
Abstract
Previous studies have indicated oxidative stress and inflammatory injury as significant contributors to the secondary damage associated with traumatic brain injury (TBI). Ursolic acid (UA) has been demonstrated to exert anti-oxidative and anti-inflammatory effects on cerebral ischemia by activating the nuclear factor-erythroid 2-related factor 2 (Nrf2) pathway. However, the effects of UA on TBI remain unclear. The aim of this study is to evaluate the potential roles of UA in the activation of the Nrf2 pathway using an experimental TBI model and the underlying mechanism. Wild-type (WT) and Nrf2(−/−) mice were divided into eight groups: (1) sham; (2) TBI; (3) TBI + vehicle; (4) TBI + 50 mg/kg UA; (5) TBI + 100 mg/kg UA; (6) TBI + 150 mg/kg UA; (7) TBI + Nrf2(−/−) + vehicle; (8) TBI + Nrf2(−/−) + UA. All mice underwent the TBI with the exception of the sham group. The neurologic outcomes of the mice were evaluated at 24 h after TBI, as well as the expression of Nrf2, NQO1, HO1,SOD, GPx, and MDA. Treatment of UA significantly ameliorated brain edema and the neurological insufficiencies after TBI. In addition, UA treatment markedly strengthened the nuclear translocation of Nrf2 protein and increased the expression of NQO1 and HO1. Moreover, UA significantly increased the expression of AKT, an Nrf2 upstream factor, suggesting that UA play a neuroprotective role through the activation of the Nrf2-ARE signal pathway. On the contrary, UA showed no neuroprotective effect on the Nrf2(−/−) mice. These data indicated that UA increases the activity of antioxidant enzymes and attenuated brain injury via Nrf2 factor.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Traumatic brain injury (TBI) is deleterious brain condition that features high disability and mortality rates, and is a threat to human health in modern society [1]. Neuronal dysfunction and death occurs after a TBI, alongside a variety of pathological mechanisms that are involved with this condition. Currently, the research findings for TBI regard increased excitatory amino acids, calcium overload, oxidative stress and inflammatory response [2, 3]. Therefore, it is critically important to identify the primary mediator in the cascade of pathological reactions that surround TBI.
Nuclear factor-erythroid 2-related factor 2 (Nrf2) is an important nuclear factor that has been of primary focus in recent years [4]. Reports show that Nrf2 is involved in a several pathological processes to include reperfusion injury, pulmonary fibrosis. And also play an important role in the nervous system [5, 6]. Nrf2 resides in the cytoplasm under basal conditions and is bound to the Kelch-like ECH-associated protein 1 (Keap1), which promotes Nrf2 to the proteasomal degradation. However, in the presence of various endogenous or exogenous inducing agents, the cysteine residues of Keap1 are modified, and promotes the release of Nrf2. Furthermore, as Nrf2 is released and enabled promoting to move within the nucleus, and cellular nuclear Nrf2 immediately binds to antioxidant response element (ARE) and activates an array of downstream protective factors [7, 8]. Heme oxygenase 1 (HO1) and NADPH: quinine oxidoreductase 1 (NQO1) are the most well-known downstream genes of Nrf2, regulating the intracellular redox balance [9]. Many studies have shown that Nrf2 activity could be modified by several upstream signaling pathway, and PI3K/AKT have been currently researched more, and phosphorylation of AKT is considered paly an ultimate role [10–12]. For example, Lee and his colleagues found that sulforaphane, a putative neuroprotective agent, activated Nrf2 through activating PI3K-Akt signaling, and in addition also found Akt-Nrf2 signaling in many other disease models [13–16].
Ursolic acid (UA) is a pentacyclic triterpenoid compound that widely exists in herbal medicines and many other edible foods including apple peels [17]. The protective effects of UA have received extensive attention as a significant anti-inflammatory, anti-oxidative and anti-tumor agent. Moreover, the protective effects of UA have been demonstrated in many diseases, such as myocardial, gastric cancer, and type 2 diabetes [18–20]. The potential benefits of UA have yet to been investigated. The goals of the current study are to examine UA treatment following ameliorated brain injury, investigate the neurological insufficiencies following deficits after TBI in mice and, determine the neuroprotective effects that are possibly associated with the activation of Nrf2.
Materials and Methods
Animals
In this study, male Imprinting Control Region (ICR) mice (28–32 g) were purchased from the Animal Center of Nanjing Medical University, Nanjing China, and Nrf2 knockout (Nrf2−/−) mice were a gift from Dr. Thomas W.Kensler (Johns Hopkins University, Baltimore, MD, USA). All experimental procedures were conformed to the Guide for the Care and Use of Laboratory Animals by the National Institutes of Health (NIH) and were approved by the Animal Care and Use Committee of Nanjing University. Mice used in the experiment were housed in a room, and were maintained on a 12:12-h light/dark cycle with free access to food and water.
