
Abstract
Previously, we reported that nicotine withdrawal (NT) significantly increased pain sensitivity in rats. Recent reports suggest that fractalkine is involved in the spinal cord neuron-to-microglia activation via CX3CR1 signaling. However, its contribution to NT-induced hyperalgesia and the underlying mechanisms have yet to be elucidated. In the present study, a rat model of NT was used to test the changes in CX3CR1 expression in the spinal cord. We also evaluated the effect of the CX3CR1 neutralizing antibody on spinal microglial activity, the expression of phosphorylated p38-mitogen-activated protein kinase (p-p38-MAPK) and heat-induced pain responses. We established a NT model via subcutaneous injection of pure nicotine (3 mg/kg), three times daily for 7 days. The expression of CX3CR1 was studied by Western blot and immunofluorescence staining. Following NT, the rats received daily intrathecal injections of CX3CR1 neutralizing antibody for 3 days. The change in paw withdrawal latency (PWL) was observed. The activation of microglia and the expression of p-p38-MAPK were investigated by Western blot and immunofluorescence staining. The expression of CX3CR1 was significantly increased after NT and co-localized with IBA-1. NT rats treated with CX3CR1 neutralizing antibody showed significantly increased PWL on day 4 after NT. Furthermore, the activation of microglia and the expression of p-p38-MAPK in the spinal cord were suppressed. These results indicate that microglial CX3CR1/p38MAPK pathway is critical for the development of pain hypersensitivity after NT.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Background
Tobacco addiction is the leading avoidable cause of death in the United States [1]. In fact, it is a global public health challenge, with an estimated one-third of all adults smoking, worldwide. Clinically, long-term smokers manifest an increased risk of lumbago and back pain. For instance, it has been reported that smokers require higher number of analgesics than non-smokers following surgery including coronary bypass and gynecological procedures [2]. Sudden discontinuation of smoking leads to a nicotine withdrawal syndrome. Our previous study has confirmed that thermal hyperalgesia was significantly increased after nicotine withdrawal in rats [3]. Nicotine withdrawal is associated with nociceptive hypersensitivity and pain. In fact, smokers undergoing withdrawal showed lower pain tolerance to cold pressor test (CPT) than non-smokers. Nicotine withdrawal may affect neurophysiologic mechanisms linked with pain, that may render abstinent smokers more sensitive to physical discomfort [4]. A better understanding of the mechanisms underlying nicotine withdrawal could potentially enable the development of new treatments.
Numerous studies have shown that chemokines play a pivotal role in pain via neuronal–glial communication, resulting in central sensitization. Fractalkine (FKN) is a structurally unique chemokine and the only member of the CX3C family of chemokines. FKN has a monogamous relationship with its receptor CX3CR1 [5]. In the dorsal horn of the spinal cord, FKN is expressed on neurons binding to its specific receptor, while CX3CR1 is mainly expressed in microglia. CX3CR1 has recently been proposed to be a neuron-to-glia signal in the spinal cord leading to microglial activation [6]. FKN activates the signaling of neuronal–microglial communication in the mediation of neuropathic and inflammatory pain processing [6, 7]. On the other hand, activated microglia in the spinal cord release a variety of mediators, including neurotransmitters, proinflammatory cytokines and chemokines [8, 9]. Microglia cells are regarded as a major source of proinflammatory cytokines [9]. Furthermore, CX3CL1/CX3CR1 signaling has been shown to mediate the induction of proinflammatory cytokines, such as TNF-α, NF-κB and IL-6 [5]. Spinal nociceptive neurons are excited by these mediators, resulting in central sensitization [10].
The central sensitization is mediated by a complex biochemical cascade following the activation of primary afferent fibers. Several protein kinases, including protein kinase A (PKA), protein kinase C (PKC), and calcium/calmodulin-dependent protein kinase II (CaMKII) play a vital role in the initiation, development and maintenance of central sensitization [11, 12]. Recent studies have established the role of p38 mitogen-activated protein kinase (p38 MAPK), a member of MAPK family, in hypersensitivity and central sensitization [13]. However, the role of p38 MAPK in the development of pain hypersensitivity after nicotine withdrawal is still unknown.
Our previous study has confirmed that thermal hyperalgesia was significantly increased after nicotine withdrawal in rats, in line with our present reports [3]. In the present study, we utilized a rat model of nicotine withdrawal, as previously described [3]. We investigated the altered expression of CX3CR1 in the spinal cord. Furthermore, we tested whether intrathecal injection of anti-CX3CR1 neutralizing antibody attenuated pain behavior and inhibited the activation of spinal microglia as well as the expression of p38 MAPK in the spinal cord induced by nicotine withdrawal.
