
Abstract
Peptidyl-prolyl cis–trans isomerase NIMA-interacting 1 (Pin1) is a unique PPIase belonging to the parvulin family, and it isomerizes peptide bond between phospho-(Ser/Thr) and Pro. Pin1 has been linked to the pathogenesis of various human diseases; however, its exact biological functions remain unclear. The aim of the present study is to explore the expression pattern of Pin1 in patients with refractory epilepsy and in a chronic pilocarpine-induced epileptic mouse model. Using Western blot, immunofluorescence and immunoprecipitation analysis, we found that Pin1 protein was mainly distributed in neurons, demonstrated by colocalization with the dendritic marker, MAP2. However, the expression of Pin1 decreased remarkably in epileptic patients and experimental mice. Furthermore, the reciprocal coimmunoprecipitation analysis showed that Pin1 interacted with NR2A and NR2B-containing NMDA receptors not AMPA receptors in epileptic mouse models. Our results are the first to indicate that the expression of Pin1 in epileptic brain tissue could play important roles in epilepsy.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Epilepsy is defined as the most common neurological disease characterized by unprovoked epileptic seizures and affects approximately 50 million people worldwide [1–4]. Antiepileptic drugs (AEDs) have been widely prescribed for the treatment of epilepsy, but there are a number of adverse side effects associated with the use of AEDs, which often lead to poor outcomes in some patients. Temporal lobe epilepsy (TLE), the most common focal epilepsy, is characterized by chronic spontaneous recurrent seizures (SRS) and constitutes up to 30–40% pharmacoresistant patients [5, 6]. Many signaling pathways are involved in seizure-induced cognitive dysfunction or neuronal apoptosis [7, 8]. So far, the molecular mechanism underlying epilepsy is still unclear.
Pin1 (peptidyl-prolyl cis–trans isomerase NIMA-interacting 1), a unique cis–trans isomerase, is an enzyme that specifically isomerizes the pSer/pThr-Pro motifs in proteins [9, 10]. Due to its plentiful protein targets, Pin1 has been involved in the regulation of many cellular processes, including proliferation, cell signaling, neuronal death, and has been reported to be closely associated with human diseases, including cancer and Alzheimer disease (AD) [9–13]. It is reported that the loss of Pin1 function is implicated in neurodegeneration [14, 15]. Pin1 could regulate important neuronal protein gephyrin [16]. Gephyrin, a postsynaptic scaffolding protein participated in clustering GABA(A) receptors at inhibitory synapses, involved in the development of TLE, and the expression of gephyrin was significantly decreased in patients with TLE [17]. However, although the crucial role of Pin1 in the pathogenesis of AD has begun to be characterized, its involvement in epilepsy is not yet clear.
So far, no study has been published about the expression of Pin1 in the brain tissue of epileptic patients. Now, we have observed that Pin1 is down-regulated in epilepsy. In addition, Pin1 could interact with NR2A and NR2B-containing NMDA receptors. Thus, our findings provide valuable information towards unveiling the mechanisms of human TLE.
Experimental Procedures
Human Brain Tissues
The present study was performed with the formal consent of the patients for the use of data and brain tissues and approved by the Ethics Committee of Chongqing Medical University. The written informed consent was obtained from all patients. The study was conducted in accordance with the Declaration of Helsinki.
Twenty-four patient neocortical tissue samples undergoing surgical therapy for intractable epilepsy and ten control samples obtained from non-TLE patients undergoing surgery for post-trauma intracranial hypertension were chosen randomly from brain tissue bank established in our lab, and the clinical data of human brain tissue bank have been reported in our previous studies [18, 19]. Diagnosis of epilepsy was confirmed in these patients according to criteria established by the International League Against Epilepsy [20]. Inclusion criteria includes: typical clinical features; brain MRI or CT found no other central nervous system (CNS) diseases; AEDs taken at least 2-years, with maximal doses of three or more AEDs and showing effective plasma concentrations; 24 h abnormal electroencephalogram (EEG); and pathological histological changes (Table 1). In these control cases, there was no history of epilepsy and exposure to AEDs and had not other neurological diseases.
