO production at 10 μM. We further studied the cytoprotection of 1-5 on PC12 cells. At the initial stage, we determined this effect on 6-OHDA-induced PC12 cells. As shown in Fig. 6c–g, except for 3, all the other tested compounds displayed toxicity at 50 μM. Then, the optimal concentration 20 μM was used in the following protection experiment. Notably, compound 2 gave the best protection at 20 μM both on H2O2 and 6-OHDA-induced PC12 cells (Fig. 6h).

Reagents and conditions: (a) Ac2O, 80 °C, 1 h, total yields of 1–4 = 77.1%; (b) (i) tert-butyldimethylsilyl chloride (TBSCl), pyridine, rt,1 h, 66.7%, (ii) Ac2O, ZnCl2, rt, 4 h, 39.6%; (c) HCOOH/H2O (9:1), THF, 0 °C, 4 h, 14.9%

Effect of 1–5 on LPS-induced NO production and H2O2 or 6-OHDA-induced PC12 cell death. a The inhibitory effect of 1–5 on LPS-induced NO production was measured at 1, 5, 10 μM. b Effect of 4 on cell viability of BV-2 cells was determined by MTT assay. Effect of 1–5 on H2O2 or 6-OHDA-induced apoptosis in PC12 cells was evaluated by MTT assay (c, d, e, f and g). h Protection against H2O2- and 6-OHDA-induced PC12 cell damage was assessed at 20 μM. The data are presented as the mean ± SD of three independent experiments. **P < 0.01 versus control; &&P < 0.001 versus control
Noncovalent Binding of Compounds 1–5 on NF-κB p50
It was shown that andrographolide and 3 showed lower binding free energies (− 7.13 and − 7.07, respectively), while 5 showed the highest (Table 1), indicating the high-affinity binding of andrographolide and 3 and low-affinity binding of 5. Moreover, our results showed that section a of studied molecules (Fig. 7a) tended to bind to the small cavity mainly formed by the Ser63, Gly65, Asn136, Gly138 residues in the active site of NF-κB p50. Section b in 1–5 was accommodated in the large cavity mainly composed by the Tyr57, Val58, Gly61 residues. The hydroxyl groups (C-3, C-19) formed hydrogen bonds with the back bone of residue Val58, which stabilized the binding mode of andrographolide in the active site of p50 (Fig. 7b). Compared to andrographolide, the introduction of acetyl group to C-19 is beneficial for the partially spatial matching between section b of 1 and the large cavity of the active site of p50. Additionally, the relatively high binding affinity of 3 was on the one hand attributed to the hydrogen bonding interaction between the hydroxyl group at C-14 and the side chain of residue Asn136, on the other hand to the spatial matching of section b and the large cavity (Fig. 7b, c). However, the esterification of hydroxyl group at C-14 (compounds 2, 4 and 5) aborted the hydrogen bonding formation, resulting in the weaker binding targeting p50.

The noncovalent binding on NF-κB p50. a Chemical structures of compounds 1–5. b The lowest binding energetic structures of andrographolide, 1 and 3. The NF-κB p50 protein was shown in cartoon, the hydrogen bonds were indicated in green dotted line, and the molecule was shown in stick. c The binding modes of andrographolide, 1 and 3 to the active site of NF-κB p50. NF-κB p50 was shown as van der Waals surface, and the positively charged residues and the hydrophobic residues were indicated in blue and green, respectively. The molecules were shown in stick (Color figure online)
Discussion
An expanding body of preclinical evidence suggested that overactivation of microglia and oxidative damage could initiate a cascade of intracellular events and eventually lead to neuronal dysfunction and death which play a critical role in neurodegenerative processes such as AD and PD [9]. In this study, we reported the neuroprotection of andrographolide in both microglia-mediated injury and oxidative damage on PC12 neurons. Over-activated microglia could release excess inflammation related mediators (NO, ROS, TNF-α, IL-β, IL-6, COX-2), which likely harm neighboring healthy cells including neurons. Therefore, BV-2 cells were activated by LPS and culture medium were harvested to stimulate PC12 cells for the determination of microglia-mediated injury. H2O2 and 6-OHDA-induced oxidative stress caused damage to major macromolecules such as lipids, proteins, and nucleic acids, resulting in apoptosis and neurotoxicity. Thus, H2O2 and 6-OHDA-induced PC12 cells were used for the evaluation of oxidative damage.