Traumatic Brain Injury Model
The TBI model was induced utilizing a modified weight-drop device [21]. Firstly mice were anesthetized with chloral hydrate (1 %, 5 ml/kg, i.p.) and then placed on a platform directly under a weight-drop device. A 1.5-cm longitudinal incision was made at the midline of the scalp exposing the skull. The left anterior frontal area (1.5 mm lateral to the midline on the midcoronal plane) was selected as the impact area (Fig. 1a), then a 200-g weight was dropped onto the skull from a height of 2.5 cm. Early respiratory support was provided to the mice to reduce apnea-related mortality. Lastly, the scalps of the mice were sutured scalp and then, the mice were returned to their home cages. Sham animals received the same operation as mentioned above with the exception the weight drop.

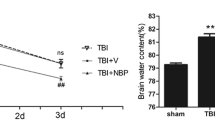
UA protectes against secondary brain injury in the model of TBI. a Display of the position of the TBI in mice model. b Neurobehavioral function was evaluated by the NSS scores at day 1 and 3 following TBI. Data represent mean ± SD (n = 6). **P < 0.01, nsP > 0.05 and #P < 0.05 versus TBI + vehicle group. c Effect of UA treatment on brain water content. Data represent mean ± SD (n = 6). *P < 0.05, nsP > 0.05, ##P < 0.01 and @P < 0.05 versus TBI group
Mouse Groups and Drug Administration
All mice were randomly distributed into the following groups: Sham, TBI, TBI + vehicle, TBI + UA (three subgroups were administrated UA at: 50, 100, and 150 mg/kg), Nrf2(−/−), TBI + Nrf2(−/−) + vehicle and TBI + Nrf2(−/−) + UA. Six mice in each group were used for the determination of brain edema, six for NSS, two for TUNEL, two for immunohistochemistry, three for western blot of nuclear protein and plasma protein, and three for western blot of total protein.
The dose applied in this study was based on previously conducted neuroprotection studies of UA in a cerebral ischemia model and a subarachnoid hemorrhage model [22, 23]. The dose of the TBI + Nrf2(−/−) + UA group was determined based on results of the neurobehavioral evaluation and brain water content in previous experiments. Ursolic acid (Sigma, MO, USA) was dissolved in 1 % dimethylsufoxide (DMSO)- phosphate-buffered saline (PBS) and injected into the mice intraperitoneally immediately following TBI. An equal volume of 0.1 % DMSO in PBS served as the vehicle control (vehicle).
Neurobehavioral Evaluation
The neurologic status of the mice was evaluated at days 1 and 3 following TBI using a 10-point neurological severity score (NSS) [24]. Neurological tests are based on the ability of the mice to perform ten different tasks in order to evaluate motor ability, balance, and alertness. The tasks consist of an exit circle, monoparesis/hemiparesis, straight walk, startle reflex, seeking behavior, beam balancing, round stick balancing, and beam walk at 1, 2, 3 cm. One point is assigned for failing to perform each of the tasks (Table 1). All neurobehavioral tests were performed by two investigators who were blinded to the experimental groups.
Brain Water Content
Brain water content was determined by utilizing a wet/dry weight ratio method as previously described [25]. The brain tissues were removed from the mice 24 h following TBI and placed on a pre-cooled operating table. Next the right and left cortical tissue was harvested after removal of the brainstem and cerebellum. Each sample was then immediately weighed to determine the wet weight (ww), and then dried at 80 °C in an oven. After drying for 72 h, the brain tissues were weighed again to obtain the dry weight (dw). The brain water content was estimated as a percentage utilizing the following formula: (ww − dw)/ww × 100 %.