A part of the excised brain tissue sample from each TLE patient and control patient was stored in liquid nitrogen until used for Western blot and immunoprecipitation analysis. The remaining part of the samples was fixed in 4% paraformaldehyde (PFA) for 24 h, embedded in paraffin and then sectioned at 10 μm for immunofluorescence analysis.
Mouse Models of Epilepsy
Forty-eight healthy adult male C57 BL/6 mice (7–8 weeks old, 20–24 g) from the Experimental Animal Center of Chongqing Medical University were kept in a 12:12h light:dark cycle with ad libitum access to food and water. All protocols were approved by the Commission of Chongqing Medical University for ethics of experiments on animals and were conducted in accordance with international standards.
The pilocarpine epileptic mouse model was made as previously reported [21]. In brief, all mice (n = 48) were injected intraperitoneally (i.p.) with 1 mg/kg methyl-scopolamine (Sigma, St. Louis, MO, USA) followed 30 min later by 320 mg/kg of pilocarpine (i.p.). Within 10 to 45 min after pilocarpine injection, mice displayed generalised clonic-tonic seizures (GCTS) that progressed to continuous convulsive activity, i.e. status epilepticus (SE). The duration of SE lasted up to 1 h and was interrupted by injection (i.p.) of 10 mg/kg diazepam to limit the extent of brain damage. Animals surviving SE typically show spontaneous recurrent seizures (SRS) within a few days and continue to display them for several weeks. The behavior of SRS was assessed using Racine’s scale [22]. Only mice experiencing SE (at grade 4 or 5) were used in further analyses. To detect the occurrence of SRS, mice underwent 24 h of daily video recording starting the day after SE and lasting for 30 days. Only those detected SRS were considered as chronic phase pilocarpine epilepsy model (as “epilepsy” in figures, n = 18). The mean number of SRS per day is 1.8 ± 0.5, and duration of each episodes of 45 ± 6.9 s. By contrast, the mice that experienced SE but did not exhibit SRS were regarded as the control group (as “control” in figures, n = 10). The electroencephalographic (EEG) activity was monitored using a 16-channel electrophysiological data acquisition system (OmniPlex, Plexon, Dallas, TX, USA) as previously described in our lab [19].
All mice (n = 28) were anesthetized by an injection (i.p.) of 3.5% chloral hydrate (1 ml/100 g). Twenty mice (epilepsy, n = 14; control, n = 6) were decapitated, and the hippocampus and cortex were isolated and stored in liquid nitrogen for Western blot or immunoprecipitation analysis. The remaining mice (epilepsy, n = 4; control, n = 4) were intracardially perfused using 0.9% saline followed by 4% PFA, and the brains were immediately removed and postfixed in PFA for 24 h, embedded in paraffin and then sectioned at 10 μm for immunofluorescence analysis.
Triple-Label Immunofluorescence and Confocal Microscopy
Immunofluorescence was carried out as previously described in our lab [18, 19]. Simply put, the tissue sections were deparaffinized, rehydrated and antigen recovery, then sections were permeabilized for 10 min with 0.4% Triton X-100 and blocked using normal goat serum (ZLI-9021; Zhongshan Golden Bridge Inc, Beijing, China) for 1 h to eliminate nonspecific staining and then incubated in a mixture of rabbit anti-Pin1 antibody (10495-1-AP; Proteintech, Wuhan, China), mouse anti-microtubule-associated protein 2 (MAP2) antibody (ZM-0380; Zhongshan Golden Bridge) and chicken anti-astrocyte marker glial fibrillary acidic protein (GFAP) antibody (Q28115; Zhongshan Golden Bridge) overnight at 4 °C. On the following day, the sections were washed using PBS and incubated with Alexa Fluor 350 goat anti-mouse IgG (A11045; Zhongshan Golden Bridge), Alexa Fluor 488 goat anti-rabbit IgG (A11008; Zhongshan Golden Bridge) and Alexa Fluor 594 goat anti-chicken IgG (A11042; Zhongshan Golden Bridge) in the dark for 2 h at 37 °C. Sections were washed again in PBS, mounted, sealed, and dried overnight. Finally, the images were captured using confocal laser scanning microscopy (Leica, Wetzlar, Germany).