Firstly, we found that culture medium (CM) from activated BV-2 cells pretreated with andrographolide could protect PC12 neurons from apoptosis. However, andrographolide had no effect on CML-induced damage, suggesting that andrographolide exerted a neuroprotective effect probably due to block the inflammatory response induced by LPS. We next measured NO concentration in the culture supernatants of BV-2 cells by Griess assay. The results showed that NO production were severely inhibited. Moreover, the protein and mRNA expressions of iNOS were also suppressed by andrographolide. In addition to NO, andrographolide also inhibited the release of neuroinflammatory mediators TNF-α, IL-6 and ROS in activated microglia. Upon phagocytosis of invading bacteria, classically activated, pro-inflammatory (M1) microglia characterized by production of high levels of pro-inflammatory cytokines (NO, ROS, TNF-α, IL-1β, IL-6, COX-2) [27] serve in the first line of defence of the innate immune system. The production of anti-inflammatory factors CD206 and Arg-1 are involved in an alternative (M2) activation state to deactivate pro-inflammatory cell, repair damage and re-establish homeostasis [28]. Though no effect on the level of Arg-1, andrographolide could markedly upregulate the mRNA expression of CD206, indicating that the anti-inflammatory effect of andrographolide, at least in part, was associated with the microglial anti-inflammatory activation.
NF-κB is a key nuclear transcription factor and has been considered as an important regulator of proinflammatory cytokine production such as NO, ROS, TNF-α and IL-6. In unstimulated cells, NF-κB is retained in the cytoplasm where it is in complex with IκB. Conversely, stimulation of microglia by LPS leads to the release and translocation of NF-κB dimers to the nucleus and further induces the transcriptional activation of specific inflammatory genes. Consistent with reported signal pathways underlying microglial activation, our results showed that andrographolide reversed the nuclear translocation of p65 subunit of NF-κB in LPS-induced BV-2 cells. Moreover, previous study had demonstrated that andrographolide with an α, β-unsaturated lactone group could react with the nucleophilic Cys62 of NF-κB p50 through a Michael addition [29]. Nrf2 is an important endogenous redox-sensitive transcription factor [30] and forms Nrf2-Keap1 complex in the cytosol under normal conditions. Stimulants, such as electrophiles or oxidants, could modify the critical cysteine residue(s) in the regulatory protein Keap1 and cause the release and nucleus translocation of Nrf2. Several studies have showed that NF-κB and Nrf2 pathway may be concomitantly involved in the anti-inflammatory mechanism [31,32,33,34]. Our data also proved that the protein expression of Nrf2 in nuclei was significantly up-regulated by treatment of andrographolide. In addition, andrographolide could elevate mRNA level of Nrf2 downstream factor-HO-1, an inducible and cytoprotective enzyme, to strengthen the anti-inflammatory response. Taken together, all these results strongly suggested that andrographolide inhibited LPS-induced microglial activation implicated in suppressing NF-κB activation and inducing Nrf2-mediated heme oxygenase-1 (HO-1) expression. In addition, the structure–activity relationship revealed that compound 3 with the relatively high binding affinity toward NF-κB was more potent on NO inhibition compared with andrographolide.
Oxidative stress disrupts the survival, proliferation, and differentiation of neurons and has been considered as one of pathogenic causes in the neuropathology of adult neurodegenerative disorders. Our results showed that andrographolide could prevented PC12 cells against H2O2- and 6-OHDA-mediated oxidative damage. We also found that the production of ROS was effectively inhibited by andrographolide, suggesting that the inhibition of ROS accumulation in neuronal cells might be the molecular basis of the neuroprotective action of andrographolide. Moreover, increasement of activity was observed by introduction of acetyl group to C-14 and C-19 of andrographolide (compound 2). However, the underlying cause remains unknown and is the main focus of our following research.
Together, our data indicated the potent neuroprotective effect of andrographolide against neuroinflammation-mediated injury and oxidative damage (Fig. 8). It is unequivocal that suppression of neuroinflammation by andrographolide involved inhibition of pro-inflammatory mediators and cytokines (NO, TNF-α, IL-6, ROS) and increasement of anti-inflammatory factor CD206. Furthermore, Nrf2/HO-1 and NF-κB pathway may work in synergy to mitigate the inflammatory reaction induced by LPS. Our discovery provided deep insights in understanding the neuroprotection of andrographolide and strong experimental evidence supporting the therapeutic potential of andrographolide in relative to neuroinflammation and neurodegenerative diseases. Finally, our study revealed that addition of acetyl group to C-3 and C-19 of andrographolide may enhance the anti-inflammatory effect, while acetylation of C-14-hydroxyl group seems unfavorable to the activity.

