
Abstract
Mitochondrial dysfunction has been proposed to be one of the earliest triggering events in isoflurane-induced neuronal damage. Lidocaine has been demonstrated to attenuate the impairment of cognition in aged rats induced by isoflurane in our previous study. In this study, we hypothesized that lidocaine could attenuate isoflurane anesthesia-induced cognitive impairment by reducing mitochondrial damage. H4 human neuroglioma cells and 18-month-old male Fischer 344 rats were exposed to isoflurane or isoflurane plus lidocaine. Cognitive function was tested at 14 days after treatment by the Barnes Maze test in male Fischer 344 rats. Morphology was observed under electron microscope, and mitochondrial transmembrane potential, electron transfer chain (ETC) enzyme activity, complex-I–IV activity, immunofluorescence and flow cytometry of annexin V-FITC binding, TUNEL assay, and Western blot analyses were applied. Lidocaine attenuated cognitive impairment caused by isoflurane in aged Fischer 344 rat. Lidocaine was effective in reducing mitochondrial damage, mitigating the decrease in mitochondrial membrane potential (ΔΨm), reversing isoflurane-induced changes in complex activity in the mitochondrial electron transfer chain and inhibiting the apoptotic activities induced by isoflurane in H4 cells and Fischer 344 rats. Additionally, lidocaine suppressed the ratio of Bax (the apoptosis-promoting protein) to Bcl-2 (the apoptosis-inhibiting protein) caused by isoflurane in H4 cells. Lidocaine proved effective in attenuating isoflurane-induced POCD by reducing mitochondrial damage.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Post-operative cognitive dysfunction (POCD) is a clinical condition characterized by alterations in memory, learning, concentration and attention after the operation or/and anesthesia [1, 2]. Many research results have indicated that an increasing number of aged patients are diagnosed with POCD. It has been reported that POCD affects approximately 41.4% of aged patients after non-cardiac operations [3], and the cognitive changes can persist for 3 months [4]. POCD disrupts the lives of patients and increases the cost of medical care, thus determining how to attenuate this cognitive dysfunction remains an urgent problem.
The underlying pathophysiology of POCD is increasingly understood, implicating a prominent role of neuroinflammation and mitochondrial dysfunction [2]. Some theories regarding the mechanism responsible for POCD highlight the process of neuroinflammation, which includes tumor necrosis factor alpha (TNF-α) and pro-inflammatory cytokines interleukin (IL)-1β and IL-6 [5]. On the other hand, mitochondrial dysfunction is increasingly considered a significant contributor to POCD [6, 7]. As mitochondrial sources are the main provider of the energy needed for neurons to survive, neurons show great vulnerability to death or injury caused by mitochondrial dysfunction [8]. As a major functional component of cells, mitochondria are of great importance in modulating the functions of cells and in determining the ultimate death-survival balance in a cellular environment as effectors and detectors [9]. Mitochondrial dysfunction can lead to a drastic decrease in the generation of adenosine triphosphate (ATP) and potentiate oxidative damage to neurons [10]. Mitochondrial fragmentation during dysfunction was associated with mitochondrial osmotic swelling, collapse of the mitochondrial membrane potential (ΔΨm) and dysfunction of the electron transport chain [11]. Isoflurane, an ordinarily used inhalational anesthetic, is of great significance during the pathological process of POCD [12, 13]. Mitochondria have been proposed to be one of the targets of isoflurane [14]. A disequilibrium of mitochondria has been considered to be one of the earliest triggering events in isoflurane-induced neuronal damage [15]. Nevertheless, the exact mechanism of POCD induced by isoflurane has not been thoroughly elucidated to date.
Lidocaine, a local anesthetic, may confer neuroprotection by reducing the release of excitatory amino acid, decelerating the ischemic transmembrane ion shift, reducing the metabolic rate of the brain, modulating the inflammatory response and preserving the cerebral blood flow [16,17,18,19,20,21]. In a previous study, we have demonstrated that lidocaine can attenuate the impairment of cognition in aged rats induced by isoflurane, but it shows no impact on the amount of IL-1β, TNF-α in the hippocampus [22]. However, the mechanism by which lidocaine attenuates the isoflurane-induced impairment of cognition in aged rats is still not known. Since isoflurane can induce mitochondrial dysfunction and cause oxidative damage [15], in this study, we hypothesized that lidocaine attenuates cognitive impairment after isoflurane anesthesia by reducing mitochondrial damage.