Western Blot and Coimmunoprecipitation
Isolated brain tissues were homogenized in RIPA buffer (P0013B; Beyotime, Haimen, China) supplemented with protease inhibitors (complete Mini, Roche, Indianapolis, USA). Protein concentrations were measured using a BCA assay (BCA01; Dingguo, Beijing, China). Protein samples (20–50 μg, depending on which protein was going to be detected) were separated on a 12% SDS–PAGE gel and electrotransferred onto a 0.45 μm PVDF membrane (Millipore, Billerica, MA, USA). Before immunodetection, the membranes were blocked with 5% nonfat dry milk in TBST for 1 h at room temperature (RT) and incubated with rabbit anti-Pin1 (10495-1-AP; Proteintech) and mouse anti-GAPDH (60004-1-Ig; Proteintech) antibodies overnight at 4 °C. The blots were washed and incubated for 1 h with HRP-conjugated anti-rabbit (SA00001-2; Proteintech) or anti-mouse (SA00001-1; Proteintech) secondary antibodies. The bands were visualized using an ECL reagent (Thermo, Marina, CA, USA) and a Fusion FX5 image analysis system (Vilber Lourmat, Marne-la-Vallée, France). Relative protein expression levels were calculated using the Quantity One software (Bio-Rad, CA, USA) with normalization to the GAPDH signal. For immunoprecipitation (IP) studies, protein extracts (approximately 100 mg) from mice hippocampus tissues were was homogenised and added IP lysis buffer (P0013; Beyotime). Equal amounts of the proteins were incubated with 2 μl of Rabbit IgG (ab199376; Abcam, Cambridge, UK) as control or 4 μl followed antibodies: Pin1, NR2A (19953-1-AP; Proteintech), NR2B (21920-1-AP; Proteintech), GluR1 (ab109450; Abcam) or GluR2 (11994-1-AP; Proteintech) overnight at 4 °C followed by incubation of 20 μl protein A/G agarose beads (Santa Cruz Biotechnology) 2 h at 4 °C. The protein–bead complex was then washed five times and collected by centrifugation, and the samples were mixed with 2× loading buffer and heated at 95 °C for 5 min. The samples were subjected to Western blotting with same set of antibodies as above.
Statistical Analysis
All of the data are shown as the mean ± standard deviation (SD) of three independent experiments and analyzed using Student’s t test and one-way ANOVA analysis to determine the levels of significance. P value less than 0.05 was considered to be statistically significant.
Results
Clinical Characteristics
There were no significant differences in age and sex distribution between the epilepsy patients and controls (P > 0.05). In this study, the average age of the epilepsy patients was 25.96 ± 11.18 (range 7–53 years) with 12 males and 12 females, and the mean time from the onset of seizures was 11.71 ± 6.91 years (range 3–30 years) (Table 1). All patients had taken three or more antiepileptic drugs (AEDs), and had recurrent seizures for at least 3 years. The control group had an average age of 23.80 ± 13.02 years (range 9–47 years), with 5 men and 5 women (Table 1).
Pin1 Expression in TLE Patients
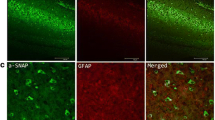
First, immunofluorescence was used to locate the distribution of Pin1 in the brain. Pin1 immunofluorescent staining was mainly observed in the cytomembrane and cytoplasm in human brain tissues (Fig. 1a). Pin1 (green) and neuron marker MAP2 (blue) were co-expressed in neurons, but not in glial fibrillary acidic protein (GFAP, a marker of astrocytes, red).

Pin1 expression in TLE patients. a Immunofluorescent labeling for Pin1 in the human neocortex. Pin1 (green) and MAP2 (blue) (not GFAP, red) are co-expressed in the neocortex of TLE patient. The scale bar 100 μm. (original magnification ×200). b The representative Western blot shows Pin1 expression in the neocortex of patients who were controls (n = 10) or had TLE (n = 24). c Three such experiments were quantified from (b) by measuring the intensity of the Pin1 proteins relative to the GAPDH control. (**P < 0.01, compared to control group). The bars indicated mean ± SD. (Color figure online)
Pin1 protein expression levels in the neocortex of TLE patients and control individuals were detected by Western blotting. As shown in Fig. 1b, compared with the controls, Pin1 expression was reduced in patients with TLE. The expression of Pin1 was normalized by calculating the intensity ratio of the bands according to the corresponding levels of GAPDH. The analysis showed that the levels of Pin1 proteins dramatically decreased by 67.3% in the neocortex compared with the control group (P < 0.01, Fig. 1c).