Methods
Animals
Healthy male Sprague–Dawley (SD) rats (Experimental Animal Center, Xuzhou Medical College, Xuzhou, China) weighing 180–200 g were housed in plastic cages and maintained on a 12:12 h light/dark circle under conditions of 24 ± 1 °C, with ad libitum access to food and water. Animals were habituated to the testing environment for at least 3 days prior to the experiment. All procedures were approved by the Animal Care and Use Committee of Xuzhou Medical College, and were consistent with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. All efforts were made to minimize the number of animals used and their suffering.
Reagents
Nicotine hydrogen tartrate salt ([2]-1-methyl-2-[3-pyridil]pyrrolidine) and mecamylamine hydrochloride were purchased from Sigma Aldrich (St Louis, MI, USA). Anti-NeuN antibody was purchased from Millipore (Billerica, MA, USA). Anti-IBA1 antibody, anti-CX3CR1 antibody, anti-CX3CR1 neutralizing antibody and Anti-IgG antibody (for control) were purchased from Abcam (Cambridge, MA, USA). Antibodies for phosphorylated-p38 MAPK and GFAP were obtained from Cell Signaling Technology (Cell Signaling Technology, USA). Anti-CX3CR1 neutralizing antibody and anti-IgG antibody injections were performed intrathecally (10 μl/10 μg). The doses of all drugs were based on the results of preliminary experiments.
Model of Nicotine Withdrawal
As described previously, we induced a rat model of nicotine withdrawal [3]. The rats were subcutaneously injected with 3 mg/kg nicotine at 7:00, 15:00 and 23:00 h daily for 7 consecutive days. In addition, 1 mg/kg of mecamylamine was subcutaneously injected 60 min after the last injection on the 7th day, in order to trigger nicotine withdrawal.
Intrathecal Infusion
The method described by Xu et al. [14] was used for the intrathecal drug injection. Briefly, the rats were anesthetized with isoflurane. The hair in the lumbar region was shaved and disinfected with 75 % (v/v) ethanol. The intervertebral spaces were widened by placing the animal on a plexiglass tube. Next, animals were injected at the L5-6 interspace using a 29-gauge microinjection syringe needle filled with CX3CR1 neutralizing antibody or anti-IgG antibody (for control), at a dose of 10 μg/10 μl, daily from day 1 to day 3 after nicotine withdrawal. The correct subarachnoid positioning of the tip of the needle was verified by a tail- or paw-flick test, that is an abrupt flick of the tail or paw when the tip of the needle reached subarachnoid space could be visualized. Rats with signs of motor dysfunction were excluded from the experiments. Motor function was evaluated by the presence of two specific behaviors [15]. (1) The placing/stepping reflex was evoked by drawing the dorsum of either hind paw across the edge of the table. This stimulus elicits an upward lifting of the paw from the surface of the table. (2) In the righting reflex, a rat placed supine shows an immediate and coordinated twisting of the body around its longitudinal axis with crossed extension of its fore and hind paws to regain its normal crouching posture.
Measurement of Thermal Hyperalgesia
Thermal hyperalgesia was determined by measuring the paw withdrawal latency (PWL) in response to radiant heat stimulation. A plantar analgesia meter (IITC Life Science Inc., Victory Blvd Woodland Hills, CA, USA) was used to provide a heat source. The method was similar to the published one [16]. Briefly, each rat was left to adapt to the testing environment for at least 1 h prior to any stimulation. The high-intensity, movable radiant heat source was placed underneath the glass and focused onto the plantar surface of each hind paw. The nociceptive endpoint in the radiant heat test was lifting or licking of the hind paw. The time from onset of radiant heat to endpoint was considered as the PWL. An automatic 30-s cutoff was used to prevent tissue damage. Thermal stimulus was delivered three times to each hind paw at 5-min intervals. The heat was maintained at a constant intensity throughout this study.