Materials and Methods
Treatment of the Cells
Cell Groups
The China Center for Type Culture Collection in WuHan provided the experimental H4 human neuroglioma cells (H4 cells), which were derived from a human glioma cell line. Dulbecco’s Modified Eagle’s Medium (DMEM, 4.5 g/L glucose, HyClone, USA) supplemented with 100 μg/mL streptomycin, 100 Units/mL penicillin (Gibco, USA) and 10% fetal bovine serum (FBS, Gibco, USA) was used to culture the H4 cells. H4 cells were placed at a density of 0.5 × 106/mL in six-well plates, and the experiments were repeated for three times with two wells for each group. The cells were divided into six groups: control (CON group), isoflurane (ISO group), isoflurane plus 40 μg/mL lidocaine (ISO + LIDO 40 μg/mL), isoflurane plus 60 μg/mL lidocaine (ISO + LIDO 60 μg/mL), isoflurane plus 80 μg/mL lidocaine (ISO + LIDO 80 μg/mL), isoflurane plus 100 μg/mL lidocaine (ISO + LIDO 100 μg/mL). Isoflurane exposure was performed as we described previously [23]. Briefly, the culture medium for the ISO group and the ISO + LIDO group was pregassed with 3% isoflurane in advance, meanwhile, the corresponding concentration of lidocaine was added to the culture medium for the lidocaine intervention group. After replacing the pretreatment medium, the H4 cells were placed in a chamber gassed with 3% isoflurane/21% O2/5% CO2, a Datex TM infrared analyzer (Capnomac, Helsinki, Finland) was used to monitor the concentrations of gas exiting from the chamber. The chamber was sealed and cultured in a 37 °C incubator for 2 h.
Treatment of the Animals
Animal Groups
The Vital River Company (Beijing, China) provided 18-month-old male Fischer 344 rats with a weight between 470 and 550 g for our experiments. The rats were grouped into three categories in a random manner: isoflurane plus lidocaine (ISO + LIDO), isoflurane alone (ISO) and the control group (CON) (n = 6). The rats in ISO and ISO + LIDO groups were treated with 1.2% isoflurane for two consecutive hours. Additionally, the ISO + LIDO rats were simultaneously treated with intravenous lidocaine (1.5 mg/kg as a bolus followed by 2 mg/kg/h during the 2 h of isoflurane treatment), as described by our group previously [22]. In the ISO + LIDO group, lidocaine treatment was administered through tail vein injection of 8 mg/mL lidocaine saline solution. To ensure the consistency of the experiment, the same quantity of saline was injected into the rats in the ISO group. Considering that the isoflurane was mixed with 21% O2/79% N2, to confirm the reliability the experiment, CON rats were exposed to 21% O2/79% N2 for two consecutive hours. The animal experiments were approved by the Sun Yat-sen Memorial Hospital Ethics Committee.
Isoflurane Anesthesia
According to a previous description [24], the experimental rats were anaesthetized by exposure to a mixture of 21% O2 plus 79% N2 and 1.2% isoflurane. Briefly, the animals were anesthetized with 1.2% isoflurane and inserted intubation to mechanically ventilation. A MouseOx™ Pulse Oximeter (Harvard Apparatus, Holliston, MA, USA) was used to continuously measure the pulse oximeter-oxygen (SpO2) and heart rate of the rats during the anesthesia process, and a CODA Monitor (Kent Scientific Corp., Torrington, CT, USA) was used to non-invasively measure the blood pressure of the rats. During anesthetization, a Datex TM infrared analyzer (Capnomac, Helsinki, Finland) was used to continuously monitor the concentrations of exhaled and inhaled gas (oxygen and isoflurane).
Barnes Maze
After all the above procedures were completed, the rats were kept for 14 days and then subjected to the Barnes maze. As we have described previously [24], the rats were placed on a round platform with 20 equally spaced holes (SD Instruments, San Diego, CA). Among all of the holes, only one was linked to the target box, which was actually a dark chamber. The rats were expected to find this very hole and enter the target box under the circumstance of bright light (200 W) and aversive noise (85 dB). The rates were subjected to a 4-day training period, with two trials per day lasting 3 min each; the second trial was started no sooner than 15 min after the first one had ended. On 5th and 12th day, the reference memory of the rats was tested to evaluate their short-term and long-term retention, respectively. Each rat was subjected to one trial on 5th and 12th day, with no other tests during the intervening 6 days. The ANY-Maze video tracking system (SD Instruments) assisted the researchers in recording number of errors for the latency of the rats in finding the right hole leading to the target box in all trials.
Harvesting of Brain Tissue
Thirty minutes after Barnes maze test, the rats were deeply anaesthetized with isoflurane and perfused transcardially with normal saline. Brains were dissected in air, the right hippocampi was immediately dissected for electronic microscopic examination, mitochondrial transmembrane potential and the left hippocampi was harvested for electron transfer chain enzyme activity, and the TUNEL assay.
Electron Microscopy
After treatment, the H4 cells were embedded in Epon812 epoxy resin and sectioned. Subsequently, toluidine blue was used to stain the sections, which were then observed under a light microscope. Similarly, after the intervention, the rats were sacrificed by cervical dislocation. Brain tissue was quickly and completely removed, and hippocampal tissue was dissected. H4 cells and brain tissues of rats were fixed and observed under an electron microscope (Hitachi FE-SEM SU8000, Japan) focus on mitochondria. Five fields were randomly selected in the electron microscopy images of each specimen, and at least 20 mitochondria were randomly selected in each field to obtain a semiquantitative score for the mitochondria according to the Flameng classification system [25, 26]. Mitochondria were graded based on scores ranging from 0 to 4 according to the degree of damage, with a higher score representing a higher degree of damage. The damage to each mitochondrion and the average score for all mitochondria were evaluated independently by at least two investigators to avoid bias.
Measurement of the Mitochondrial Transmembrane Potential (ΔΨm)
After treatment, 5 μg/mL JC-1® (Molecular Probes, Leiden, The Netherlands) was applied to the H4 cells and rat brain tissue [27], which were then analyzed using a Becton–Dickinson FACScan® flow cytometer (Becton–Dickinson, Oxford, UK). The results were analyzed using CellQuest® software. The mitochondrial transmembrane potential was evaluated by flow cytometry to detect JC-1 aggregates/JC-1 monomers.