Pin1 Expression in a Pilocarpine-Induced Epilepsy Mouse Models
Pin1 immunofluorescent staining was mainly observed in the cytomembrane and cytoplasm in mice hippocampus (Fig. 2a) and cortex (Fig. 2b). Neuron marker MAP2 (blue) and Pin1 (green) were co-expressed in neurons, but not in glial fibrillary acidic protein (GFAP, a marker of astrocytes, red).

Immunofluorescent labeling for Pin1 in the mouse models. Pin1 (green) and MAP2 (blue) (not GFAP, red) are co-expressed in the hippocampus (a) and the cortex b of mice. The scale bar 100 μm. (original magnification ×200). (Color figure online)
To extend the results from the analysis of human tissues and exclude the possibility that altered Pin1 expression may be caused by AEDs in patients with epilepsy, Pin1 expression levels in the hippocampus and cortex of epilepsy and control mice were detected by Western blot. We observed significantly decreased Pin1 expression in both the hippocampus (by 75%, P < 0.01, Fig. 3a, b) and cortex (by 62.5%, P < 0.05, Fig. 3c, d) in epileptic mice, respectively, compared with the controls (n = 6). Our results indicate that Pin1 may be involved in epileptic seizures.

Pin1 expression in a pilocarpine-induced epilepsy mouse models. The representative Western blot images show Pin1 expression in the hippocampus (a) and the cortex c of mice with SRS (n = 14) or no SRS (n = 6). b, d Three such experiments were quantified from a, c by measuring the intensity of Pin1 proteins relative to the GAPDH control (*P < 0.05, **P < 0.01, compared to no SRS control group). The bars indicated mean ± SD. e Representative hippocampal EEG recordings in the pilocarpine model during a spontaneous seizure in the chronic phase of epilepsy
The bursts of spiking activity lasting more than 15 s, followed by depressed background activity, could be detected in the SRS groups with an EEG recording, but could not be detected in the non-SRS groups. The EEG recording showed that the pilocarpine epileptic mice were characterized by high-frequency and large-amplitude polyspikes (Fig. 3e).
Pin1 Could Interact with NMDA Receptors in the Hippocampus of Mouse Models
Pin1 could interact with postsynaptic density protein-95 (PSD95), a key scaffold protein that anchors NMDA receptors (NMDARs) [23]. We speculate that Pin1 could interact with NMDARs to negatively affect NMDARs signaling. The results indicated that coimmunoprecipitation of Pin1 and NR2A/2B by anti-Pin1 antibodies (Fig. 4a, b) demonstrated the interaction of Pin1 and NR2A/2B, and the interaction was validated by reciprocal coimmunoprecipitation of these proteins by anti-NR2A/2B antibodies (Fig. 4a, b). However, GluR1 and GluR2-containing AMPA receptors were not precipitated by anti-Pin1 antibody (Fig. 4c, d). These data suggest that Pin1 may form a complex with NMDARs and that the Pin1/NMDARs complex may play an important role in regulating epileptic seizures.

Pin1 interacted with NMDA receptors in the hippocampus of mouse models. a–d Coimmunoprecipitation was used to survey the interaction between Pin1 and NR2A/NR2B-containing NMDA receptors or GluR1/GluR2-containingAMPA receptors
Discussion
In this study, we found that Pin1 levels significantly decreased in patients with TLE and epileptic mice. Coimmunoprecipitation studies showed that Pin1 could interact with NR2A and NR2B-containing NMDA receptors. Therefore, we proposed that Pin1 could play an important role in regulating epileptic seizures and may be involved in the development of refractory epilepsy in humans.