Immunohistochemistry
In deep anesthesia, rats were intracardially perfused with saline followed by 4 % paraformaldehyde (PFA). After the perfusion, the L4–6 segments of the spinal cord were removed, fixed in 4 % PFA for 12 h at 4 °C, and immersed in 30 % sucrose in phosphate buffer at 4 °C for cryoprotection. Transverse spinal sections (30 μm) were cut in a cryostat and processed for immunofluorescence. All of the sections were blocked with 10 % donkey serum in 0.01 M PBS (pH 7.4), with 0.3 % Triton X-100 for 2 h at room temperature, and incubated overnight at 4 °C with the following primary antibodies: rabbit anti-CX3CR1 (1:100), rabbit anti-phosphorylated-p38MAPK (1:50), mouse anti-NeuN (1:400), mouse anti-GFAP (1:400), goat anti-IBA1 (1:400). The sections were then washed three times with PBS and incubated with the specific secondary antibodies raised in donkey serum [conjugated to Alexa Fluor 488 (1:200) or 594 (1:200), Invitrogen, Carlsbad, CA, USA] for 2 h at room temperature. For double immunofluorescence staining, the sections were incubated with a mixture of anti-CX3CR1 (1:100) antibodies with the IBA-1, microglial marker (1:400), the GFAP, astrocytic marker (1:400), and the NeuN, neuronal marker (1:400) overnight at 4 °C. Double immunolabeling of p-p38MAPK/IBA1 sections was performed by incubation in a mixture of p-p38 MAPK (1:50) with the IBA1 (1:400) overnight at 4 °C, followed by incubation with a mixture of Alexa 594-conjugated rabbit IgG, Alexa 488-conjugated mouse IgG and Alexa 488-conjugated goat IgG (1:200; Invitrogen, Carlsbad, CA, USA). All sections were cover-slipped with a mixture of 90 % glycerin in 0.01 M PBS, followed by confocal microscopy (FV1000, Olympus Co. Ltd, Tokyo, Japan).
Western Blot
The L4–6 spinal cords from studied rats were isolated, and frozen in liquid nitrogen or stored at 80 °C for subsequent procedures. The tissues were homogenized in a lysis buffer (Bio-Rad Laboratories, Hercules, CA, USA) containing a cocktail of protease inhibitor and phosphatase inhibitors (Sigma Aldrich, St Louis, MI, USA). The homogenates were centrifuged at 12,000×g for 15 min at 4 °C. The supernatants were used as cytosolic proteins for Western blot. Equal amounts of protein (80 μg) were loaded in each lane and separated by 10 % sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE) and transferred onto a polyvinylidene fluoride membrane (PVDF, Millipore, Billerica, MA, USA). The membranes were blocked in 3 % bovine serum albumin (BSA) for 2 h at room temperature and rinsed three times in Washing Buffer for 5 min each, and incubated overnight at 4 °C with rabbit anti-CX3CR1 (1:500), rabbit anti-p-p38 MAPK (1:1000), mouse anti-β-actin (1:1000) antibody, respectively. They were further incubated with HRP-conjugated goat anti-rabbit IgG (1:1500) or goat anti-mouse secondary antibody IgG (1:1500), respectively, for 1 h at room temperature. The blots were rinsed in Washing Buffer four times for 8 min, followed by visualization using electrochemiluminescence (ECL) and exposure to X-ray film (Roche, Indianapolis, IN, USA) for 1–10 min. The scanned images were imported into Adobe Photoshop software (Adobe, CA, USA). Scanning densitometry was used for semiquantitative analysis of the data.
Experimental Groups
Part 1: The rats were randomized into three groups (n = 24): normal group (normal), saline group (saline) and nicotine withdrawal group (NT). Animal behavior was observed for 7 consecutive days after nicotine withdrawal. After behavioral assessments, from the first day of nicotine withdrawal, three rats in each group were sacrificed for 7 consecutive days, with the L4–6 spinal cords isolated for Western blot, while the other three rats in each group were sacrificed only on day 4 after nicotine withdrawal, followed by the removal of L4–6 spinal cords for immunofluorescence assay.
Part 2: To study the effect of intrathecal injection of anti-CX3CR1 neutralizing antibody on nicotine withdrawal-induced pain and p-p38MAPK expression, rats were divided into four groups (n = 12) as following: normal group (normal), nicotine withdrawal group (NT), nicotine withdrawal + anti-IgG antibody (for control) group (NT + control IG), and nicotine withdrawal + anti-CX3CR1group (NT + anti-CX3CR1). Behavioral assessment was performed for 7 consecutive days following nicotine withdrawal. On day 4 after behavioral evaluation, six rats in each group were sacrificed, and their L4–6 spinal cord were isolated for either Western blot or immunofluorescence analysis (n = 6 each).
Statistical Analysis
The data are expressed as the mean ± standard deviation (SD). Time courses measured for PWL was analyzed by two-way ANOVA with Bonferroni’s post hoc test. Western blot and immunofluorescence analysis were performed by one-way ANOVA, followed by Dunnett’s multiple comparison post hoc test. Data were analyzed using SPSS 16.0 (Chicago, IL, USA). P < 0.05 was considered statistically significant.
Results
Spinal CX3CR1 is Upregulated in Microglia After Nicotine Withdrawal
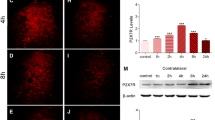
Western blot showed that the CX3CR1 expression was significantly increased in the spinal cord from day 1 to 7 after nicotine withdrawal (Fig. 1a), compared with that of normal and saline rats. The increase started on day 1, peaked on day 4. Our immunofluorescence result revealed that CX3CR1 was predominantly distributed in the superficial layers (laminae I-II), and the expression of CX3CR1 was increased in the nicotine withdrawal rats compared with normal and saline rats on day 4 (Fig. 1b).