Western Blot Analysis
The animals were decapitated 24 h post-injury and brain tissues were collected for western blot analysis as previously described [26, 27]. Decapitation occurred immediately following anesthetization with chloral hydrate (1 %, 10 ml/kg, i.p.) and the left brains were rapidly separated within 5 min. The specimens were immediately frozen, and stored at −80 °C until the initiation of protein extraction. Protein extraction was carried out utilizing a Total Protein Extraction Kit and Nuclear-Cytosol Extraction Kit (Beyotime Biotech, Nantong, China) according to the protocol provided by the manufacturer. Equal amounts (50 µg) of protein was separated by 10 or 15 % sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to polyvinylidene fluoride (PVDF) membranes (Millipore, Bedford, MA, USA). The membranes were blocked in the blocking buffer [5 % nonfat dry milk in Tris-buffered saline/0.05 % Tween 20 (TBST)] for 2 h at room temperature. The membranes were then incubated with primary antibodies at 4 °C overnight respectively according to the following dilution ratio: Nrf2 (1:1000, Abcam), NQO1 (1:1000, Abcam), HO1 (1:1000, Abcam), p-AKT (1:1000, Cell Signaling Technology), β-actin (1:5000, Bioworld) and H3 (1:1000, Bioworld). After incubation with the primary antibody, the membranes were washed three times with Tris-buffered saline with 0.1 % Tween 20 (TBST), and then blotted with secondary antibody (1:5000, Bioworld) for 2 h. Next the membrane was washed with TBST, and then prepared for exposure., The immunoreactive bands were then detected using chemiluminescence (ECL kit, Millipore) upon exposure to X-ray films. The quantification of band intensity was performed using the UN-Scan-It 6.1 software (Silk Scientific, Orem, UT, USA). The data was normalized to β-actin and H3 and expressed as a percentage of the protein level as expressed in the sham group.
Immunohistochemical Staining
Consecutive coronal sections were cut at 4-μm intervals from approximately bregma-1.0–3.0 mm in order to collect the lesioned cortex for immunohistochemistry. Endogenous peroxidase was blocked using 3 % H2O2/methanol following routine deparaffinization. The sections were then incubated in blocking buffer (10 % normal goat serum in PBS) for 30 min to prevent nonspecific binding. After blocking, the sections were then incubated with primary antibodies against Nrf2 (1:100, Abcam) overnight at 4 °C. After being washed three times in PBS for 5 min respectively the sections were incubated with horseradish peroxidase-conjugated IgG (1:500; Santa Cruz Biotechnology) for 60 min. Nrf2 was visualized by utilizing 3,3′-diaminobenzidine (DAB)/H2O2 solution. Cell nuclei were counterstained with hematoxylin. In the process of cell counting, six random fields in each coronal section were chosen using the high power magnification (×400). The experiment was performed by two investigators who were blinded to the grouping.
Nissl Staining
The detection method was carried out according to the specification of the Nissl Staining Solution (Beyotime Biotech). Four micrometer sections were hydrated in 1 % toluidine blue for Nissl staining at 50 °C for 20 min, and rinsed with double distilled water. The sections were then dehydrated and mounted with Permount. A large cell body, with abundant cytoplasm, and with substantially significant levels of Nissl body represents a normal neuron. However some other cell forms such as a shrunken cell body, condensed nuclei, reduced or disappearance Nissl body represents a damaged cell. Cell counting was conducted as described above. A total of six sections (×400) from each group were used for quantification. The experiment was performed by two investigators who blinded to the grouping.
Determination of Glutathione Peroxidase (GPx), Superoxide Dismutase (SOD) and Malondialdehyde (MDA)
Protein was extracted from the lesioned hemisphere according to the instructions provided by the manufacturer (Nanjing Jiancheng Biochemistry Co., Nanjing, China). GPx, SOD, and MDA content in the total protein were measured using commercial kits (Nanjing Jiancheng Biochemistry Co.) and conducted according to the instructions provided by the manufacturer. Absorbance values were collected using a spectrophotometer. Protein concentrations were determined by applying the Bradford method. The activity of GPx, SOD, and MDA was expressed as U/mg protein, U/mg protein, and nmol/mg protein, respectively.
Terminal Deoxynucleotidyl Transferase-Mediated dUTP Nick End Labeling (TUNEL) Analysis
Apoptotic cells were determined using a TUNEL detection kit (Roche, Indianapolis, IN, USA) according to the instructions provided by the manufacturer. The sections underwent deparaffinization with Xylene first, followed by dehydration with Gradient ethanol (100, 95, 90, 80, 70 % respectively). Next, the sections were digested for 15 min in proteinase K, and then washed with PBS twice. The sections were then incubated at 37 °C with labeling solution that contains TUNEL reaction fluid for 1 h. After being washed with PBS (3 × 10 min), the sections were then blocked with 10 % goat serum in 0.1 M Tris for 15 min. DNA was visualized by treating the tissue with a 1:40 dilution of streptavidin peroxidase (HRP), and then stained with DAB. The presence of distinctive morphological features of apoptosis, to include cell shrinkage with condensed nuclei, classified cells as apoptotic. Apoptotic cells were observed using an optical microscope by an investigator blinded to the grouping process. The apoptotic index was used to denote the extent of brain damage, which was defined as the average percentage of TUNEL-positive cells in six cortical microscopic fields (×400) in each section. The final average percentage of TUNEL-positive cells of the six sections was regarded as the sample data. All of the observation and counting processes were performed by two investigators who were blinded to the grouping.