Electron Transfer Chain (ETC) Enzyme Activity Assay
The activity of ETC enzymes was determined based on the activity of the specific donor–acceptor oxidoreductase [9, 28]. The mitochondria isolated from H4 cells and brain tissues of rats were solubilized in 2% cholic acid and then diluted to a mitochondrial protein concentration of 100 μg/mL. The final experimental buffer contained 0.2% BSA, 5 mM 3-(N-morpholino) propanesulfonic acid, 2 mM EDTA, 220 mM d-mannitol and 70 mM sucrose with a pH value of 7.4.
Complex I (NADH-Ubiquinone Oxidoreductase) Activity Assay
In the complex I activity reaction, 20 μg/mL mitochondrial protein prepared previously was acquired and added to a spectrophotometer cuvette containing 2 mM antimycin A, 50 mM KH2PO4, 0.15 mg/mL asolectin, 0.1% BSA, 0.2 mM NADH and 0.1 mM EDTA, followed by the addition of 75 mM decylubiquinone as an inhibitor. Measurements of the alterations in NADH absorbance were conducted at 340 nm (ε = 6.22 mM−1 cm−1).
Complex II (Succinate-Ubiquinone Oxidoreductase) Activity Assay
In the complex II activity reaction, 20 μg/mL mitochondrial protein was acquired and added to a spectrophotometer cuvette containing 5 mM NaN3, 50 mM KH2PO4, 0.5 mM duroquinone, 25 mM dichlorophenolindophenol, 0.1 mM EDTA, and 0.1% BSA, followed by the addition of 20 mM succinate as an inhibitor. Measurements of the alterations in dechlorophenolindophenol absorbance were conducted at 600 nm (ε = 21 mM−1 cm−1).
Complex III (Ubiquinone-Cytochrome c Reductase) activity Assay
In the complex III activity reaction, 5 μg/mL mitochondrial protein was acquired and added to a spectrophotometer cuvette containing 60 μM oxidized cytochrome c, 50 mM KH2PO4, 0.1 mM EDTA, 5 mM NaN3, and 0.1% BSA, with 100 μM of the inhibitor decylubiquinol. Measurements of the alterations in cytochrome c absorbance were conducted at 550 nm (ε = 18.5 mM−1 cm−1).
Complex IV (Cytochrome c Oxidase) Activity Assay
The assay mixture was acquired in the complex IV activity reaction containing 20 μg/mL mitochondrial protein, 40 μM reduced cytochrome c, 0.15 mg/mL asolectin, and 50 mM KH2PO4, supplemented with 1 μg/mL of mitochondrial protein inhibitor. Measurements of the alterations in cytochrome c absorbance were conducted at 550 nm (ε = 18.5 mM−1 cm−1).
Immunofluorescence Flow Cytometry of Annexin V-FITC Binding
Annexin V-FITC binding to phosphatidylserine was considered a sensitive measurement of neural cell apoptosis [29]. In brief, annexin V-FITC (0.6 µg/mL) and propidium iodide (Sigma) (10 µg/mL) were used for dual staining of H4 cells (0.5 × 106 cells/mL), and the stained cells were then diluted in binding buffer (140 mmol NaCl, 10 mmol HEPES/NaOH, pH 7.4, 2.5 mmol CaCl2) at room temperature for 5 min. The cells were analyzed using a Becton–Dickinson FACScan flow cytometer and then programmed with CellQuest software. The flow cytometer was also used to determine cell apoptosis via detection of Annexin V-FITC/PI.
TUNEL Assay
Formalin was used to buffer the hippocampus of the rats, which was then embedded in paraffin for TUNEL staining as described previously [24]. Based on the protocol proposed by the manufacturer, an in situ cell death detection kit (POD; Roche Diagnostics Corp, Indianapolis, IN, USA) was used for the TUNEL staining. Brown nuclei were counted in 10 randomly selected fields of each section under the microscope. The apoptotic index was determined by calculating the proportion of brown nuclei in the CA1 region of rats’ hippocampus.
Western Blot Analysis
The Western blot analysis was performed according to previously conducted studies [30]. After exposure to 3% isoflurane (with or without added 100 μg/mL lidocaine) for two consecutive hours, the H4 cells were detached by 30 s of sonication and scraping in lysis buffer. The 12% gel electrophoresis was used to separate 50 μg of total cell protein, which was then transferred onto nitrocellulose membranes (Bio-Rad Laboratories, Hercules, CA). Bcl-2 (Cell Signaling Technology, MA, USA). Alternately, a monoclonal antibody against Bax (Cell Signaling Technology, MA, USA) was used for incubation of the blots, and horseradish peroxidase-conjugated secondary antibody (Cell Signaling Technology, MA, USA) was subsequently used to probe these blots. The ECL-PLUS system was used to detect the immunoreactive bands, and β-actin (Cell Signaling Technology, MA, USA) served as the loading control in the images of the bands captured using alpha Image software (Alpha Innotech, Santa Clara, CA, USA).