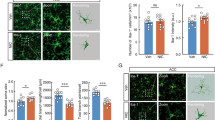
Expression levels of CX3CR1s were increased, and exclusively co-localized in microglia in the spinal cord. a Western blot using rabbit anti-CX3CR1 antibody (1:500) showed an increase in the level of CX3CR1 protein in the spinal cord at different times after nicotine withdrawal. The increase commenced on day 1 and peaked by day 4. The times course of change in CX3CR1s protein was similar to that of PWL (n = 3,*P < 0.05, **P < 0.01 vs normal group; # P < 0.05, ## P < 0.01 vs saline group). b There was a significant increase in the number of CX3CR1 immunoreactive cells in the dorsal horn of nicotine withdrawal rats compared with those in normal and saline rats on day 4 (n = 3, **P < 0.01 vs normal group; ## P < 0.01 vs saline group; Scale bar 50 μm). c Double staining shows that CX3CR1 (red) is co-localized with IBA1 (green), a marker for microglia, but not with NeuN (green), a marker for neurons, or GFAP (green), a marker for astrocytes. IBA1/CX3CR1 dual-labeled cells are shown in yellow. Scale bars 50 μm. The antibody concentrations for confocal imaging are as follows: rabbit anti-CX3CR1 (1:100), mouse anti-NeuN (1:400), mouse anti-GFAP (1:400), and goat anti-IBA1 (1:400)
To investigate the cell distribution of CX3CR1 in the dorsal horn following nicotine withdrawal, we performed double immunostaining of CX3CR1 with three major spinal nerve cell-specific markers, including NeuN (neuronal marker), GFAP (astrocyte marker), and IBA-1 (microglial marker). Confocal images identified that CX3CR1 was co-localized with IBA-1 only (Fig. 1c). These results demonstrated that the spinal CX3CR1 was upregulated after nicotine withdrawal, and was predominately expressed in activated microglia.
Anti-CX3CR1 Antibody Injection Attenuates Thermal Hyperalgesia Caused by Nicotine Withdrawal
We found that the animal models manifested significantly decreased PWL from day 1 to day 4 after nicotine withdrawal. PWL of animal models was significantly reduced at day 4 after nicotine withdrawal compared with that of normal rats (P < 0.01). To further confirm if the suppression of spinal CX3CR1 inhibited pain-related behavior following nicotine withdrawal, we intrathecally injected a CX3CR1-neutralizing antibody (10 μg/10 μl) or anti-IgG antibody (10 μg/10 μl) daily from day 1 to day 3 after nicotine withdrawal. Compared with NT + control IG group, NT rats treated with CX3CR1-neutralizing antibody, at a dose of 10 μg/10 μl, significantly attenuated nicotine withdrawal-induced pain-related behaviors simultaneously (Fig. 2). These results indicate that CX3CR1 may be involved in the maintenance of nicotine withdrawal-induced pain-related behaviors.

Intrathecal administration of anti-CX3CR1 neutralizing antibody (10 μg/10 μl) attenuated nicotine withdrawal-induced hyperalgesia. The PWL of nicotine withdrawal rats was increased after injection of CX3CR1 neutralizing antibody. Treatment of control IgG (10 μl) failed to alter PWL in nicotine withdrawal rats. Six rats were included in each group. *P < 0.05, **P < 0.01 versus NT + control IgG group. # P < 0.05, ## P < 0.01 versus normal group. Each arrow shows the corresponding time points of each administration
Intrathecal Injection of Anti-CX3CR1 Antibody Inhibits the Activation of Microglia in the Spinal Cord after Nicotine Withdrawal
On the day 4 after nicotine withdrawal, confocal images showed that IBA-1 immunoreactivity in the spinal cord was significantly increased in NT rats compared with that in normal controls (Fig. 3A-a, A-b). Activated microglial cells exhibited hypertrophic morphological changes such as cellular hypertrophy and retraction of cytoplasmic processes (Fig. 3A-b). After intrathecal injection of anti-CX3CR1 antibody, (10 μg/10 μl), on days 1, 2 and 3 after nicotine withdrawal, the number of IBA-limmunoreactive cells was dramatically decreased in NT + anti-CX3CR1 rats compared with NT rats that received anti-IgG on day 4 (Fig. 3A-c, A-d). The morphology of spinal microglia restored to baseline structure, showing small, compact somata bearing long, thin, and ramified processes (Fig. 3A-c).