Statistical Analysis
All data was reported as the mean ± SD. The SPSS 19.0 software package was used to perform all statistical analyses. Comparisons between two groups were performed using the Student’s t test, comparisons between multiple groups was performed using analysis of variance (ANOVA). A P value less than 0.05 (P < 0.05) was considered to be statistically significant. Each experiment was repeated at a minimum of three times.
Results
UA Improved the Neurobehavioral Function and Alleviated Cerebral Edema in the Brain Following TBI
To determine in the effectiveness of UA in neuroprotection, Neurological Severity Scores (NSS) were obtained to evaluate the neurobehavioral function in the mice following TBI to determine UA effectiveness in neuroprotection. The ability of each mouse to perform ten different tasks was evaluated to determine motor function, balance, and alertness. Each mouse was trained for all tasks 1 day period with all mice trained for all tasks 1 day prior to administering TBI. One point was allocated for failing to perform each task. Here something special needed to be point out that we did not showed the NSS results of sham group. We found the NSS scores in the sham group were almost zero in the experiment, and moreover considering that we only wanted to compare improvement of the animals treated with the different doses of UA to the TBI controls.
The group of mice that were administrated TBI + 50 mg/kg UA exhibited a declining trend for improving neurobehavioral performance, however, there was no statistically significant difference compared to the TBI + vehicle group of mice (P > 0.05 versus TBI + vehicle group) (Fig. 1b). The mice induced with TBI + 100 mg/kg UA had NSS that were significantly improved compared to the NSS of the vehicle-treated mice on days 1 and 3 (P < 0.05 versus TBI + vehicle group). Improved neuroprotection was not indicated by larger doses of UA, for example, there was no obvious difference between mice treated with 100 mg/kg of UA compared with the other groups.
The brain water content was examined as an indicator of brain edema and was measured at 24 h following TBI to confirm the role of UA post-TBI. As shown in Fig. 1c, the brain water content was increased in the TBI group as compared to the sham group (P < 0.01). There was no significant difference appeared between the TBI group and TBI + 50 mg/kg UA group (P > 0.05). Mice treated with 100 and 150 mg/kg UA exhibited reduced brain edema compared to the TBI + vehicle group (P < 0.01 and P < 0.05). These results are consistent with the NSS of the mice administered 100 mg/kg UA that featured superior neuroprotection, and was thus applied as the treatment dose in the following experiments.
UA Inhibits Neuronal Apoptosis After TBI
To further investigate the protective effects of UA, neuron morphology was examined using Nissl staining to investigate the protective effects of UA. Neurons in the sham group were clear and intact (Fig. 2a), however, many neurons in the TBI and TBI + vehicle group were damaged, and exhibited extensive changes, to include sparse cell arrangement, oval or triangular nuclei, shrunken cytoplasm, and swollen cell bodies (Fig. 2b, c). We also observed an improvement in the neuronal morphology and cytoarchitecture in the TBI + UA group (Fig. 2d).

Ursolic acid suppresses neural apoptosis induced by TBI. a–d, i Nissl staining of cortical tissue sections display neuronal damage in mice subjected to TBI, including sparse cell arrangement and shrunken cytoplasm and swollen cell bodies, which were alleviated by UA treatment (100 mg/kg). e–h, j Apoptosis of neural cells surrounding the injury site was evaluated by TUNEL staining. Data represent mean ± SD (n = 6 per group). ***P < 0.001 versus sham group; nsP > 0.05 versus TBI group; #P < 0.05 and ###P < 0.001 versus TBI + vehicle group. Scale bar 50 µm
To examine the neural cells of injured brain tissue and investigate the mechanistic basis for the effects of UA, TUNEL (terminal deoxynucleotidyl transferase dUTP nick end labeling) staining was utilized. TUNEL-positive cells were detected at a low frequency in the brains of mice in the sham group (Fig. 2e). The apoptotic index was significantly increased in the TBI group (P < 0.001 versus sham group) and there was no significant difference between the TBI group and TBI + vehicle group (P > 0.05) (Fig. 2f, g). However, neuronal degeneration was significantly decreased in the UA injected groups at all doses (P < 0.001 versus TBI + vehicle group) (Fig. 2h).