Statistical Analysis
All data were expressed as mean ± S.D. and statistically analyzed using SPSS20.0. One-way ANOVA was applied followed by the least significant difference (LSD) post hoc test for inter-group comparisons. Two-way repeated measures analysis of variance followed by Tukey test were used for comparisons of the data of training sessions of Barnes maze test. Statistical significance was confirmed when p < 0.05.
Results
Lidocaine Attenuates Cognitive Impairment Induced by Isoflurane in Aged Fischer 344 Rats
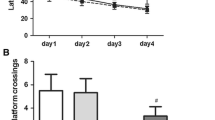
The time required for all the rats to find the right hole to the target box significantly decreased on day 4 of training compared with day 1, which indicated that all the rats achieved performance development during the training course (Fig. 1). However, rats in the isoflurane group required a longer time to identify the target holes and selected a greater number of incorrect holes before finding the correct holes compared to the rats in the control group when assessed 1 or 8 days after spatial learning training in the Barnes maze (Fig. 2). The ISO + LIDO group showed no significant difference in comparison to the CON group in the tests, suggesting that lidocaine was effective in attenuating the cognitive impairment induced by isoflurane in aged rats. This result is consistent with our previous findings [22].

Performance during the training phase of the Barnes maze test: at 14 days after isofluane exposure with or without injection of lidocaine, a series of training trials were conducted in a Barnes maze. It would be good to see if the fourth day results are significantly different. The data are presented as the mean ± S.D. (n = 6). *P < 0.05 in comparison to the data obtained in the first trial on training day 1

Performance during the memory phase of Barnes maze test: the time to identify the target holes (a) and the number of incorrect holes searched before identifying the correct hole connected to the target box (b) in the Barnes maze test were tabulated. Subsequently, the rats were subjected to a reference memory test day 1 and 8. The data are presented as mean ± S.D. (n = 6). *P < 0.05 compared with the CON group. ^P < 0.05 compared with the ISO group
Lidocaine Reduces the Mitochondrial Structure Damage Induced by Isoflurane in H4 Cells and Fischer 344 Rat Hippocampus
The pathological features of the mitochondrial ultrastructure were observed in H4 cells and rat brain tissues. The morphology of the mitochondria changed with the influence of isoflurane. In the control group, the mitochondrial structure was normal, and intact (Fig. 3A-CON,B-CON), mitochondrial score was 0.69 ± 0.42 in H4 cell and 0.55 ± 0.11 in rat brain tissues. In the isoflurane group, the mitochondrial were swollen and lost its inner structure (Fig. 3A-ISO, B-ISO), mitochondrial score was 2.51 ± 0.73 in H4 cell and 2.84 ± 0.50 in rat brain tissues. Lidocaine reduced the above mitochondrial damage induced by isoflurane (Fig. 3A-ISO + LIDO,B-ISO + LIDO), mitochondrial score was 0.58 ± 0.37 in H4 cell and 0.80 ± 0.54 in rat brain tissues (p < 0.05, ISO + LIDO group vs. ISO group).

Lidocaine is effective in reducing isoflurane-induced mitochondrial structure injuries in H4 cells and Fischer 344 rat hippocampus. The cells (A-CON, A-ISO, A-ISO + LIDO) and hippocampus were harvested after the Barnes maze test (B-CON, B-ISO, B-ISO + LIDO) for observation under microscope. Lidocaine was effective in reducing mitochondrial injury. The objects indicated by red arrows are mitochondria (magnification: × 40,000, scale bar in each panel = 1 μm) (Color figure online)
Lidocaine Reduces the Decrease in Isoflurane-Induced Membrane Potential (ΔΨm) of Mitochondria in H4 Cells and Fischer 344 Rat Hippocampus
The JC-1 staining results demonstrated that isoflurane could destroy the membrane potential of mitochondria and that lidocaine significantly reduced this damage in H4 cells (80 μg/mL LIDO group and 100 μg/mL LIDO group) and the hippocampus of Fischer 344 rats (100 μg/mL LIDO group) (Fig. 4).

Lidocaine is effective in reducing the decrease in isoflurane-induced membrane potential (ΔΨm) of mitochondria in H4 cells and Fischer 344 rat hippocampus. a. Lidocaine dose-dependently reduced the decrease in ΔΨm induced by isoflurane. b Lidocaine reduced the decrease in ΔΨm induced by isoflurane. The data are expressed as mean ± S.D. (n = 6). *P < 0.05 compared with the CON group. ^P < 0.05 compared with the ISO group
Lidocaine Reverses Isoflurane-Induced Changes in the Activity of Mitochondrial Electron Transfer Chain (ETC) Complexes in H4 Cells and Hippocampus of Fischer 344 Rats
The results of the activity of ETC complexes obtained for the rat hippocampus and H4 cells presented consistent results. Mitochondrial respiratory chain complex I and II activities were significantly increased by isoflurane, and this increase was attenuated by lidocaine. The activity of complex III increased by isoflurane, but it’s not statistically significant. The activity of complex IV was inhibited by isoflurane, and after lidocaine treatment, the activity was increased (Fig. 5). Although the changing trend of the four respiration chain enzyme complexes was different, the changes followed a certain rule: lidocaine could reverse the activity changes of the complexes in the mitochondrial respiratory chain induced by isoflurane.