Intrathecal injection of anti-CX3CR1 neutralizing antibody (10 μg/10 μl) suppressed the expression of IBA-1 (A). Immunofluorescence revealed an extensive decrease in microglial (IBA-1) immunoreacivity in the dorsal horn of NT rats by intrathecal anti-CX3CR1 neutralizing antibody (A-c). Treatment of control IgG failed to alter the expression of microglia (IBA-1) (A-d). In Normal and NT + anti-CX3CR1 rats, the morphology of spinal microglia displayed resting state features showing small, compact somata bearing long, thin, and ramified processes (A-a, A-c). However, in NT and NT + control IG rats, spinal microglia exhibited activated phenotype showing cellular hypertrophy and retraction of processes (A-b, A-d). B Quantification of IBA-1 immunoreactive cells in the dorsal horn (n = 3) using goat anti-IBA1 ab (1:400). # P < 0.05, ## P < 0.01 versus normal group; **P < 0.01 versus control IgG group. Scale bar 50 μm
Intrathecal Injection of Anti-CX3CR1 Antibody Suppresses p-p38 Expression in the Spinal Cord after Nicotine Withdrawal
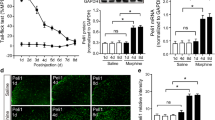
Our immunofluorescence results revealed that the number of p-p38 positive cells was increased in the dorsal horn of NT rats compared with that in normal rats on day 4. The number of p-p38 immunoreactive cells was significantly decreased by intrathecal treatment of neutralizing CX3CR1 antibody compared with that in control group by day 4 (Fig. 4a). The Western blot showed that the p-p38 was increased in NT rats compared with that in normal rats on day 4 after withdrawal. Following neutralizing CX3CR1 antibody injection from day 1 to day 3, the level of p-p38 protein expression was significantly reduced in the spinal cord of NT rats on day 4 (Fig. 4c). Furthermore, we also found that p-p38 (red) staining was co-localized with IBA1-positive cells and CX3CR1 (green) (Fig. 4e and f).

Expression levels of p-p38MAPK were inhibited by intrathecal injection anti-CX3CR1 neutralizing antibody (10 μg/10 μl). a Confocal imaging shows the expression of p-p38 immunoreactive cells was suppressed by intrathecal anti-CX3CR1 neutralizing antibody compared with the intrathecal injection of the control IgG (rabbit anti-p-p38MAPK (1:50). b Quantification of p-p38 immunoreative cells in the dorsal horn (n = 3). c Western blot shows a clear upregulation of p-p38 protein level in NT rats. Blockade of CX3CR1 with a neutralizing antibody (rabbit anti-p-p38MAPK (1:1000) reduced the p-p38 protein level in NT rats on day 4. d Quantification of p-p38 protein level in the spinal cord. p-p38 levels were normalized against β-actin (n = 3). e Double staining shows that p-p38 (red) co-localized with IBA1 (green), a marker for microglia. f Co-localization of the green signal (CX3CR1) and the red signal (p-p38). # P < 0.05, ## P < 0.01 versus normal group; **P < 0.01 versus control IgG group. Scale bar 50 μm. The antibody concentrations for confocal imaging are as follows: rabbit anti-p-p38 MAPK (1:50) and goat anti-IBA1 (1:400)
Discussion
The current study demonstrates that (a) CX3CR1 is upregulated in microglia of the spinal cord from NT rats after nicotine withdrawal; (b) Intrathecal injection of anti-CX3CR1 neutralizing antibody significantly attenuated nicotine withdrawal-induced nociceptive behaviors; (c) Blockade of CX3CR1 suppressed nicotine withdrawal-induced spinal microglial activation as well as p-p38 MAPK expression in the spinal cord. The findings confirm that neurophysiological mechanisms modulate the pain associated with nicotine withdrawal.
Over 4000 different chemical substances have been identified in tobacco. Animal experiments and clinical studies suggested that nicotine is the main component involved in pain modulation [17, 18]. Nicotine is capable of modulating the activity of sensory neurons and the transmitter substances from sensory neurons [19]. Previous studies suggested that nicotine reduced the number of in vitro cultured microglia [20, 21]. Nicotine plays a robust anti-inflammatory role through alpha-7-nicotinic acetylcholine receptors (α7nAchR) in microglia and provides significant neuroprotection [21]. Our results indicated that nicotine withdrawal was associated with nociceptive hypersensitivity and the activated microglia cells in the spinal cord of rats were significantly increased in number. Clinically, various types of chronic pain, including back pain, carpal tunnel syndrome and complex regional pain syndrome, are worsened in chronic smokers [22]. After smoking cessation, the postoperative pain in these patients is more severe compared with non-smokers. Subsequently, they need more painkillers for pain management. The reason for the discrepancy in these results is unclear. It is plausible that this result is associated with the anti-nociceptive effect of nicotine. However, long-term exposure to nicotine induces upregulation and inactivation of nAChRs, resulting in a decreased release of inhibitory neurotransmitters. Therefore, pain sensitivity is increased in long-term smokers who quit smoking.