UA Attenuated Oxidative Stress After TBI
The activity of superoxide dismutase (SOD) as well as glutathione peroxidase (GPx) and malondialdehyde (MDA), which are indicators of lipid peroxidation and antioxidant activity, respectively, were examined in the brain tissue to determine whether the neuroprotective effect of UA was associated with an ability to attenuate the oxidative stress caused by TBI. SOD and GPx were significantly lower in the TBI and TBI + vehicle groups compared with the sham group (P < 0.01 and P < 0.01, respectively), while the SOD and GPx levels nearly returned to normal following the administration of UA (P < 0.001 and P < 0.05, respectively) (Fig. 3a, b). MDA levels increased following TBI (P < 0.001 versus sham group). Conversely, the administration of UA reduced the elevation of MDA significantly (P < 0.001 versus TBI + vehicle group; Fig. 3c).

Ursolic acid (100 mg/kg) reduced oxidative stress in the brain following TBI. Oxidative stress was assessed by a SOD activity, b GPx activity and c MDA level. Data represent mean ± SD (n = 6 per group). **P < 0.01 and ***P < 0.001 versus sham group; nsP > 0.05 versus TBI group; ###P < 0.001 and #P < 0.05 versus TBI + vehicle group
UA Promoted Translocation of Nrf2 from Cytoplasm to Nucleus and Enhanced Nrf2-ARE Binding
To investigate the activation of Nrf2 following UA treatment, we conducted western blot analysis. The results indicated that UA treatment significantly increased the expression of nuclear Nrf2 while decreasing the expression of cytoplasmic Nrf2, as compared with the vehicle treatment. These results suggest that the administration of UA promoted the translocation of Nrf2 from the cytoplasm to the nucleus (Fig. 4a). These results were also confirmed by the immunohistochemistry (Fig. 4b).

Effects of UA (100 mg/kg) on Nrf2, NQO1, and HO1 protein expression after TBI. Mice brain tissues were collected 1 day after TBI in different groups. a The expression of cytoplasmic Nrf2 and nuclear Nrf2 in the ipsilateral cortex was evaluated by western blotting at 24 h after injury. b Nrf2 expression was assessed by immunohistochemistry. As compared with sham group, the TBI group presented a morphology with Nrf2 concentrated in the nucleus, and after treatment with UA, this concentration, as evident by the morphology, was more apparent. c The expression of NQO1 and HO1proteins was upregulated after TBI. Ursolic acid further increased their expression in brain tissue. Data represent mean ± SD (n = 3 per group). *P < 0.05 and **P < 0.01 versus sham group; nsP > 0.05 versus TBI group; ##P < 0.01 and ###P < 0.001 versus TBI + vehicle group. Scale bar 50 µm
The expression of the downstream factors in the Nrf2 pathway, NQO1 and HO1, were also investigated (Fig. 4c). Western blot results indicated that NQO1 and HO1 proteins were upregulated following TBI (P < 0.05 versus sham group). Moreover, the administration of UA further enhanced NQO1 and HO1 protein expression compared with the vehicle group (P < 0.001 versus TBI + vehicle group). These results indicated that UA induced expression of Nrf2 downstream factors through the activation of the Nrf2-ARE signal pathway.
UA Activated the AKT Signaling After TBI
To investigate the mechanism of Nrf2 activatied by UA, we further studied the upstream factor of Nrf2, AKT. It has been well documented that the AKT signaling was involved in TBI-induced apoptosis, and activated form of phosphorylated AKT (p-AKT) played an important neuroprotection. The results indicated that the contents of AKT increased significantly after TBI (P < 0.05) (Fig. 5). However, there was no significant difference between TBI group and TBI + vehicle group (P > 0.05) (Fig. 5). After administration of UA, p-AKT expression increased significantly compared with TBI + vehicle group (P < 0.05) (Fig. 5). These results indicated that UA activated the Nrf2 pathway may be by activating the upstream factor AKT.

Effects of UA (100 mg/kg) on p-AKT expression. Data represent mean ± SD (n = 3 per group). *P < 0.05 versus sham group; nsP > 0.05 versus TBI group; #P < 0.05 versus TBI + vehicle group
Knockout of Nrf2 Partly Reversed Neuroprotection of UA After TBI
The presence of oxidative stress and neuronal apoptosis in Nrf2(−/−) mice was evaluated to confirm the enhancement of antioxidant enzyme activities and the reduction of oxidative stress by UA via the Nrf2-ARE signaling pathway. The experimental results revealed that knockout of Nrf2 partly reversed the neuroprotective aspects of UA (Fig. 6a–d, P < 0.0001, P < 0.01 versus TBI + Nrf2(+/+) + UA group) and these effects were accompanied by the decrease of oxidation factors such as SOD, GPX and the increase of oxidation products such as MDA (Fig. 6e–g, P < 0.001, P < 0.0001, P < 0.001 versus TBI + Nrf2(+/+) + UA group). However the administration of UA in Nrf2 knockout mice did not appear to be significantly important in modulating neurological function nor oxidation (P > 0.05 versus TBI + Nrf2(−/−) + UA).