In H4 cells and Fischer 344 rat hippocampus, the isoflurane-induced activity of the complexes in the mitochondrial electron transfer chain is altered by lidocaine. a H4 cells were treated with or without a 2-h 3% isoflurane exposure with or without lidocaine. b. Male experimental Fischer 344 rats aged 18 months were treated with or without a 2-h 1.2% isoflurane exposure with or without injection of lidocaine. The activity of complex I–IV in the mitochondrial respiratory chain was assayed. The data are expressed as the mean ± S.D. (n = 6). *P < 0.05 compared with the CON group. ^P < 0.05 compared with the ISO group
Lidocaine Inhibits the Apoptotic Activity Induced by Isoflurane in H4 Cells and Hippocampus of Fischer 344 Rats
The results showed that (Fig. 6) compared with the control group, the numbers of the apoptotic cells in the isoflurane group increased and that lidocaine caused a dose-dependent reduction of this increase in apoptosis (P < 0.05, compared with the isoflurane group). As shown in Fig. 7, the proportion of TUNEL-positive among total nuclei in the rat hippocampus was significantly increased in the ISO group compared with the CON group. This tendency toward an increase was suppressed with lidocaine treatment.

Lidocaine is effective in inhibiting H4 cell isoflurane-induced apoptotic activities. The apoptotic rate of cells after intervention was then examined by immunofluorescence flow cytometry. The data are expressed as mean ± S.D. (n = 6). *P < 0.05 compared with the CON group. ^P < 0.05 compared with the ISO group

In Fischer 344 rat hippocampus, the apoptotic activity induced by isoflurane is inhibited by lidocaine treatment. Aa Control group, Ab isoflurane group, Ac isoflurane + lidocaine group. The graphs in B display the data quantitation. The data are presented as mean ± S.E.M. (n = 6). *P < 0.05 compared with the CON group. ^P < 0.05 compared with the ISO group (Color figure online)
Lidocaine Suppresses the H4 Cell Isoflurane-Induced Ratio of Bax to Bcl-2 Protein
As shown in Fig. 8, 3% isoflurane for 2 h significantly increased the Bax/Bcl-2 ratio, and this increase was significantly attenuated by lidocaine.

Lidocaine is effective in suppressing the H4 cell isoflurane-induced ratio of Bax to Bcl-2 protein. After intervention, the total protein of H4 cells was then isolated for Western blot analysis. β-actin served as a loading control. a Images from the Western blot analysis showed how Bcl-2 and Bax proteins were impacted by the treatment with 3% isoflurane plus lidocaine or 3% isoflurane alone. b The ratio of Bax to Bcl-2 protein was affected by treatment with lidocaine and isoflurane. The data are expressed as mean ± S.E.M. (n = 6). *P < 0.05 compared with the CON group. ^P < 0.05 compared with the ISO group
Discussion
According to the results of the present study, lidocaine is effective in attenuating isoflurane-induced POCD by reducing mitochondrial damage. In the aged Fischer 344 rat model of cognitive impairment caused by isoflurane-induced anesthesia, lidocaine-treated rats took less time and made fewer mistakes in identifying the target holes than rats treated with isoflurane alone. Our results demonstrated that lidocaine was effective in reducing the mitochondrial damage induced by isoflurane, including: structure, ΔΨm, complexs activities of ETC, apoptotic activities, and mitochondrial apoptosis-related proteins (Bax/Bcl-2).
The pathophysiology under PCOD focuses on the roles of neuroinflammation and the stress response [2, 31]. Our findings expand the possible mechanism underlying POCD by showing that mitochondrial damage could make a significant contribution to cognitive impairment. Mitochondrial abnormalities play an indispensable role in network processes such as cognition impairment [32]. Because the most sufficient energy supply for neuron survival is dependent on mitochondrial sources, the brain, an important aerobic organ, is extremely vulnerable to mitochondrial dysfunction. Mitochondrial dysfunction was one of the earliest triggering events in isoflurane-induced neuronal damage [33]. Moreover, studies have suggested that mitochondrial dysfunction is involved in different neurodegenerative disorders, including Alzheimer’s, Parkinson’s, and Huntington’s diseases and amyotrophic lateral sclerosis [33]. The impaired mitochondria could exhibit an altered morphology, decreased membrane potential and disrupted function of electron transport chain (ETC) complexes, which are considered to play key role in the incidence of POCD [31]. According to the findings of the present study, mitochondrial damage in H4 cells treated with isoflurane and the cognitive impairment observed in rats after isoflurane anesthesia further confirm that mitochondrial damage could make an important role in the POCD process. Moreover, we found that lidocaine could attenuate the mitochondrial damage induced by isoflurane.