Pin1 has been involved to the pathogenesis of various human diseases, including Alzheimer disease (AD), Parkinson disease (PD), cancer, and asthma [9, 10], and its activity is diminished in AD, indicating Pin1 has neuroprotective role [24–26]. In dendritic spines and shafts, Pin1 suppresses protein synthesis required to sustain the late phase of long-term potentiation (LTP) [26]. Pin1 restrains PSD95 ability to complex with NMDARs by interacting with PSD95, thus negatively affecting NMDAR signaling and spine morphology. The findings further emphasize the emerging role of Pin1 as a critical modulator of synaptic transmission [23]. So far, the molecular and cell biological mechanisms by which Pin1 acts in epilepsy are largely unexplored. Immunofluorescence studies in TLE patients have revealed that Pin1 protein is mainly distributed in neurons as shown by co-localization with the dendritic marker, MAP2 (Fig. 1a). The results in experimental mice also showed that Pin1 protein was largely expressed in neurons of hippocampus and cortex (Fig. 2). Using Western blotting analysis, we found that the expression of Pin1 significantly decreased in the neocortex of TLE patients than non-epileptic controls (Fig. 1). To eliminate the effects of AEDs on these experimental results, we verified the results using a chronic pilocarpine epileptic mouse model. The results showed that Pin1 expression dramatically decreased in the hippocampus and cortex of epileptic mice display spontaneous recurrent seizures (SRS) compared with no SRS controls (Fig. 3a–d). Therefore, these results provide direct evidence that epileptic seizures down-regulate Pin1 expression.
Abnormally synchronized synaptic transmission in the brain leads to epilepsy. The inhibitory and excitatory neurotransmitters are important modulators of hyper-excitability in epilepsy [27]. NMDARs are important regulators of the glutamatergic synaptic transmission in the CNS, and the function of NMDARs is regulated by multiple physiological and pathological processes [28]. During epileptic seizures, the increased number of cell surface NMDARs has important implications for cell injury and neuron death, and NMDAR blockade can provide neuroprotection against SE [29–31]. AMPA receptors (AMPARs) are distributed widely in CNS and are present in all areas relevant to epilepsy, and GluR1/2 is the predominant subtype of AMPARs in the hippocampus. The overactivation of AMPARs could have a role in the progression of hippocampal damage in TLE and has been identified as suitable targets in epilepsy therapy [32]. Pin1 has been recently considered as a key modulator of synaptic transmission [23], and Pin1 influenced PSD95/NR2B complex formation, an effect associated with a decrease in spine density and NMDAR-mediated synaptic transmission. In addition, the expression of NR2B was significant increased in Pin1−/− mice [23], and a direct interaction could strongly alter NMDARs surface trafficking [33]. As shown in Fig. 4a, b, we unveiled that Pin1 could interact with NR2A and NR2B-containing NMDA receptors. However, GluR1 and GluR2-containing AMPA receptors were not precipitated by anti-Pin1 antibody (Fig. 4c, d). It is reported that Pin1 interacts with PSD95 to affect negatively NMDAR function, and PSD95 promotes NR2B receptor surface expression [34]. It can be speculated that Pin1 may competitively influence the formation of PSD95/NMDARs complex by interacting with NMDARs not AMPARs, and subsequently affect the number of cell surface NMDARs and NMDARs-mediated synaptic transmission, then conclusively regulate seizure activities. The reduction of Pin1 expression could partly decrease surface NMDARs by promoting NMDARs internalization and then regulate hyper-excitability in epilepsy. The detailed mechanisms of whether Pin1 influences NMDARs internalization and how NMDARs are regulated remain to be illuminated in the future studies, and this will provide a promising strategy to modulate NMDARs signaling in physiological and pathological conditions.
The data from human brain tissues presented in our study have some deficiency. For ethical reasons and limitations, we could not acquire normal brain tissues from humans, so we used histologically normal brain tissue obtained from temporal lobectomy performed to treat head trauma [18, 19]. We could not also use normal hippocampal tissue as control samples. Therefore, comparisons of hippocampal Pin1 expression between epilepsy patients and control cases could not be conducted. Meanwhile, the brain tissue samples of the neocortex with TLE could only be obtained from the drug-resistant epileptic patients. For this reason, a pilocarpine-induced epileptic mouse model that has been generally used as a model for human TLE has obvious advantages in repeating the results of epileptic patients [21, 35, 36]. Thus, we executed our study on brain tissues of both TLE patients and experimental mice using two complementary methods to further investigate the expression of Pin1 and the potential mechanism of TLE. Our study in an epileptic mouse model also provides direct evidence that seizure activities alter Pin1 expression levels in the hippocampus and cortex (Fig. 3).