Knockout of Nrf2 partly reversed neuroprotection of UA (100 mg/kg) after TBI. a, c Nissl staining and b, d TUNEL staining displays enhanced cell damage in Nrf2 gene knockout mice, and indicates the partially reversed neuroprotective aspects of UA. Data represent mean ± SD (n = 6 per group). $$$$P < 0.0001, $$P < 0.01,TBI + Nrf2(−/−) + UA versus TBI + Nrf2(+/+) + UA; and nsP > 0.05, TBI + Nrf2(−/−) + UA versus TBI + Nrf2(−/−). e–g The illustrations showed that UA failed to decrease oxidative stress in Nrf2 gene knockout mice. Data represent mean ± SD (n = 6 per group), $$$P < 0.001, $$$$P < 0.0001, TBI + Nrf2(−/−) + UA versus. TBI + Nrf2(+/+) + UA; nsP > 0.05, TBI + Nrf2(−/−) + UA versus TBI + Nrf2(−/−). Scale bar 50 µm
Discussion
In the present study, we investigated the neuroprotective of UA following TBI in mice. Ursolic acid treatment improved neurological function, alleviated cerebral edema, suppressed neuronal apoptosis and reversed the oxidative stress state induced by the TBI.
Previous studies have demostrated the significance of oxidative stress that is associated with the pathological processes of the secondary brain injury [2, 3, 28]. Mechanisms that govern endogenous antioxidants were damaged following TBI, and caused lipid peroxidation, protein nitration, and DNA damage and immediately produced high levels of reactive oxygen species (ROS) [29, 30]. With the continuous production of ROS, antioxidant enzymes will gradually exhausted such as superoxide dismutase, catalase, and GPx gradually become exhausted [3, 31]. The oxidative degradation of lipids, referred to as lipid peroxidation, will increase the permeability of membranes, and lead to cell damage. Ursolic acid treatment following TBI reduced MDA levels in this study and increased antioxidant enzymes including SOD and GPx activity compared to vehicle-treated mice. These results suggest that UA exhibits antioxidant effects.
Previous studies have demonstrated the roles of the Nrf2 in neuroprotection by limiting inflammation and oxidative stress following TBI [26, 27]. Ursolic acid treatment activated and promoted the nuclear translocation of Nrf2 in this study, and increased Phase (II) enzymes, to include NQO1, following TBI. These results provide evidences of UA neuroprotective effects. Conversely, Nrf2(−/−) mice exhibited greater neuronal degeneration and oxidative stress, even in the presence of UA, suggesting that UA dose exhibit a neuroprotective role in the Nrf2 (−/−) mice.
Fewer literatures have mentioned the relationship between UA and Nrf2 [32, 33]. The present study demonstrated the effect of UA on activating Nrf2 is clear, the specific mechanisms have never been reported. In the present study, we preliminary detected the expression of AKT, an Nrf2 upstream factor, founding that UA can activate AKT, suggesting a possible activation mechanism.
For the present study, we selected 24 h as the observation time based on our previous studies [34], which showed that the expression of Nrf2 was the highest at 24 h after TBI. Miller and colleagues believed that Nrf2 had the highest expression at 48 and 72 h [35]. We considered that the reason for this difference may be related to the experimental model and detection methods, thus we conducted the experiments herein to validate these findings.
In summary, the present study demonstrated that UA attenuated the oxidative stress by enhancing the expression and activity of antioxidant enzymes in a TBI model. The administration of UA resulted in further activation of the Nrf2-ARE pathway, with the Nrf2 knockout mice having exacerbated oxidative stress compared with wild-type mice. Our results revealed that UA exerts neuroprotection effects by activating Nrf2 and downstream proteins against oxidative stress.