Our findings suggest that the mitochondria played an important role in the pathophysiological mechanism of POCD induced by isoflurane. Because mitochondria are of great importance in numerous biological processes, including cell apoptosis, intracellular signaling, energy generation and the respiratory activities of cells [7, 34], the damage to these dynamic organelles can alter performance in many ways. An abnormal mitochondrial morphology is one obvious sign of their damage. The normal structure of mitochondria disappeared and vacuoles appeared in H4 cells treated with isoflurane and in the hippocampi of cognitively impaired rats after isoflurane anesthesia. Our findings confirm that mitochondria damage is induced by isoflurane. Functionally, in our study, the mitochondrial membrane potential (ΔΨm) decreased, mitochondrial ETC complex dysfunction was observed, and apoptosis increased in H4 cells treated with isoflurane and in the hippocampi of cognitively impaired rats after isoflurane anesthesia. Additionally, the ratio of Bax (the protein that promotes apoptosis) to Bcl-2 (the protein that inhibits apoptosis) increased in H4 cells treated with isoflurane. The decrease in ΔΨm revealed a change in the mitochondrial outer membrane permeability (MOMP), which is mainly subjected to the mediation of Bcl-2 and Bax protein. Opening of mitochondrial permeability transition pore (mPTP) and changes in calcium buffering make the cytosol accessible to proteins in the space between mitochondrial membranes, including the mitochondrial-to-cytosol release of cytochrome c, which then contributes to the activation of caspases that function to promote cellular apoptotic activities [35, 36]. Thus, mitochondrial dysfunction was induced by isoflurane and, in turn, triggered the mitochondrial apoptosis pathway. In contrast, there has been an association between mitochondrial ETC complex dysfunction and nerve cell dysfunction as causal factors of many chronic age-related neurodegenerative diseases [37, 38]. Studies have shown that the abnormality of any complex enzyme (including complex I, II, III and IV) would affect the function of ECT [39]. In our study, the activity of complex I and II increased, complex III did not change, and complex IV decreased after isoflurane treatment both in cells and in the animal experiment. Complex IV is the terminal complex of the ECT, the reduced activity of which has been consistently observed in post-mortem disease samples from Alzheimer’s patients [40]. Furthermore, inhibition of the activity of complex IV could increase Ca2+-independent glutamate release, which has been shown to be associated with excitotoxic cell death [38]. According to the findings of the present experiment, complex IV activity declined in response to isoflurane, which indicated that the excitotoxic cell death in neurons might result from exposure to isoflurane. Additionally, except for complex IV, the activity of I, II and III did not decreased on effect of isoflurane, suggesting that the target of mitochondrial damage induced by isoflurane might be complex IV.
Lidocaine has been reported to have neuroprotective functions by reducing the release of ischemic excitotoxin [41]. Additionally, excitotoxic death might result from mitochondrial damage and excessive mitochondrial calcium accumulation [42]. According to the findings of the present experiment, lidocaine was effective in attenuating mitochondrial damage induced by isoflurane, specifically in reducing the mitochondrial structure damage and the decline in mitochondrial membrane potential (ΔΨm), as well as in reversing isoflurane-induced changes in complex activity in the mitochondrial electron transfer chain and inhibiting the apoptotic activities induced by isoflurane. The potential mechanism underlying lidocaine-attenuated mitochondrial damage induced by isoflurane might be associated with a reduction of excitotoxin release, even excitotoxic cell death, induced by isoflurane. However, some existing studies have reported results that are inconsistent with our findings. Lidocaine have been reported to be involved in mitochondrial dysfunction and implicated in the intrinsic mitochondrial death pathway [42]. The different results may be explained by the concentration of lidocaine used. Michael [43] found that a concentration of lidocaine of 2.3 mM (0.06%) and higher caused neuronal death in that study. Robert [44] indicated that lidocaine concentrations of 3–6 mM (0.08–0.16%) induced apoptotic activities that may result from overexpressed Bcl-2 or from the deficiency of caspase-9. In our study, the maximum lidocaine concentration was 100 μg/mL (0.37 mM, 0.01%), which was lower than the above effective apoptotic concentration. The neuroprotective effects of lidocaine found in this study may indicate multiple effects of lidocaine other than Na+ blockade and have important implication for the prevention and treatment of POCD.
Our study has several limitations. First, we found that complex IV might be the target of mitochondrial damage induced by isoflurane, but we did not explore the effect of lidocaine and isoflurane on complex IV in terms of whether it is related to Ca2+-independent glutamate release leading to excitotoxic cell death. Second, we found that lidocaine was only effective in attenuating isoflurane-induced POCD by reducing mitochondrial damage, but the underlying mechanism is still unclear and requires further analysis.
In conclusion, our study provides evidence that lidocaine is effective in attenuating isoflurane-induced POCD by reducing mitochondrial damage in vitro and vivo. The finding of our study may have pragmatic implications stimulating a larger number of studies on the exact mechanisms underlying the neurotoxicity of isoflurane anesthesia and target interventions in isoflurane-induced POCD.