Diagram summarizing the neuroprotection of andrographolide
Abbreviations
- DCFH-DA:
-
2,7-Dichlorofluorescein diacetate
- HO-1:
-
Heme oxygenase-1
- H2O2 :
-
Hydrogen peroxide
- iNOS:
-
Inducible nitric oxide synthase
- IL-6:
-
Interleukin-6
- LDH:
-
Lactate dehydrogenase
- LPS:
-
Lipopolysaccharide
- MTT:
-
3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- NF-κB:
-
Nuclear factor kappa B
- NO:
-
Nitric oxide
- Nrf2:
-
NF-E2-related factor-2
- 6-OHDA:
-
6-Hydroxydopamine
- ROS:
-
Reactive oxygen species
- TNF-α:
-
Tumor necrosis factor alpha
References
Mattson MPN (2000) Apoptosis in neurodegenerative disorders. Nat Rev Mol Cell Biol 1:120–129
Lees AJ, Hardy J, Revesz T (2009) Parkinson’s disease. Lancet 373:2055–2066
Dirnagl U, Iadecola C, Moskowitz M (1999) Pathobiology of ischemic stroke: an integrated view. Trends Neurosci 22:391–397
Hanisch UK (2002) Microglia as a source and target of cytokines. Glia 40:140–155
Streit WJ (2002) Microglia as neuroprotective, immunocompetent cells of the CNS. Glia 40:133–139
Fu R, Shen Q, Xu P, Luo JJ, Tang Y (2014) Phagocytosis of microglia in the central nervous system diseases. Mol Neurobiol 49:1422–1434
Neumann H, Kotter MR, Franklin RJM (2008) Debris clearance by microglia: an essential link between degeneration and regeneration. Brain 132:288–295
Block ML, Zecca L, Hong JS (2007) Microglia-mediated neurotoxicity: uncovering the molecular mechanisms. Nat Rev Neurosci 8:57–69
Barnham KJ, Masters CL, Bush AI (2004) Neurodegenerative diseases and oxidative stress. Nat Rev Drug Discov 3:205–214
Fang YZ, Yang S, Wu G (2002) Free radicals, antioxidants, and nutrition. Nutrition 18:872–879
Floyd RA (1999) Antioxidants, oxidative stress, and degenerative neurological disorders. Proc Soc Exp Biol Med 222:236–245
Sheeja K, Guruvayoorappan C, Kuttan G (2007) Antiangiogenic activity of Andrographis paniculata extract and andrographolide. Int Immunopharmacol 7:211–221
Wang T, Liu B, Zhang W, Wilson B, Hong JS (2004) Andrographolide reduces inflammation-mediated dopaminergic neurodegeneration in mesencephalic neuron-glia cultures by inhibiting microglial activation. J Pharmacol Exp Ther 308:975–983
Misra P, Pal NL, Guru PY, Katiyar JC, Srivastava V, Tandon JS (1992) Anti-malarial activity of Andrographis paniculata (kalmegh) against Plasmodium berghei NK 65 in mastomys natalensis. Int J Pharmacogn 30:263–274
Gupta S, Choudhry MA, Yadava JNS, Srivastava V, Tandon JS (1990) Anti-diarrheal activity of diterpenes of Andrographis paniculata (kalmegh) against Escherichia coli enterotoxin in vivo models. Int J Crude Drug Res 28:273–283
Chen JX, Xue HJ, Ye WC, Fang BH, Liu YH, Yuan SH, Yu P, Wang YQ (2009) Activity of andrographolide and its derivatives against influenza virus in vivo and in vitro. Biol Pharm Bull 32:1385–1391
Chiou WF, Lin JJ, Chen CF (1998) Andrographolide suppresses the expression of inducible nitric oxide synthase in macrophage and restores the vasoconstriction in rat aorta treated with lipopolysaccharide. Br J Pharmacol 125:327–334
Li J, Huang W, Zhang H, Wang X, Zhou H (2007) Synthesis of andrographolide derivatives and their TNF-α and IL-6 expression inhibitory activities. Bioorg Med Chem Lett 17:6891–6894
Tzeng YM, Lee YC, Cheng WT, Shih HN, Wang HC, Rao YK, Lee MJ (2012) Effects of andrographolide and 14-deoxy-11,12-didehydroandrographolide on cultured primary astrocytes and PC12 cells. Life Sci 90:257–266
Wang Y, Ruan W, Mi J, Xu J, Wang H, Cao Z, Saavedra JM, Zhang L, Lin H, Pang T (2018) Balasubramide derivative 3C modulates microglia activation via CaMKKβ-dependent AMPK/PGC-1α pathway in neuroinflammatory conditions. Brain Behav Immun 67:101–117