References
Maas AI, Stocchetti N, Bullock R (2008) Moderate and severe traumatic brain injury in adults. Lancet Neurol 7:728–741
Werner C, Engelhard K (2007) Pathophysiology of traumatic brain injury. Br J Anaesth 99:4–9
Cornelius C, Crupi R, Calabrese V, Graziano A, Milone P, Pennisi G, Radak Z, Calabrese EJ, Cuzzocrea S (2013) Traumatic brain injury: oxidative stress and neuroprotection. Antioxid Redox Signal 19:836–853
Kobayashi MY, Yamamoto M (2006) Nrf2-Keap1 regulation of cellular defense mechanisms against electrophiles and reactive oxygen species. Adv Enzyme Regul 46:113–140
Kandhare AD, Bodhankar SL, Mohan V, Thakurdesai PA (2015) Effect of glycosides based standardized fenugreek seed extract in bleomycin-induced pulmonary fibrosis in rats: decisive role of Bax, Nrf2, NF-kappaB, Muc5ac, TNF-alpha and IL-1beta. Chemi Biol Interact 237:151–165
Wang B, Zhu X, Kim Y, Li J, Huang S, Saleem S, Li RC, Xu Y, Dore S, Cao W (2012) Histone deacetylase inhibition activates transcription factor Nrf2 and protects against cerebral ischemic damage. Free Radic Biol Med 52:928–936
de Vries HE, Witte M, Hondius D, Rozemuller AJ, Drukarch B, Hoozemans J, van Horssen J (2008) Nrf2-induced antioxidant protection: a promising target to counteract ROS-mediated damage in neurodegenerative disease? Free Radic Biol Med 45:1375–1383
Itoh K, Tong KI, Yamamoto M (2004) Molecular mechanism activating Nrf2-Keap1 pathway in regulation of adaptive response to electrophiles. Free Radic Biol Med 36:1208–1213
Zhang M, An C, Gao Y, Leak RK, Chen J, Zhang F (2013) Emerging roles of Nrf2 and phase II antioxidant enzymes in neuroprotection. Prog Neurobiol 100:30–47
Endo H, Nito C, Kamada H, Yu F, Chan PH (2006) Akt/GSK3beta survival signaling is involved in acute brain injury after subarachnoid hemorrhage in rats. Stroke 37:2140–2146
Yap TA, Garrett MD, Walton MI, Raynaud F, de Bono JS, Workman P (2008) Targeting the PI3K-AKT-mTOR pathway: progress, pitfalls, and promises. Curr Opin Pharmacol 8:393–412
Xu Y, Duan C, Kuang Z, Hao Y, Jeffries JL, Lau GW (2013) Pseudomonas aeruginosa pyocyanin activates NRF2-ARE-mediated transcriptional response via the ROS-EGFR-PI3K-AKT/MEK-ERK MAP kinase signaling in pulmonary epithelial cells. PLoS One 8:e72528
Gong YQ, Huang W, Li KR, Liu YY, Cao GF, Cao C, Jiang Q (2016) SC79 protects retinal pigment epithelium cells from UV radiation via activating Akt-Nrf2 signaling. Oncotarget 7:60123–60132
Lee YJ, Jeong HY, Kim YB, Lee YJ, Won SY, Shim JH, Cho MK, Nam HS, Lee SH (2012) Reactive oxygen species and PI3K/Akt signaling play key roles in the induction of Nrf2-driven heme oxygenase-1 expression in sulforaphane-treated human mesothelioma MSTO-211 H cells. Food Chem Toxicol 50:116–123
Baskaran R, Poornima P, Priya LB, Huang CY, Padma VV (2016) Neferine prevents autophagy induced by hypoxia through activation of Akt/mTOR pathway and Nrf2 in muscle cells. Biomed Pharmacother 83:1407–1413
Liu DY, Zhang YL, Wei YZ, Liu GY, Liu YF, Gao QM, Zou LP, Zeng WJ, Zhang N (2016) Activation of AKT pathway by Nrf2/PDGFA feedback loop contributes to HCC progression. Oncotarget. doi:10.18632/oncotarget.11700
Cui T, Li JZ, Kayahara H, Ma L, Wu LX, Nakamura K (2006) Quantification of the polyphenols and triterpene acids in chinese hawthorn fruit by high-performance liquid chromatography. J Agric Food Chem 54:4574–4581
Yang Y, Li C, Xiang X, Dai Z, Chang J, Zhang M, Cai H, Zhang H, Zhang M, Guo Y, Wu Z (2014) Ursolic acid prevents endoplasmic reticulum stress-mediated apoptosis induced by heat stress in mouse cardiac myocytes. J Mol Cell Cardiol 67:103–111
Wang X, Zhang F, Yang L, Mei Y, Long H, Zhang X, Zhang J, Qimuge S, Su X (2011) Ursolic acid inhibits proliferation and induces apoptosis of cancer cells in vitro and in vivo. J Biomed Biotechnol 2011:419343