References
Rudolph JL, Schreiber KA, Culley DJ, McGlinchey RE, Crosby G, Levitsky S, Marcantonio ER (2010) Measurement of post-operative cognitive dysfunction after cardiac surgery: a systematic review. Acta Anaesthesiol Scand 54:663–677
Skvarc DR, Berk M, Byrne LK, Dean OM, Dodd S, Lewis M, Marriott A, Moore EM, Morris G, Page RS, Gray L (2018) Post-operative cognitive dysfunction: an exploration of the inflammatory hypothesis and novel therapies. Neurosci Biobehav Rev 84:116–133
Monk TG, Weldon BC, Garvan CW, Dede DE, van der Aa MT, Heilman KM, Gravenstein JS (2008) Predictors of cognitive dysfunction after major noncardiac surgery. Anesthesiology 108:18–30
Price CC, Garvan CW, Monk TG (2008) Type and severity of cognitive decline in older adults after noncardiac surgery. Anesthesiology 108:8–17
Kohl BA, Deutschman CS (2006) The inflammatory response to surgery and trauma. Curr Opin Crit Care 12:325–332
Haxaire C, Turpin FR, Potier B, Kervern M, Sinet PM, Barbanel G, Mothet JP, Dutar P, Billard JM (2012) Reversal of age-related oxidative stress prevents hippocampal synaptic plasticity deficits by protecting d-serine-dependent NMDA receptor activation. Aging Cell 11:336–344
Golpich M, Amini E, Mohamed Z, Azman AR, Mohamed IN, Ahmadiani A (2017) Mitochondrial dysfunction and biogenesis in neurodegenerative diseases: pathogenesis and treatment. CNS Neurosci Ther 23:5–22
Golpich M, Amini E, Hemmati F, Ibrahim NM, Rahmani B, Mohamed Z, Raymond AA, Dargahi L, Ghasemi R, Ahmadiani A (2015) Glycogen synthase kinase-3 beta (GSK-3beta) signaling: implications for Parkinson’s disease. Pharmacol Res 97:16–26
O’Toole JF, Patel HV, Naples CJ, Fujioka H, Hoppel CL (2010) Decreased cytochrome c mediates an age-related decline of oxidative phosphorylation in rat kidney mitochondria. Biochem J 427:105–112
Sullivan PG, Rabchevsky AG, Waldmeier PC, Springer JE (2005) Mitochondrial permeability transition in CNS trauma: cause or effect of neuronal cell death? J Neurosci Res 79:231–239
Jeong SY, Seol DW (2008) The role of mitochondria in apoptosis. BMB Rep 41:11–22
Xie Z, Culley DJ, Dong Y, Zhang G, Zhang B, Moir RD, Frosch MP, Crosby G, Tanzi RE (2008) The common inhalation anesthetic isoflurane induces caspase activation and increases amyloid beta-protein level in vivo. Ann Neurol 64:618–627
Xie Z, Dong Y, Maeda U, Alfille P, Culley DJ, Crosby G, Tanzi RE (2006) The common inhalation anesthetic isoflurane induces apoptosis and increases amyloid beta protein levels. Anesthesiology 104:988–994
Zhang Y, Dong Y, Wu X, Lu Y, Xu Z, Knapp A, Yue Y, Xu T, Xie Z (2010) The mitochondrial pathway of anesthetic isoflurane-induced apoptosis. J Biol Chem 285:4025–4037
Sanchez V, Feinstein SD, Lunardi N, Joksovic PM, Boscolo A, Todorovic SM, Jevtovic-Todorovic V (2011) General anesthesia causes long-term impairment of mitochondrial morphogenesis and synaptic transmission in developing rat brain. Anesthesiology 115:992–1002
Fried E, Amorim P, Chambers G, Cottrell JE, Kass IS (1995) The importance of sodium for anoxic transmission damage in rat hippocampal slices: mechanisms of protection by lidocaine. J Physiol 489(Pt 2):557–565
Sakabe T, Maekawa T, Ishikawa T, Takeshita H (1974) The effects of lidocaine on canine cerebral metabolism and circulation related to the electroencephalogram. Anesthesiology 40:433–441
MacGregor RR, Thorner RE, Wright DM (1980) Lidocaine inhibits granulocyte adherence and prevents granulocyte delivery to inflammatory sites. Blood 56:203–209
Fujitani T, Adachi N, Miyazaki H, Liu K, Nakamura Y, Kataoka K, Arai T (1994) Lidocaine protects hippocampal neurons against ischemic damage by preventing increase of extracellular excitatory amino acids: a microdialysis study in Mongolian gerbils. Neurosci Lett 179:91–94
Rasool N, Faroqui M, Rubinstein EH (1990) Lidocaine accelerates neuroelectrical recovery after incomplete global ischemia in rabbits. Stroke 21:929–935
Bilotta F, Stazi E, Zlotnik A, Gruenbaum SE, Rosa G (2014) Neuroprotective effects of intravenous anesthetics: a new critical perspective. Curr Pharm Des 20:5469–5475
Lin D, Cao L, Wang Z, Li J, Washington JM, Zuo Z (2012) Lidocaine attenuates cognitive impairment after isoflurane anesthesia in old rats. Behav Brain Res 228:319–327
Lin D, Feng C, Cao M, Zuo Z (2011) Volatile anesthetics may not induce significant toxicity to human neuron-like cells. Anesth Analg 112:1194–1198
Lin D, Zuo Z (2011) Isoflurane induces hippocampal cell injury and cognitive impairments in adult rats. Neuropharmacology 61:1354–1359
Flameng W, Borgers M, Daenen W, Stalpaert G (1980) Ultrastructural and cytochemical correlates of myocardial protection by cardiac hypothermia in man. J Thorac Cardiovasc Surg 79:413–424
Li HB, Yue ZD, Zhao HW, Wang L, Fan ZH, He FL, Dong XQ, Liu FQ (2018) Pathological features of mitochondrial ultrastructure predict susceptibility to Post-TIPS hepatic encephalopathy. Can J Gastroenterol Hepatol 2018:4671590