Our results indicate that CX3CR1 is upregulated after nicotine withdrawal, which is consistent with previous studies involving other models, including neuropathic and inflammatory pain. In the present study, Western blot data demonstrated that CX3CR1 protein expression was gradually increased from day 1 to day 4 in the spinal cord following nicotine withdrawal and peaked on day 4. The time course of the CX3CR1 upregulation in the spinal cord is consistent with the development of hyperalgesia behavioral response. PWL was significantly reduced on day 4 after nicotine withdrawal. In addition, our study demonstrated that CX3CR1 was expressed in spinal microglia, and that the number of CX3CR1-positive cells was increased in the NT rats. Blockade of CX3CR1 significantly attenuated the existing pain hypersensitivity induced by nicotine withdrawal. These data suggest that CX3CR1 upregulation may be involved in the maintenance of nicotine withdrawal induced pain-related behaviors.
Microglia are the major immune cells in the central nervous system. Accumulating evidence suggests that microglial cells in the spinal cord play a crucial role in facilitating pain states [23]. Activated microglia release a variety of mediators, such as proinflammatory cytokines and the neuronal chemokine FKN. FKN acts on microglial CX3CR1 receptors, further leading to the activation of microglia in the spinal dorsal horn. Such reciprocal interactions between glia and neurons contribute to the activation of a neuro–glial amplification loop, leading to central sensitization and exaggeration of pain signals [24]. According to several studies using somatosensory behavioral tests, the intrathecal injection of FKN evokes dose-dependent thermal hyperalgesia in naïve rats [23]. In the spinal cord, concomitant with microglial activation, CX3CR1 expression is up-regulated after chronic constriction injury to the sciatic nerve [7]. Recently, FKN administration has been shown to increase abdominal electromyographic responses to noxious colorectal distension in control rats, and this hypersensitivity was further prevented by intrathecal injection of minocycline [24]. Thus, it is plausible that blocking CX3CR1 can inhibit microglia activation, and reduce the release of neuronal and glial-excitatory substances, and eventually, attenuate nociceptive hypersensitivity of the rats after nicotine withdrawal. Intrathecal injection of FKN leads to pain hypersensitivity in naïve rats, which can be reversed by intrathecal injection of an antibody blocking either CX3CR1 or FKN [7, 25], and CX3CR1 knock-out mice fail to develop nociception induced by FKN [7]. These results showed that FKN induces pain hypersensitivity by acting on its receptor CX3CR1. On the contrary, Holmes et al. [26] reported that intra-neural injection of FKN into the sciatic nerve of mice robustly delayed the development of allodynia following spared nerve injury (SNI). The CX3CR1 knockout mice displayed increased allodynia after SNI. The reason for these different results is unclear. It is possible that differences in strains and sites of FKN action might contribute to the results obtained.
It has been reported that p38 is activated in spinal microglia under different pain conditions. Importantly, p38 activation also contributes to the development of pain hypersensitivity [27]. Several studies explored the upstream mechanisms associated with p38 activation in spinal microglia. Although microglial receptor TLR4 (toll-like receptor 4) was reported to be one of the cellular events upstream of p38 MAPK, it is unlikely that TLR4 was the only microglial receptor associated with p38 MAPK activation [28]. Remarkably, a recent study has established that intrathecal infusion of FKN not only induces hyperalgesia behavior but also activates p38 in spinal microglia. FKN activated p38MAPK in the microglia of spinal cord samples [28]. Conversely, a neutralizing antibody against CX3CR1 reduces both p38 activation and nociceptive hypersensitivity [28]. In the present study, we observed a similar effect of intrathecal administration of anti-CX3CR1- neutralizing antibody which suppressed p-p38 MAPK activation in nicotine withdrawal rats. These findings suggest that FKN-CX3CR1-p38 cascade mediates the development of hyperalgesia after nicotine withdrawal.
Conclusions
We have provided novel evidence that spinal CX3CR1 may play an important role in the maintenance of nicotine withdrawal-induced pain-related behaviors. We observed an increase of nociceptive sensitivity, CX3CR1 protein expression and immunoreactivity, microglia and p-p38 levels in spinal cord sections from rats following nicotine withdrawal. Intrathecal injection of anti-CX3CR1 antibody attenuates thermal hyperalgesia induced by nicotine withdrawal, possibly through a p-p38 pathway. CX3CR1 and p-p38 were co-localized with the microglial marker IBA-1, which suggests that the activated microglia paralleled the activation of the CX3CR1/p38 signaling pathway. These findings open new perspectives in the understanding of the mechanisms underlying hyperalgesia associated with nicotine withdrawal in rats. Thus, the blockade of this chemokine signaling in the spinal cord may play a vital role in the management of pain hypersensitivity following nicotine withdrawal.