Lee J, Lee HI, Seo KI, Cho HW, Kim MJ, Park EM, Lee MK (2014) Effects of ursolic acid on glucose metabolism, the polyol pathway and dyslipidemia in non-obese type 2 diabetic mice. Indian J Exp Biol 52:683–691
Flierl MA, Stahel PF, Beauchamp KM, Morgan SJ, Smith WR, Shohami E (2009) Mouse closed head injury model induced by a weight-drop device. Nat Protoc 4:1328–1337
Li L, Zhang X, Cui L, Wang L, Liu H, Ji H, Du Y (2013) Ursolic acid promotes the neuroprotection by activating Nrf2 pathway after cerebral ischemia in mice. Brain Res 1497:32–39
Zhang T, Su J, Guo B, Zhu T, Wang K, Li X (2014) Ursolic acid alleviates early brain injury after experimental subarachnoid hemorrhage by suppressing TLR4-mediated inflammatory pathway. Int Immunopharmacol 23:585–591
Beni-Adani L, Bassan M, Gibney G, Berenneman DE, Gozes J, Shohami E (2000) Activity-dependent neurotrophic protein is neuroprotective in a mouse model of closed head injury [abstract]. J Pharmacol Exp Ther 296:57–63
Wei W, Wang H, Wu Y, Ding K, Li T, Cong Z, Xu J, Zhou M, Huang L, Ding H, Wu H (2015) Alpha lipoic acid inhibits neural apoptosis via a mitochondrial pathway in rats following traumatic brain injury. Neurochem Int 87:85–91
Xu J, Wang H, Ding K, Zhang L, Wang C, Li T, Wei W, Lu X (2014) Luteolin provides neuroprotection in models of traumatic brain injury via the Nrf2-ARE pathway. Free Radic Biol Med 71:186–195
Ding K, Wang H, Xu J, Li T, Zhang L, Ding Y, Zhu L, He J, Zhou M (2014) Melatonin stimulates antioxidant enzymes and reduces oxidative stress in experimental traumatic brain injury: the Nrf2-ARE signaling pathway as a potential mechanism. Free Radic Biol Med 73:1–11
Rodriguez-Rodriguez A, Egea-Guerrero JJ, Murillo-Cabezas F, Carrillo-Vico A (2014) Oxidative stress in traumatic brain injury. Curr Med Chem 21:1201–1211
Smith JA, Park S, Krause JS, Banik NL (2013) Oxidative stress, DNA damage, and the telomeric complex as therapeutic targets in acute neurodegeneration. Neurochem Int 62:764–775
Ansari MA, Roberts KN, Scheff SW (2008) A time course of contusion-induced oxidative stress and synaptic proteins in cortex in a rat model of TBI. J Neurotrauma 25:513–526
Hou Z, Luo W, Sun X, Hao S, Zhang Y, Xu F, Wang Z, Liu B (2012) Hydrogen-rich saline protects against oxidative damage and cognitive deficits after mild traumatic brain injury. Brain Res Bull 88:560–565
Liu W, Tan X, Shu L, Sun H, Song J, Jin P, Yu S, Sun M, Jia X (2012) Ursolic acid inhibits cigarette smoke extract-induced human bronchial epithelial cell injury and prevents development of lung cancer. Molecules 17:9104–9115
Ma JQ, Ding J, Zhang L, Liu CM (2015) Protective effects of ursolic acid in an experimental model of liver fibrosis through Nrf2/ARE pathway. Clin Res Hepatol Gastroenterol 39:188–197
Yan W, Wang HD, Feng XM, Ding YS, Jin W, Tang K (2009) The expression of NF-E2-related factor 2 in the rat brain after traumatic brain injury. J Trauma 66:1431–1435
Miller DM, Wang JA, Buchanan AK, Hall ED (2014) Temporal and spatial dynamics of nrf2-antioxidant response elements mediated gene targets in cortex and hippocampus after controlled cortical impact traumatic brain injury in mice. J Neurotrauma 31:1194–1201
Acknowledgments
This study was grant-supported by the National Natural Science Fund of China (Grant No. 81371357).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare no potential conflicts of interest.
Rights and permissions
About this article

Cite this article
Ding, H., Wang, H., Zhu, L. et al. Ursolic Acid Ameliorates Early Brain Injury After Experimental Traumatic Brain Injury in Mice by Activating the Nrf2 Pathway. Neurochem Res 42, 337–346 (2017). https://doi.org/10.1007/s11064-016-2077-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11064-016-2077-8