The present study provided direct evidence that Pin1 is down-regulated in patients with epilepsy and in mouse models. Furthermore, immunoprecipitation studies showed that Pin1 could interact with NMDARs. Our findings may contribute to the understanding of Pin1/NMDAR complex involved in the epileptogenesis and also suggest new therapeutic targets for the treatment of epilepsy, particularly refractory epilepsy. Although the abnormal expression of Pin1 developed after epilepsy, the detailed mechanisms underlying Pin1 remain to be elucidated in the future.
References
Rajendran S, Iyer A (2008) Epilepsy: addressing the transition from pediatric to adult care. Adolesc Health Med Ther 7:77–87
Duncan JS, Sander JW, Sisodiya SM, Walker MC (2006) Adult epilepsy. Lancet 367:1087–1100
Vezzani A, French J, Bartfai T, Baram TZ (2011) The role of inflammation in epilepsy. Nat. Rev Neurol 7:31–40
Moshe SL, Perucca E, Ryvlin P, Tomson T (2015) Epilepsy: new advances. Lancet 385:884–898
Spencer S, Huh L (2008) Outcomes of epilepsy surgery in adults and children. Lancet Neurol 7:525–537
Javidan M (2012) Electroencephalography in mesial temporal lobe epilepsy: a review. Epilepsy Res Treat 2012:637430
Cendes F, Kobayashi E, Lopes-Cendes I (2005) Familial temporal lobe epilepsy with auditory features. Epilepsia 10:59–60
Henshall DC, Engel T (2013) Contribution of apoptosis-associated signaling pathways to epileptogenesis: lessons from Bcl-2 family knockouts. Front Cell Neurosci 7:110
Lu KP, Zhou XZ (2007) The prolyl isomerase PIN1: a pivotal new twist in phosphorylation signalling and disease. Nat Rev Mol Cell Biol 8:904–916
Lee TH, Pastorino L, Lu KP (2011) Peptidyl-prolyl cis–trans isomerase Pin1 in ageing, cancer and Alzheimer disease. Expert Rev Mol Med 13:e21
Girardini JE, Napoli M, Piazza S et al (2011) A Pin1/mutant p53 axis promotes aggressiveness in breast cancer. Cancer Cell 20:79–91
Liou YC, Zhou XZ, Lu KP (2011) Prolyl isomerase Pin1 as a molecular switch to determine the fate of phosphoproteins. Trends Biochem Sci 36:501–514
Yuan WC, Lee YR, Huang SF et al (2011) A Cullin3-KLHL20 Ubiquitin ligase-dependent pathway targets PML to potentiate HIF-1 signaling and prostate cancer progression. Cancer Cell 20:214–228
Driver JA, Lu KP (2010) Pin1: a new genetic link between Alzheimer’s disease, cancer and aging. Curr Aging Sci 3:158–165
Rudrabhatla P, Pant HC (2010) Phosphorylation-specific peptidyl-prolyl isomerization of neuronal cytoskeletal proteins by Pin1: implications for therapeutics in neurodegeneration. J Alzheimers Dis 19:389–403
Zita MM, Marchionni I, Bottos E et al (2007) Zacchi, Post-phosphorylation prolyl isomerisation of gephyrin represents a mechanism to modulate glycine receptors function. EMBO J 26:1761–1771
Fang M, Shen L et al (2011) Downregulation of gephyrin in temporal lobe epilepsy neurons in humans and a rat model. Synapse 65:1006–1014
Zhu BL, Zha JS, Long Y et al (2016) Increased expression of copine VI in patients with refractory epilepsy and a rat model. J Neurol Sci 360:30–36
Lai YJ, Hu XT, Chen GJ, Wang XF, Zhu BL (2016) Down-regulation of adenylate kinase 5 in temporal lobe epilepsy patients and rat model. J Neurol Sci 366:20–26
Kwan P, Arzimanoglou A, Berg AT et al (2010) French, Definition of drug resistant epilepsy: consensus proposal by the ad hoc task force of the ILAE commission on therapeutic strategies. Epilepsia 51:1069–1077
Vignoli T, Nehlig A, Massironi SG et al (2012) Consequences of pilocarpine-induced status epilepticus in immunodeficient mice. Brain Res 1450:125–137