Abbreviations
- NT:
-
Nicotine withdrawal
- CX3CR1:
-
Chemokine CX3C motif receptor
- SD:
-
Sprague–Dawley
- p-p38-MAPK:
-
Phosphorylation of p38-mitogen-activated protein kinase
- CPT:
-
Cold pressor test
- PKA:
-
Protein kinase A
- PKC:
-
Protein kinase C
- CaMKII:
-
Calcium/calmodulin-dependent protein kinase II
- p38 MAPK:
-
p38-mitogen-activated protein kinase
- IBA1:
-
Ionized calcium binding adapter molecule-1
- NeuN:
-
Neuronal nucle
- GFAP:
-
Glial fibrillary acidic protein
- PWL:
-
Paw withdrawal latency
- PBS:
-
Phosphate-buffered saline
- PFA:
-
Paraformaldehyde
- PVDF:
-
Polyvinylidene fluoride membrane
- SNI:
-
Spared nerve injury
- FKN:
-
Fractalkine
- TLR4:
-
Toll-like receptor 4
References
Baiamonte BA, Valenza M, Roltsch EA, Whitaker AM, Baynes BB, Sabino V, Gilpin NW (2014) Nicotine dependence produces hyperalgesia: role of corticotropin-releasing factor-1 receptors (CRF1Rs) in the central amygdala (CeA). Neuropharmacology 77:217–223. doi:10.1016/j.neuropharm.2013.09.025
Shi Y, Weingarten TN, Mantilla CB, Hooten WM, Warner DO (2010) Smoking and pain: pathophysiology and clinical implications. Anesthesiology 113(4):977–992. doi:10.1097/ALN.0b013e3181ebdaf9
Liu Z, Liu XW, Lu SF, Yu AL, Zhang ZW (2014) Effect of nicotine withdrawal on pain sensitivity in rats to mechanical stimulation and thermal stimulation. Eur Rev Med Pharmacol Sci 18(18):2759–2765
Nakajima M, Al’Absi M (2014) Nicotine withdrawal and stress-induced changes in pain sensitivity: a cross-sectional investigation between abstinent smokers and nonsmokers. Psychophysiology 51(10):1015–1022. doi:10.1111/psyp.12241
Bazan JF, Bacon KB, Hardiman G, Wang W, Soo K, Rossi D, Greaves DR, Zlotnik A, Schall TJ (1997) A new class of membrane-bound chemokine with a CX3C motif. Nature 385(6617):640–644. doi:10.1038/385640a0
Lindia JA, McGowan E, Jochnowitz N, Abbadie C (2005) Induction of CX3CL1 expression in astrocytes and CX3CR1 in microglia in the spinal cord of a rat model of neuropathic pain. J Pain 6(7):434–438. doi:10.1016/j.jpain.2005.02.001
Clark AK, Yip PK, Grist J, Gentry C, Staniland AA, Marchand F, Dehvari M, Wotherspoon G, Winter J, Ullah J, Bevan S, Malcangio M (2007) Inhibition of spinal microglial cathepsin S for the reversal of neuropathic pain. Proc Natl Acad Sci USA 104(25):10655–10660. doi:10.1073/pnas.0610811104
O’Callaghan JP, Miller DB (2010) Spinal glia and chronic pain. Metabolism 59(Suppl 1):S21–S26. doi:10.1016/j.metabol.2010.07.011
Rostene W, Kitabgi P, Parsadaniantz SM (2007) Chemokines: A new class of neuromodulator? Nat Rev Neurosci 8(11):895–903. doi:10.1038/nrn2255
Cao H, Zhang YQ (2008) Spinal glial activation contributes to pathological pain states. Neurosci Biobehav Rev 32(5):972–983. doi:10.1016/j.neubiorev.2008.03.009
Zimmermann M (2001) Pathobiology of neuropathic pain. Eur J Pharmacol 429(1–3):23–37
Fang L, Wu J, Lin Q, Willis WD (2002) Calcium-calmodulin-dependent protein kinase II contributes to spinal cord central sensitization. J Neurosci 22(10):4196–4204
Zhang FE, Cao JL, Zhang LC, Zeng YM (2005) Activation of p38 mitogen-activated protein kinase in spinal cord contributes to chronic constriction injury-induced neuropathic pain. Acta Physiol Sin 57(5):545–551
Xu JJ, Walla BC, Diaz MF, Fuller GN, Gutstein HB (2006) Intermittent lumbar puncture in rats: a novel method for the experimental study of opioid tolerance. Anesth Analg 103(3):714–720. doi:10.1213/01.ane.0000226100.46866.ea
Yamamoto T, Yaksh TL (1992) Comparison of the antinociceptive effects of pre- and posttreatment with intrathecal morphine and MK801, an NMDA antagonist, on the formalin test in the rat. Anesthesiology 77(4):757–763