Tyther R, O’Brien J, Wang J, Redmond HP, Shorten G (2003) Effect of sevoflurane on human neutrophil apoptosis. Eur J Anaesthesiol 20:111–115
Krahenbuhl S, Talos C, Wiesmann U, Hoppel CL (1994) Development and evaluation of a spectrophotometric assay for complex III in isolated mitochondria, tissues and fibroblasts from rats and humans. Clin Chim Acta 230:177–187
Susin SA, Zamzami N, Castedo M, Daugas E, Wang HG, Geley S, Fassy F, Reed JC, Kroemer G (1997) The central executioner of apoptosis: multiple connections between protease activation and mitochondria in Fas/APO-1/CD95- and ceramide-induced apoptosis. J Exp Med 186:25–37
Spierings D, McStay G, Saleh M, Bender C, Chipuk J, Maurer U, Green DR (2005) Connected to death: the (unexpurgated) mitochondrial pathway of apoptosis. Science 310:66–67
Cascella M, Muzio MR, Bimonte S, Cuomo A, Jakobsson JG (2018) Postoperative delirium and postoperative cognitive dysfunction: updates in pathophysiology, potential translational approaches to clinical practice and further research perspectives. Minerva Anestesiol 84:246–260
Petschner P, Gonda X, Baksa D, Eszlari N, Trivaks M, Juhasz G, Bagdy G (2018) Genes Linking Mitochondrial Function, Cognitive Impairment and Depression are Associated with Endophenotypes Serving Precision Medicine. Neuroscience 370:207–217
Zhang Y, Xu Z, Wang H, Dong Y, Shi HN, Culley DJ, Crosby G, Marcantonio ER, Tanzi RE, Xie Z (2012) Anesthetics isoflurane and desflurane differently affect mitochondrial function, learning, and memory. Ann Neurol 71:687–698
Verdin E, Hirschey MD, Finley LW, Haigis MC (2010) Sirtuin regulation of mitochondria: energy production, apoptosis, and signaling. Trends Biochem Sci 35:669–675
Ward MW, Rego AC, Frenguelli BG, Nicholls DG (2000) Mitochondrial membrane potential and glutamate excitotoxicity in cultured cerebellar granule cells. J Neurosci 20:7208–7219
Sharma J, Johnston MV, Hossain MA (2014) Sex differences in mitochondrial biogenesis determine neuronal death and survival in response to oxygen glucose deprivation and reoxygenation. BMC Neurosci 15:9
Trushina E, McMurray CT (2007) Oxidative stress and mitochondrial dysfunction in neurodegenerative diseases. Neuroscience 145:1233–1248
Kilbride SM, Gluchowska SA, Telford JE, O’Sullivan C, Davey GP (2011) High-level inhibition of mitochondrial complexes III and IV is required to increase glutamate release from the nerve terminal. Mol Neurodegener 6:53
Baker BM, Haynes CM (2011) Mitochondrial protein quality control during biogenesis and aging. Trends Biochem Sci 36:254–261
Parker WJ, Parks J, Filley CM, Kleinschmidt-DeMasters BK (1994) Electron transport chain defects in Alzheimer’s disease brain. Neurology 44:1090–1096
Taylor CP, Burke SP, Weber ML (1995) Hippocampal slices: glutamate overflow and cellular damage from ischemia are reduced by sodium-channel blockade. J Neurosci Methods 59:121–128
Pivovarova NB, Stanika RI, Watts CA, Brantner CA, Smith CL, Andrews SB (2008) Reduced calcium-dependent mitochondrial damage underlies the reduced vulnerability of excitotoxicity-tolerant hippocampal neurons. J Neurochem 104:1686–1699
Johnson ME, Uhl CB, Spittler KH, Wang H, Gores GJ (2004) Mitochondrial injury and caspase activation by the local anesthetic lidocaine. Anesthesiology 101:1184–1194
Werdehausen R, Braun S, Essmann F, Schulze-Osthoff K, Walczak H, Lipfert P, Stevens MF (2007) Lidocaine induces apoptosis via the mitochondrial pathway independently of death receptor signaling. Anesthesiology 107:136–143
Funding
This work was funded by grants from the National Science Foundation of China (Grant No. 81201022), National Science Foundation of Guangdong Province (Grant No. 2018A0303130195).
Author information
Authors and Affiliations
Contributions
All authors listed have made great contribution to this study. JL and DL managed the experiment design, performed the experiments and wrote the paper. XZ, SY and HX performed the cell experiments and analyzed the data. JL, MG and YY performed the animal experiments and analyzed the data. ZH performed English quality revision and critical revision.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that there is no personal or institutional conflict of interest related to the presented research and its publication.
Ethical Approval
All procedures performed in studies involving animals were in accordance with the ethical standards of the institution or practice at which the studies were conducted.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Daowei Lin is the first corresponding author. Zhiquan Huang is the second corresponding author.
Rights and permissions
About this article

Cite this article
Li, J., Zhu, X., Yang, S. et al. Lidocaine Attenuates Cognitive Impairment After Isoflurane Anesthesia by Reducing Mitochondrial Damage. Neurochem Res 44, 1703–1714 (2019). https://doi.org/10.1007/s11064-019-02799-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11064-019-02799-0