Kim J, Yang M, Son Y, Jang H, Kim D, Kim JC, Kim SH, Kang MJ, Im HI, Shin T, Moon C (2014) Glial activation with concurrent up-regulation of inflammatory mediators in trimethyltin-induced neurotoxicity in mice. Acta Histochem 116:1490–1500
Vo TS, Ngo DH, Ta QV, Wijesekara I, Kong CS, Kim SK (2012) Protective effect of chitin oligosaccharides against lipopolysaccharide-induced inflammatory response in BV-2 microglia. Cell Immunol 277:14–21
Chen JC, Ho FM, Chao PDL, Chen CP, Jeng KCG, Hsu HB, Lee ST, Wu WT, Lin WW (2005) Inhibition of iNOS gene expression by quercetin is mediated by the inhibition of IκB kinase, nuclear factor-kappa B and STAT1, and depends on heme oxygenase-1 induction in mouse BV-2 microglia. Eur J Pharmacol 521:9–20
Sirion U, Kasemsook S, Suksen K, Piyachaturawat P, Suksamrarn A, Saeeng R (2012) New substituted C-19-andrographolide analogues with potent cytotoxic activities. Bioorg Med Chem Lett 22:49–52
Schaftenaar G, Vlieg E, Vriend G (2017) Molden 2.0: quantum chemistry meets proteins. J Comput Aided Mol Des 31:789–800
Cosconati S, Forli S, Perryman AL, Harris R, Goodsell DS, Olson AJ (2010) Virtual screening with AutoDock: theory and practice. Expert Opin Drug Discov 5:597–607
Perego C, Fumagalli S, De Simoni MG (2011) Temporal pattern of expression and colocalization of microglia/macrophage phenotype markers following brain ischemic injury in mice. J Neuroinflamm 8:174
Orihuela R, McPherson CA, Harry GJ (2016) Microglial M1/M2 polarization and metabolic states. Br J Pharmacol 173:649–665
Nguyen VS, Loh XY, Wijaya H, Wang J, Lin Q, Lam Y, Wong WSF, Mok YK (2015) Specificity and inhibitory mechanism of andrographolide and its analogues as antiasthma agents on NF-κB p50. J Nat Prod 78:208–217
Joshi G, Johnson JA (2012) The Nrf2-ARE pathway: a valuable therapeutic target for the treatment of neurodegenerative diseases. Recent Pat CNS Drug Discov 7:218–229
Chen J, Yin W, Tu Y, Wang S, Yang X, Chen Q, Zhang X, Han Y, Pi R (2017) L-F001, a novel multifunctional ROCK inhibitor, suppresses neuroinflammation in vitro and in vivo: involvement of NF-κB inhibition and Nrf2 pathway activation. Eur J Pharmacol 806:1–9
Park SY, Kim YH, Kim Y, Lee SJ (2012) Aromatic-turmerone’s anti-inflammatory effects in microglial cells are mediated by protein kinase A and heme oxygenase-1 signaling. Neurochem Int 61:767–777
Seo JY, Pyo E, Park J, Kim JS, Sung SH, Oh WK (2017) Nrf2-mediated HO-1 induction and antineuroinflammatory activities of halleridone. J Med Food 20:1091–1099
Sun GY, Chen Z, Jasmer KJ, Chuang DY, Gu Z, Hannink M, Simonyi A (2015) Quercetin attenuates inflammatory responses in BV-2 microglial cells: role of MAPKs on the Nrf2 pathway and induction of heme oxygenase-1. PLoS ONE 10:e0141509
Funding
This work was financially supported by a grant from the National Natural Science Funds (No. 81602994) and the Scientific Startup Foundation for Doctors of Northwest A&F University (No. 2452015353).
Author information
Authors and Affiliations
Contributions
JG conceived of the study; YX, DT and HW performed experiments; JW executed MD simulations; YX analyzed data and wrote the paper.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Xu, Y., Tang, D., Wang, J. et al. Neuroprotection of Andrographolide Against Microglia-Mediated Inflammatory Injury and Oxidative Damage in PC12 Neurons. Neurochem Res 44, 2619–2630 (2019). https://doi.org/10.1007/s11064-019-02883-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11064-019-02883-5