Hargreaves K, Dubner R, Brown F, Flores C, Joris J (1988) A new and sensitive method for measuring thermal nociception in cutaneous hyperalgesia. Pain 32(1):77–88
Flood P, Daniel D (2004) Intranasal nicotine for postoperative pain treatment. Anesthesiology 101(6):1417–1421
Simons CT, Cuellar JM, Moore JA, Pinkerton KE, Uyeminami D, Carstens MI, Carstens E (2005) Nicotinic receptor involvement in antinociception induced by exposure to cigarette smoke. Neurosci Lett 389(2):71–76. doi:10.1016/j.neulet.2005.07.025
Dussor GO, Leong AS, Gracia NB, Kilo S, Price TJ, Hargreaves KM, Flores CM (2003) Potentiation of evoked calcitonin gene-related peptide release from oral mucosa: a potential basis for the pro-inflammatory effects of nicotine. Eur J Neurosci 18(9):2515–2526
Lutz JA, Kulshrestha M, Rogers DT, Littleton JM (2014) A nicotinic receptor-mediated anti-inflammatory effect of the flavonoid rhamnetin in BV2 microglia. Fitoterapia 98:11–21. doi:10.1016/j.fitote.2014.06.012
Guan YZ, Jin XD, Guan LX, Yan HC, Wang P, Gong Z, Li SJ, Cao X, Xing YL, Gao TM (2014) Nicotine inhibits microglial proliferation and is neuroprotective in global ischemia rats. Mol Neurobiol. doi:10.1007/s12035-014-8825-3
Hsu C, Harden RN, Houle T (2002) Nicotine and caffeine intake in complex regional pain syndrome. J Back Musculoskelet Rehabil 16(1):33–38
Liu PY, Lu CL, Wang CC, Lee IH, Hsieh JC, Chen CC, Lee HF, Lin HC, Chang FY, Lee SD (2012) Spinal microglia initiate and maintain hyperalgesia in a rat model of chronic pancreatitis. Gastroenterology 142(1):165–173. doi:10.1053/j.gastro.2011.09.041 (e162)
Clark AK, Malcangio M (2014) Fractalkine/CX3CR1 signaling during neuropathic pain. Front Cell Neurosci 8:121. doi:10.3389/fncel.2014.00121
Sun S, Cao H, Han M, Li TT, Pan HL, Zhao ZQ, Zhang YQ (2007) New evidence for the involvement of spinal fractalkine receptor in pain facilitation and spinal glial activation in rat model of monoarthritis. Pain 129(1–2):64–75. doi:10.1016/j.pain.2006.09.035
Holmes FE, Arnott N, Vanderplank P, Kerr NC, Longbrake EE, Popovich PG, Imai T, Combadiere C, Murphy PM, Wynick D (2008) Intra-neural administration of fractalkine attenuates neuropathic pain-related behaviour. J Neurochem 106(2):640–649. doi:10.1111/j.1471-4159.2008.05419.x
Ji RR, Suter MR (2007) p38 MAPK, microglial signaling, and neuropathic pain. Mol Pain 3:33. doi:10.1186/1744-8069-3-33
Hu JH, Yang JP, Liu L, Li CF, Wang LN, Ji FH, Cheng H (2012) Involvement of CX3CR1 in bone cancer pain through the activation of microglia p38 MAPK pathway in the spinal cord. Brain Res 1465:1–9. doi:10.1016/j.brainres.2012.05.020
Acknowledgments
This work was supported by grants from National Natural Science of China (81471134). Our great thanks also go to the critical scientific opinions of Prof. Li-Cai Zhang from Xuzhou Medical College.
Authors’ Contributions
ZWZ conceived and designed the study. YHD performed the animal surgery, behavioral testing and data analysis. WHS carried out the immunohistochemistry and Western blot. GNX, ALY and QHW participated in behavioral testing and immunohistochemistry experiments. All authors read and approved the final manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors report no financial or other conflict of interest relevant to the subject of this manuscript.
Rights and permissions
About this article

Cite this article
Ding, Y., Shi, W., Xie, G. et al. CX3CR1 Mediates Nicotine Withdrawal-Induced Hyperalgesia via Microglial P38 MAPK Signaling. Neurochem Res 40, 2252–2261 (2015). https://doi.org/10.1007/s11064-015-1715-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11064-015-1715-x