Racine RJ (1972) Modification of seizure activity by electrical stimulation. II. Motor seizure. Electroencephalogr Clin Neurophysiol 32:281–294
Antonelli R, De Filippo R, Middei S et al (2016) Pin1 modulates the synaptic content of NMDA receptors via prolyl-isomerization of PSD-95. J Neurosci 36:5437–5447
Butterfield DA, Poon HF, St Clair D et al (2006) Markesbery, redox proteomics identification of oxidatively modified hippocampal proteins in mild cognitive impairment: insights into the development of Alzheimer’s disease. Neurobiol Dis 22:223–232
Sultana R, Boyd-Kimball D, Poon HF et al (2006) Oxidative modification and downregulation of Pin1 in Alzheimer’s disease hippocampus: a redox proteomics analysis. Neurobiol Aging 27:918–925
Keeney JTR, Swomley AM, Harris JL et al (2012) Cell cycle proteins in brain in mild cognitive impairment: insights into progression to Alzheimer disease. Neurotox Res 22:220–230
Van Liefferinge J, Massie A, Portelli J et al (2013) Are vesicular neurotransmitter transporters potential treatment targets for temporal lobe epilepsy. Front Cell Neurosci 7:139
Ladépêche L, Dupuis JP, Groc L (2014) Surface trafficking of NMDA receptors: gathering from a partner to another. Semin Cell Dev Biol 27:3–13
Naylor DE, Liu H, Niquet J, Wasterlain CG (2013) Rapid surface accumulation of NMDA receptors increases glutamatergic excitation during status epilepticus. Neurobiol Dis 54:225–238
Frasca A, Aalbers M, Friqerio F et al (2011) Misplaced NMDA receptors in epileptogenesis contribute to excitotoxicity. Neurobiol Dis 43:507–515
Seo DW, Lopez-Merza ML, Allen S et al (2009) Contribution of a mitochondrial pathway to excitotoxic neuronal necrosis. J Neurosci Res 87:2087–2094
Lee K, Goodman L, Fourie C, Schenk S, Leitch B, Montgomery JM (2016) AMPA receptors as therapeutic targets for neurological disorders. Adv Protein Chem Struct Biol 103:203–261
Groc L, Bard L, Choquet D (2009) Surface trafficking of N-methyl-D-aspartate receptors: physiological and pathological perspectives. Neuroscience 158:4–18
Lau CG, Zukin RS (2007) NMDA receptor trafficking in synaptic plasticity and neuropsychiatric disorders. Nat Rev Neurosci 8:413–426
Cai ZX, Li S, Li S (2016) Antagonist targeting microRNA-155 protects against lithium-pilocarpine-induced status epilepticus in C57BL/6 mice by activating brain-derived neurotrophic factor. Front Pharmacol 7:129
Otsuka S, Ohkido T, Itakura M et al (2016) Dual mechanisms of rapid expression of anxiety-related behavior in pilocarpine-treated epileptic mice. Epilepsy Res 123:55–67
Acknowledgements
This study was supported by grants from the National Natural Science Foundation of China (No. 31500821) and the National Clinical Key Specialty Construction Foundation of China. We are grateful for the support of The Affiliated Children’s Hospital of Capital Institute of Pediatrics, The First Affiliated Hospital of PLA General Hospital and The First Affiliated Hospital of Chongqing Medical University, which supplied patient brain tissues. We also thank all patients and their families for their participation in this study.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no conflicts of interest
Rights and permissions
About this article

Cite this article
Tang, L., Zhang, Y., Chen, G. et al. Down-regulation of Pin1 in Temporal Lobe Epilepsy Patients and Mouse Model. Neurochem Res 42, 1211–1218 (2017). https://doi.org/10.1007/s11064-016-2158-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11064-016-2158-8